

Abstract
Many factors contribute to nervous system dysfunction and failure to regenerate after injury or disease. Here, we describe a previously unrecognized mechanism for nervous system injury. We show that neuronal injury causes rapid, irreversible, and preferential proteolysis of the axon initial segment (AIS) cytoskeleton independently of cell death or axon degeneration, leading to loss of both ion channel clusters and neuronal polarity. Furthermore, we show this is caused by proteolysis of the AIS cytoskeletal proteins ankyrinG and βIV spectrin by the calcium-dependent cysteine protease calpain. Importantly, calpain inhibition is sufficient to preserve the molecular organization of the AIS both in vitro and in vivo. We conclude that loss of AIS ion channel clusters and neuronal polarity are important contributors to neuronal dysfunction after injury, and that strategies to facilitate recovery must preserve or repair the AIS cytoskeleton.
Introduction
Factors contributing to nervous system dysfunction after injury or disease include glial scarring, axon degeneration, demyelination, axon outgrowth inhibitors, and neuronal cell death (Yiu and He, 2003; Benn and Woolf, 2004; Franklin and ffrench-Constant, 2008). Proteolysis of the axonal cytoskeletal proteins αII spectrin, βII spectrin, and ankyrinB by calpain, a calcium-dependent cysteine protease, has been proposed to be a major contributor to axon degeneration (Harada et al., 1997; Yoshida and Harada, 1997; White et al., 2000; Czogalla and Sikorski, 2005; Bevers and Neumar, 2008). Indeed, αII spectrin breakdown products are biomarkers for nervous system injury (Pike et al., 2004; Pineda et al., 2007). Although these cytoskeletal components are localized throughout the axon, there is a unique cytoskeleton associated with the most proximal segment of the axon (i.e., the axon initial segment or AIS) comprised of βIV spectrin and ankyrinG (ankG) (Kordeli et al., 1995; Berghs et al., 2000). However, the consequence of nervous system injury for this specialized cytoskeleton has not been explored.
The AIS functions as both a physiological and physical bridge between axonal and somatodendritic domains. It is responsible for the final integration of synaptic inputs and initiation and modulation of the action potential (AP) due to the high density of voltage-gated Na+ (Nav) and K+ (Kv) channels (Khaliq and Raman, 2006; Palmer and Stuart, 2006; Meeks and Mennerick, 2007; Goldberg et al., 2008; Kole et al., 2008; Shah et al., 2008). These ion channels are restricted to the AIS through interactions with ankG, which links AIS membrane proteins to the underlying βIV spectrin scaffold. βIV spectrin tethers ankG to the plasma membrane and actin cytoskeleton (Garrido et al., 2003; Pan et al., 2006; Dzhashiashvili et al., 2007; Hedstrom et al., 2007; Yang et al., 2007). Mice lacking ankG in Purkinje neurons fail to cluster AIS ion channels, develop ataxia, and fail to initiate APs (Zhou et al., 1998; Jenkins and Bennett, 2001; Pan et al., 2006). Similarly, βIV spectrin mutant mice develop ataxia, tremors, and deafness (Komada and Soriano, 2002; Lacas-Gervais et al., 2004; Yang et al., 2004). These behavioral deficits are coincident with progressive loss of AIS Nav channel immunoreactivity (Uemoto et al., 2007; Yang et al., 2007).
Morphologically the AIS is the transition zone from the soma to the axon compartments, and the AIS cytoskeleton functions to maintain neuronal polarity and proper axonal trafficking (Winckler et al., 1999; Song et al., 2009). For example, loss of ankG causes axons to acquire molecular and structural characteristics of dendrites including spines (Hedstrom et al., 2008). Together, these observations emphasize the importance of the AIS cytoskeleton in normal nervous system function.
Since genetic loss of the AIS cytoskeleton disrupts neuronal function and organization, we considered whether nervous system injury also disrupts this essential axonal domain. Using in vitro and in vivo injury models, we elucidate a new mechanism for neuronal injury: rapid, preferential, and irreversible proteolysis of the AIS cytoskeleton by calpain, resulting in disrupted neuronal polarity and loss of the ion channel clusters necessary for AP initiation.
Materials and Methods
Animals.
Rats and mice were housed at the University of Connecticut Health Center and Baylor College of Medicine. All experiments were approved by institutional animal care and use committees and were performed in accordance with all National Institutes of Health guidelines for the humane treatment of animals.
Antibodies.
The polyclonal βIV spectrin antibody was previously described (Ogawa et al., 2006). The polyclonal ankG antibody was kindly provided by Dr. V. Bennett (Duke University, Durham, NC), and mouse monoclonal anti-ankG was purchased from Zymed Laboratories. Mouse monoclonal pan-neurofascin and Nav channel antibodies were previously described (Schafer et al., 2004; Hedstrom et al., 2008). The chicken polyclonal MAP2 antibody was purchased from EnCor Biotechnology. The mouse monoclonal αII spectrin antibody was purchased from Millipore Bioscience Research Reagents. The mouse monoclonal β-actin and GFAP antibodies were purchased from Sigma. The mouse monoclonal NeuN and calpastatin antibodies were purchased from Millipore. All fluorescent secondary antibodies were purchased from Invitrogen except for AMCA-conjugated anti-chicken antibody (Accurate Chemical).
Tissue culture.
Primary cortical neurons were prepared from E18 rat embryos. Embryonic cortices were dissected and collected in HBSS (Invitrogen) followed by a 30 min trypsinization (0.25%) at 37°C. Cells were collected by centrifugation (600 × g for 3 min), resuspended in plating media [Neurobasal (Invitrogen) supplemented with 10% FBS (Mediatech)], and triturated with a fire-polished Pasteur pipette. The cell suspension was left to settle for 3 min, and the subsequent supernatant was filtered through a 70 μm cell strainer (BD Biosciences). Cells were then plated on cover glass coated with 1 mg/ml poly-l-lysine (Sigma) and 10 μg/ml laminin (Invitrogen) at 1 × 106 cells/35 mm dish. Neurons were incubated in a humidified 5% CO2 incubator at 37°C. After 2–3 h, the media was exchanged to maintenance media (Neurobasal, 2% B27 (Invitrogen), 0.5 mm l-glutamine; 1× Pen-Strep) with addition of 25 μm l-glutamate. On day in vitro 4 (DIV4), the media was replaced with maintenance media without l-glutamate. Cells were fed every 3 d by replacing half the media with fresh maintenance media. In some cases, primary cortical neurons were transfected with membrane-bound green fluorescent protein (GFP) by LipoFectamine 2000 (Invitrogen) as previously described for primary hippocampal neurons (Hedstrom et al., 2007).
Oxygen-glucose deprivation.
Deprivation experiments were performed as previously described with minor modifications (Uliasz and Hewett, 2000). Primary cortical neurons (DIV10) were transferred to an anaerobic chamber (Forma Scientific, Marietta, OH) containing a gas mixture of 5% CO2, 10% H2, 85% N2 (≤0.2% O2). Once in the chamber, half the media was taken out of each well and stored for future use (conditioned media). The remaining media was aspirated and replaced by washing cultures three times with deoxygenated, glucose-free balanced salt solution [BSS0: 116 mm NaCl, 5.4 mm KCl, 0.8 mm MgSO4 1 mm NaH2PO4, 26.2 mm NaHCO3, 1.8 mm CaCl2, 0.01 mm glycine, 2 mm glutamax, and 1× MEM amino acids (Invitrogen)]. Cultures were then placed in a 37°C incubator within the chamber for up to 3 h. Control cultures were washed 3 times in BSS containing 20 mm glucose (BSS20) and maintained at 37°C in normoxic conditions for up to 3 h. Cultures were subsequently removed from the chamber and media was replaced with half conditioned media and half fresh maintenance media for up to 72 h.
For pharmacological experiments, the following compounds were used: MDL 28170 (MDL; Calbiochem), CA-074 (CA; Calbiochem), MK-801 (Sigma). Stock solutions were made in either DMSO (MDL and CA) or water (MK-801). These stock solutions were diluted into BSS and then added to cultures after the third wash (MDL: 0–100 μm; CA: 0–100 μm; MK-801 10 μm). Following deprivation, fresh drug was added to the maintenance media at the appropriate concentration. Vehicle controls (0 μm drug; DMSO or water) were included for each experiment.
Calpastatin overexpression.
Recombinant adenovirus containing calpastatin and/or EGFP were gifts from D.S. Park (Sedarous et al., 2003). Primary cortical neurons (DIV7) were infected for 6 h in maintenance media (multiplicity of infection = 20). Cultures were washed once and left in maintenance media until deprivation experiments (DIV10). Infection efficiency was quantified in sister cultures by counting the number of gfp-positive cells versus total Hoechst-positive nuclei. Cell death and immunofluorescence were quantified as described below.
In vitro cell death assay and quantification. Cell death was analyzed in vitro using propidium iodide (PI; Invitrogen). PI was added to cultures 24 h after deprivation at 10 μg/ml. Cells were incubated at 37°C for 10 min followed by fixation in ice-cold 1% or 4% PFA for 30 min followed by subsequent Hoechst labeling (Invitrogen) and immunostaining (see below). For the majority of the quantification, three 10× fields of view were collected per coverslip (2–3 coverslips per experiment). In some cases, PI was quantified using ten 40× fields of view. For each field, the total numbers of PI and Hoechst-positive cells were counted using ImageJ software (National Institutes of Health). For each coverslip, the number of PI-positive cells in each view was pooled and expressed as a percentage of Hoechst-positive cells.
Immunofluorescence and quantification.
Cultured neurons were fixed in 1% (ankG or Nav channel staining) or 4% PFA (all other staining) and immunolabeled as previously described (Hedstrom et al., 2007). For quantification of in vitro immunofluorescence, the same fields of view used for PI quantification (see above) were used for quantifying AIS immunofluorescence (βIV spectrin, ankG, pan-Nav, or pan-neurofascin). An AIS was defined as a fluorescent segment of at least 10 μm in length and is expressed as a percentage of total Hoechst-positive cells. In some cases in which immunofluorescence was faint (pan-Nav and pan-neurofascin), AIS and Hoechst-positive cells were quantified using ten 40× fields of view per coverslip.
For in vivo experiments, animals were killed and brains were rapidly dissected and flash frozen in 2-methylbutane pre-cooled over dry ice. Brains were stored at −80°C until sectioning. Brains were sectioned using a cryostat (15 μm) and mounted on glass slides. Subsequently, sections were fixed with 1% (ankG or Nav channel staining) or 4% PFA. Immunolabeling was performed as previously described (Schafer et al., 2004). In a few cases, immunostaining was performed using brains from perfused animals. Briefly, anesthetized animals were perfused with 4% PFA. Brains were dissected and post-fixed in 4% PFA for 4 h followed by equilibration in 20% sucrose, sectioning, and immunolabeling. In vivo immunofluorescence was quantified using five 20× fields collected for each ipsilateral and contralateral cortex of each animal. In some cases tissue sections were labeled using fluorescent NeuroTrace (Invitrogen). The number of βIV spectrin-positive AIS, NeuN-positive cells, and Hoechst-positive cells were counted for each field. A βIV spectrin-positive AIS was defined as a fluorescent segment of at least 10 μm in length. Both βIV spectrin and NeuN staining were expressed as a percentage of total Hoechst-positive cells. Data are representative of 4 animals per condition. For staining of retinas, tissue from rats experiencing optic nerve crush was kindly provided by Drs. Yang Yang and Larry Benowitz (Children's Hospital, Boston, MA).
Calpain cleavage assay.
Rat or mouse brain homogenates were prepared from freshly dissected tissue. Brains were homogenized in ice-cold buffer containing 0.32 m sucrose and 10 mm Tris-HCl (pH 7.5) (10 ml/gm wet brain weight). Crude brain homogenates were subsequently centrifuged to remove debris and nuclei (600 × g for 10 min). The resulting supernatant was used for protein estimation using the BCA method (Pierce Biotechnology). For the cleavage assay, equal volumes of supernatant (8–10 mg/ml) were aliquoted into separate tubes for each condition. To each supernatant an equal volume of cleavage buffer [final concentration: 0–10 mm CaCl2, 20 mm Tris-HCl pH 7.5, 25 mm NaCl, 1 mm DTT] was added with or without protease inhibitors [Final Concentration: 0–100 μm MDL; 0–10 μm calpastatin (Calbiochem); 0–100 μm calpeptin (Calbiochem); 0–100 μm ALLN (Calbiochem);0–100 μm CA; 0–100 μm GM-6001 (Calbiochem); 0–100 μm Q-VD-OPH (MP Biomedicals)]. The solution was incubated at 37°C for 30 min. The reaction was quenched with an equal volume of 2× reducing sample buffer and Western blot analysis was performed as previously described (Schafer et al., 2004).
Middle cerebral artery occlusion.
Surgeries were performed as previously described (Ardelt et al., 2005; McCullough et al., 2005). Briefly, the middle cerebral artery was occluded in male C57BL/6 mice or Sprague Dawley rats (Charles River Laboratory). The animals were awakened from anesthesia to confirm neurological deficits associated with occlusion. After 90 min, the animals were briefly re-anesthetized for removal of the suture. Animals were killed for analyses at various times post-occlusion (3–72 h from the onset of 90 min occlusion). Separate non-survival cohorts were performed to confirm equivalency of blood flow reduction (laser Doppler) and to ensure physiological variables (pO2/pCO2/blood pressure) were equivalent between groups. During all surgical procedures, animals were maintained at 37°C. Immunofluorescence was performed as described above (see Immunofluorescence and Quantification). Infarction was confirmed by staining brain sections with Fluoro-Jade B (Millipore Bioscience Research Reagents), cresyl violet (Sigma), and/or anti-NeuN (Davoli et al., 2002; Türeyen et al., 2004; Duckworth et al., 2005).
For Western blot analysis, brains were rapidly dissected and flash frozen in 2-methylbutane pre-cooled over dry-ice. Brains were stored at −80°C until homogenization. Immediately before homogenization, brains were divided into contralateral and ipsilateral hemispheres. Each hemisphere was homogenized separately in ice-cold 0.32 m sucrose and 5 mm sodium phosphate (pH 7.4) containing 10 mm EDTA, 0.5 mm sodium phenylmeythylsufonyl fluoride, 2 μg/ml aprotinin, 1 μg/ml leupeptin, 2 μg/ml antipain, and 10 μg/ml benzamidine (10 ml/mg wet brain weight). Crude hemisphere homogenates were centrifuged to remove debris and nuclei (600 × g for 10 min). To ensure equal protein loading, protein concentrations were estimated from the resulting supernatant and resolving sample buffer was added to a final concentration of 2 mg/ml. Samples were boiled (5 min) or heated at 37°C (15 min) and subjected to Western blot analysis as previously described (McCullough et al., 2005).
In some cases, rats subjected to MCAO received MDL, MK-801, or vehicle. MDL or vehicle [PEG 300/EtOH (9:1)] was administered IV as previously described (Markgraf et al., 1998). Briefly, administration began at the time of reperfusion with a 20 mg/kg bolus (1 ml final volume) via an indwelling catheter. After which time, the catheter was connected to an infusion pump (Kent Scientific) and animals were infused for an additional 6 h (3.33 mg/kg/h, total volume 5 ml). Animals received MK-801 (1 mg/kg) or vehicle (phosphate buffered saline; PBS) IP 30 min before the onset of occlusion (Gerriets et al., 2003). After the first dose, the animals received a dose of MK-801 (1 mg/kg) or vehicle at 3 and 6 h after reperfusion. All animals were killed 24 h after occlusion for immunofluorescence, Western blot analysis, and/or infarct assessment.
Oxygen-glucose deprivation and live labeling of the AIS.
Primary hippocampal neurons were obtained from E18 timed pregnant Sprague Dawley rat pups. For this, dissected hippocampi were trypsinized (0.25%) in Hank's buffered salt solution (HBSS) (Invitrogen) for 15 min at 37°C, triturated, and centrifuged at 800 × g for 4 min. Cells were then resuspended in maintenance media (Neurobasal with 2% B-27 supplement and 2 mm Glutamax) and plated on photo-etched glass coverslips (23 mm, square; Electron Microscopy Sciences) at 1–5 × 105 cells/well.
For live labeling of the AIS, a mouse monoclonal antibody [A12/18; (Hedstrom et al., 2008)] recognizing an extracellular epitope of neurofascin was used in all the experiments. Conditioned media was removed from DIV 9–10 hippocampal neurons and stored for later use. After this, cells were washed twice in HBSS and incubated with primary antibody (1:200 dilution, 30 min, 37°C) in fresh maintenance media. This was followed by 3 washes in HBSS and incubation with secondary antibody (1:500, 30 min, 37°C). After final washes in HBSS, fresh and conditioned media was added to the cells in a 1:1 ratio. Images of immunostained neurons (red fluorescence) were acquired on a Zeiss Imager Z1 microscope. Etched grid numbers on the coverslips allowed for tracking of the same neurons after OGD induced injury. OGD was carried out on DIV 10 neurons as described previously. Modifications included use of modular incubation chamber (Billups-Rothenberg) and use of Neurobasal-A (Invitrogen) instead of glucose free-BSS. The neurons were fixed and immunostained for AIS (βIV spectrin) and dendritic (MAP2) markers 8–10 d after OGD.
Photolytic calcium uncaging and imaging of the AIS.
For local calcium uncaging, a cell permeant acetoxymethyl (AM) ester derivative of the caged compound o-nitrophenyl EGTA (NP-EGTA AM, Invitrogen) was used at a final concentration of 25 μm. The change in intracellular calcium concentration was documented using the cell permeant AM ester of calcium green-1 (CG-1 AM, 10 μm, Invitrogen). First, neurons were live labeled with the AIS marker A12/18 as described above. This was followed by a simultaneous incubation with NP-EGTA AM and CG-1 AM (45 min at room temperature) in the dark. For this, HBSS containing 1.2 mm CaCl2 was used as the loading solution. Also, Pluronic F-127 (20% w/v, Invitrogen) was added to the loading solution for better cellular uptake of these compounds (final concentration <0.02%). Stock solutions (5 mm) of NP-EGTA AM and CG-1 AM were prepared in DMSO. After loading, the neurons were washed and replenished with fresh maintenance media.
For photolysis and subsequent imaging of neurofascin, the AIS was first identified under low magnification (20×) using Alexa-594 secondary antibodies. A pinhole diaphragm was then closed so that the light cone of the UV lamp was focused only onto the AIS at higher magnification (40×). Calcium was then photolytically uncaged by 2 consecutive 500 ms UV flashes. Fluorescence images of the labeled AIS and intracellular calcium were acquired immediately before and after photolysis and subsequently at 5 min intervals. To measure changes in fluorescence, we calculated the ratio of the average fluorescence intensity for 6 control and 6 uncaged AIS at 30 min after uncaging to the fluorescence intensity immediately before uncaging at t = 0.
Microscopy and image analysis.
Staining was visualized and images were collected using Axiovert 200M, Axio-observer Z1, and Axio-imager Z1 microscopes (Carl Zeiss MicroImaging) fitted with AxioCam digital cameras (Carl Zeiss MicroImaging). Images were taken using 10× (0.45 NA), 20× (0.8 NA), 40× (1.0 NA), 40× (0.75 NA), or 63× (1.4 NA) objectives. AxioVision (Carl Zeiss MicroImaging) acquisition software was used for collection of images. Measurement of fluorescence intensity was performed using ImageJ (NIH). In some images, contrast and brightness were subsequently adjusted using Photoshop (Adobe). No other processing of the images was performed. All figures were assembled using CorelDraw.
Statistics.
Before statistical analyses, all percentage data were transformed as previously described (Uliasz and Hewett, 2000). A one-way ANOVA was performed. If p < 0.05, subsequent post hoc analyses were performed (Newman–Keuls). All statistical analyses and graphs were compiled using GraphPad Prism software.
Results
Neuronal injury disrupts the AIS
To determine whether the AIS is disrupted following injury, we subjected mice to ischemic injury by middle cerebral artery occlusion (MCAO). The MCA was occluded using a nylon suture as previously described (McCullough et al., 2005). After 90 min of occlusion, the block was removed to allow blood to reperfuse the affected hemisphere. Animals were killed at various times after the onset of MCAO (time post-occlusion) for immunohistochemistry and immunoblot analyses. In the uninjured (contralateral) side of the cerebral cortex neurons were identified as brightly stained Nissl-positive cells (Fig. 1 A, red), most of which were associated with robust βIV spectrin immunoreactivity at the AIS (Fig. 1 A, green; inset and arrow). In contrast, in the injured (ipsilateral) cortex there was remarkable loss of βIV spectrin staining at the AIS within 6 h after onset of occlusion. Further, there was an abrupt transition from neurons with disrupted initial segments to those with preserved βIV spectrin staining (Fig. 1 B, dashed line). Similar to βIV spectrin, ankG and Nav channel immunoreactivity at the AIS were also undetectable in the injured cortex (Fig. 2). Thus, one consequence of ischemic neuronal injury is the rapid disruption of the molecular organization of the AIS.
Figure 1.
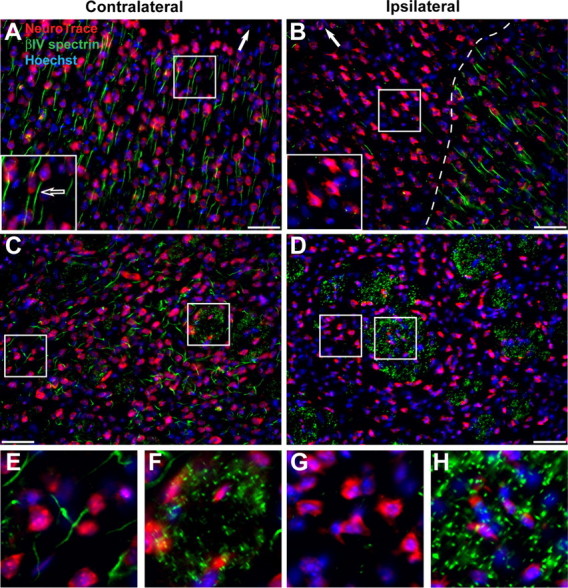
Ischemic injury causes rapid disruption of the AIS cytoskeleton, but not nodes of Ranvier. A, Coronal, contralateral sections of cortex from 6 h MCAO treated mouse, labeled with a Nissl stain (NeuroTrace, red) to detect neuronal cell bodies, Hoechst (blue) to label nuclei, and anti-βIV spectrin (green) to label the AIS. The white arrow indicates the location of the pial surface. The inset shows a higher magnification of a neuron with a βIV spectrin-positive AIS (arrow). B, Ipsilateral, injured cortex from 6 h MCAO treated mouse. Labeled as in A. The dashed line delineates the border of AIS staining. The inset shows higher magnification of neurons without βIV spectrin immunoreactivity. Note that a few neurons in the ischemic region retain their AIS βIV spectrin immunoreactivity. C, Coronal, contralateral sections of Striatum from 6 h MCAO treated mouse, labeled as in A. Boxes indicate regions of higher magnification shown in E, F. D, Ipsilateral, injured striatum from 6 h MCAO treated mouse. Labeled as in A. Boxes indicate regions of higher magnification shown in G, H. Note the complete absence of AIS βIV spectrin immunoreactivity, but preserved nodal βIV spectrin staining. Scale bar, 50 μm. E, F, Higher magnification of contralateral striatum shown in C to illustrate βIV spectrin immunostaining at the AIS (E) and nodes of Ranvier (F). G, H, Higher magnification of ipsilateral striatum shown in D to illustrate loss of βIV spectrin immunostaining from the AIS (G) and nodes of Ranvier (H). Scale bars: A–D, 50 μm.
Figure 2.

Nav channels are lost from the AIS following ischemic injury. A, B, After MCAO, contralateral (left) and ipsilateral (right) regions of rat cortex were immunolabeled for Nav channels (Pan-Nav, red), βIV spectrin (A, green) or ankG (B), and Hoechst to label nuclei (blue). The sections shown are from a brain collected 24 h after MCAO. Arrows indicate labeled AIS in layer 2/3 cortex. Scale bar, 50 μm.
Since nodes of Ranvier and axon initial segments share a common cytoskeletal organization (Kordeli et al., 1995; Berghs et al., 2000), we assumed that nodes would also be disrupted by ischemic injury. To test this possibility, we examined nodal and AIS βIV spectrin immunoreactivity in contralateral and ipsilateral striatum since this region is strongly affected by MCAO and has many neuronal cell bodies and bundles of myelinated axons. Strikingly, whereas βIV spectrin labeled the AIS (Fig. 1 C,E) and nodes (Fig. 1 C,F) in the contralateral striatum, only the AIS was disrupted in the ipsilateral striatum (Fig. 1 D,G). In the ipsilateral striatum large bundles of myelinated axons were robustly labeled for βIV spectrin indicating preserved nodes of Ranvier (Fig. 1 D,H). Together, these results show that despite a common molecular organization, the AIS cytoskeleton is preferentially susceptible to disruption after injury.
We next considered whether early disruption of the AIS cytoskeleton is a common phenomenon in different kinds of nervous system injury. To test this possibility, we examined the AIS of retinal ganglion cells (RGCs) 5 d after optic nerve crush injury. Whereas control NeuN-stained RGCs (red) had robust AIS βIV spectrin immunoreactivity [(Fig. 3 A,C, green) we used βIV spectrin as an indicator of the AIS cytoskeleton since its localization depends on ankG (Yang et al., 2007)], RGCs with injured axons lost virtually all AIS βIV spectrin immunostaining (Fig. 3 B,D). Together, these results suggest a common consequence of neuronal injury is the preferential disruption of the AIS cytoskeleton.
Figure 3.

Optic nerve crush results in loss of βIV spectrin staining at the AIS. A, Immunostaining of control retina for βIV spectrin (green), NeuN (red), and Hoechst (blue). B, Retina corresponding to the crushed optic nerve immunostained for βIV spectrin (green), NeuN (red), and Hoechst (blue). C, Higher magnification of the boxed region shown in A reveals intact AIS (green). D, Higher magnification of the boxed region shown in B demonstrates the loss of βIV spectrin immunostaining. Scale bars: A, B, 50 μm.
The AIS cytoskeleton is rapidly proteolyzed following injury
How does neuronal injury cause disruption of the AIS? One possible explanation is that like ankB and αII spectrin, ankG and βIV spectrin are susceptible to proteolysis after injury. To test this possibility, we performed immunoblot analyses of brain homogenates from sham, or MCAO-treated mice with antibodies against various AIS components (βIV spectrin, ankG, Nav channels, and neurofascin) or αII spectrin, a spectrin previously shown to be proteolyzed following ischemic injury (White et al., 2000; Czogalla and Sikorski, 2005). We identified distinct breakdown products associated with the AIS cytoskeleton. In each ipsilateral hemisphere, we found low molecular weight fragments from βIV spectrin (∼45 kDa) and ankG (∼95 kDa and 72 kDa) (Fig. 4 A, asterisks indicate breakdown products, arrows indicate full-length proteins). These bands could be detected at every time point analyzed (3 h–72 h post-occlusion). Coincident with their proteolysis, we also observed reductions in the amounts of full-length βIV spectrin and ankG (Fig. 4 A, arrows). In contrast to these AIS cytoskeletal proteins, neurofascin, a CAM highly enriched at the AIS, showed no detectable loss of the full-length protein or appearance of proteolytic fragments (Fig. 4 A). On the other hand, when homogenates were probed with an antibody against Nav channels, low molecular weight fragments could be detected in the ipsilateral hemisphere (Fig. 4 A, bracket and asterisk). However, these were not prominent until 12 h post-occlusion suggesting these proteins are proteolyzed after the AIS cytoskeleton. Importantly, immunoblots for αII spectrin demonstrated a clear ∼150 kDa breakdown product associated with each ipsilateral hemisphere (Fig. 4 A, asterisk). This product could not be detected in the contralateral hemisphere, sham control, or naive mouse brain homogenates and is the product of proteolysis by the calcium-dependent protease calpain (Siman et al., 1984). Together, these results indicate that components of the AIS cytoskeleton are rapidly proteolyzed following injury.
Figure 4.

βIV spectrin and ankyrinG are proteolyzed after neuronal injury. A, Immunoblots of brain homogenates made from naive mouse brain (MBr) and ipsilateral (I) and contralateral (C) hemispheres of sham and occluded animals (time post-occlusion = 3, 6, 12, 24, or 72 h). Immunoblots were prepared using antibodies against αII spectrin, AIS cytoskeleton (ankG and βIV spectrin), and AIS membrane proteins (Nav channels and Neurofascin). β-actin was used as a loading control. Arrows indicate full-length forms of each protein and asterisks indicate major breakdown products (BDP). Breakdown products can be observed as early as 3 h after the onset of occlusion. The lower immunoblots (breakdown products) shown for βIV spectrin and ankG correspond to exposure times that were 3 and 10 times longer, respectively, than the immunoblots showing full-length proteins. Data are representative of 3 animals/occlusion time point. B, Mouse brain homogenate was incubated at 25°C for 30 min in the presence (1 mm CaCl2) or absence (−CaCl2) of calcium together with 0–100 μm of the calpain inhibitor MDL 28170. Homogenates were then analyzed by immunoblot to detect βIV spectrin, ankyrinG, or as a control for calpain mediated proteolysis αII spectrin. The control condition (0 μm drug) was incubated with vehicle alone (DMSO). Full-length proteins are indicated by arrows while breakdown products are indicated by asterisks.
Increased Ca2+ causes proteolysis of the AIS cytoskeleton
What is the mechanism underlying proteolysis of the AIS cytoskeleton? Exposing naive brain homogenate in vitro to increasing amounts of Ca2+ caused a concentration dependent increase in the proteolysis of βIV spectrin and ankG (Fig. 5 A), and their proteolytic fragments had molecular weights identical to those observed after in vivo ischemic injury (Fig. 4 A). Thus, proteolysis of the AIS cytoskeleton occurs under conditions of elevated Ca2+.
Figure 5.

Ca2+ dependent disruption of the AIS cytoskeleton. A, The AIS cytoskeleton is proteolyzed under conditions of elevated Ca2+. Naive mouse brain homogenate (MBr) was incubated at 25°C for 30 min in the presence of increasing concentrations of calcium (0–5 mm CaCl2). There is a concentration-dependent increase in proteolysis of full-length AIS and axonal cytoskeletal components. Full-length proteins are designated by an arrow and breakdown products are designated by asterisks. These breakdown products are the same molecular weights as observed following MCAO (n = 3). B, Live imaging of 10 DIV hippocampal neuron AIS using anti-neurofascin (A12/18) and after photolysis of NP-EGTA to uncage Ca2+ only at the AIS. AIS were imaged at 10 min intervals. Scale bar, 10 μm. C, The ratio of fluorescence intensity 30 min after Ca2+ uncaging (F (t) = 30) to the fluorescence intensity immediately before Ca2+ uncaging (F (t) = 0). 6 control and 6 uncaged cells were used for this analysis. Error bars, SD.
Since ankG and βIV spectrin proteolysis occurs under conditions of elevated Ca2+, we next sought to determine whether a local, AIS restricted increase in Ca2+ concentration could also disrupt the molecular organization of the AIS. To do this we live-labeled the AIS using antibodies against an extracellular epitope of neurofascin and fluorescent conjugated secondary antibodies to visualize the AIS by fluorescence microscopy. We then loaded the labeled neurons with NP-EGTA, to permit photolytic uncaging of Ca2+. Finally, after identifying the AIS by neurofascin immunofluorescence, we uncaged Ca2+ only at the AIS and monitored the fluorescence intensity of neurofascin for 30 min. We found that in contrast to control initial segments in the same cultures, uncaging of Ca2+ at the AIS resulted in a rapid decrease in NF immunofluorescence (Fig. 5 B,C). These observations are consistent with the idea that Ca2+-dependent proteolysis of ankG and βIV spectrin leads to loss of AIS membrane proteins.
Calpain proteolyzes the AIS cytoskeleton
Alpha II spectrin proteolysis after injury depends on the calcium-dependent cysteine protease calpain (Siman et al., 1984). To test whether calpain also proteolyzes βIV spectrin and ankG, we incubated naive brain homogenates in 1 mm Ca2+ and increasing concentrations of different calpain inhibitors including MDL 28170, calpastatin, calpeptin, and ALLN (Fig. 4 B, supplemental Fig. S1, available at www.jneurosci.org as supplemental material; calpeptin and ALLN not shown). The addition of calpain inhibitors resulted in a concentration dependent reduction in both βIV spectrin and ankG proteolysis. However, some reports suggest that calpain inhibitors lack specificity and cross-inhibit cathepsins, another class of calcium-dependent cysteine proteases (Wang and Yuen, 1997; Donkor, 2000; Neffe and Abell, 2005; Cuerrier et al., 2007). To exclude cathepsins and other proteases as mediators of βIV spectrin and ankG proteolysis, we tested CA-074, a cathepsin B inhibitor (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), Q-VD-OPH, an inhibitor of caspases (data not shown) and GM-6001, an inhibitor of matrix metalloproteases (data not shown). Unlike calpain inhibitors, these inhibitors all failed to attenuate proteolysis of βIV spectrin or ankG at all concentrations tested (0–100 μm). As an important control, when these same samples were tested for αII spectrin proteolysis, production of the calpain-mediated ∼150 kDa breakdown product was inhibited by addition of calpain inhibitors but not other protease inhibitors (Fig. 4 B; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Together, these data strongly suggest that proteolysis of βIV spectrin and ankG is mediated by calpain.
AIS disruption in vitro
The considerable neuronal damage induced in the MCAO model made it difficult to determine whether AIS disruption is a consequence of cell death, or a specific early response to neuronal injury. Therefore, to elucidate the chronology and molecular mechanism underlying AIS disassembly after neuronal injury, we analyzed AIS disruption in a culture model of ischemic injury: oxygen-glucose deprivation (OGD). At DIV10, we deprived cortical neuron cultures of oxygen (0–0.02%) and glucose (0 mm) for up to 3 h. After deprivation, cultures were returned to normoxia and glucose-containing media was added back to the cultures. Cell death and AIS integrity were then analyzed 20–24 h later. Cells deprived of oxygen and glucose for 1 h were indistinguishable from control cultures. However, similar to our in vivo results, 2 h of OGD resulted in a >80% decrease in neurons with βIV spectrin immunoreactivity at the AIS (Fig. 6 A). Comparable reductions in ankG, Nav channel, and neurofascin immunoreactivity were also observed with an identical time course (Fig. 6 B; quantification for βIV spectrin not shown). We also performed OGD experiments using hippocampal neurons and obtained similar results (data not shown).
Figure 6.
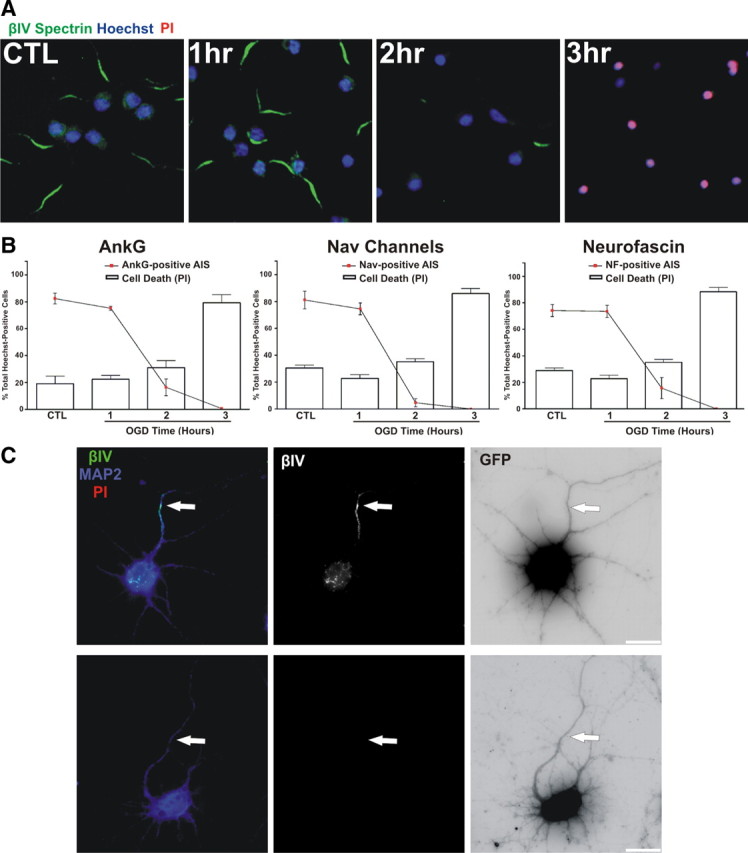
AIS disassembly is rapid and occurs before cell death. A, Cultured cortical neurons (DIV10) were deprived of oxygen and glucose for increasing times, followed by return to normal oxygen and glucose levels for 24 h. 24 h after OGD the molecular integrity of the AIS was analyzed by immunostaining neurons with anti-βIV spectrin (green). Nuclei are indicated by Hoechst staining (blue). Cell death was assessed using propidium iodide (PI, red). B, Quantification of AIS integrity (lines) and cell death (bars) as a percentage of total Hoechst-positive cells (±SEM). The AIS was labeled for ankG, Nav channels, neurofascin, or βIV spectrin (data not shown). Asterisks (red, AIS; black, cell death) denote mean values significantly different from control (CTL; p < 0.05). N = 9 in 3 independent experiments. C, Loss of the AIS does not cause axon degeneration. Neuronal process integrity was analyzed by transfecting neurons with GFP (GFP fluorescence is inverted) and staining for MAP2 (blue) and βIV spectrin (green and middle panel). The top panels show a control neuron. After 2 h of OGD (lower 3 panels), live neurons (PI-negative, red) lose their AIS, but retain neuronal processes (MAP2, blue and GFP). The axon is indicated by an arrow. Note: βIV spectrin staining (green) was labeled using a far-red fluorophore and is pseudocolored green in these images for visualization. Scale bar, 20 μm.
Are cells dead after 2 h of OGD, and does this explain the loss of AIS proteins? Staining cultures with propidium iodide (PI), an indicator of cell death, showed that significant neuron death did not occur until 3 h of OGD (Fig. 6 A,B, bars). Cell viability as assessed by the MTT assay yielded similar results (data not shown). Alternatively, the loss of AIS βIV spectrin immunoreactivity could be a consequence of rapid and general axon degeneration. Therefore, we examined neuronal morphology by transfecting neurons with GFP and by staining for MAP2, a dendritic marker that has previously been shown to be proteolyzed and redistributed in neurons following ischemic injury (Pettigrew et al., 1996; Buddle et al., 2003; Bevers and Neumar, 2008). After 2 h of OGD, loss of AIS immunofluorescence occurred without axonal or dendritic retraction or degeneration (Fig. 6 C). Together, these results demonstrate that proteolysis of the AIS cytoskeleton and loss of clustered AIS proteins (e.g., Nav channels and neurofascin) is an early consequence of neuronal injury, occurs independently of cell death, and is not a consequence of axon degeneration.
AIS disruption is irreversible and causes loss of neuronal polarity
Since neuronal injury results in loss of the AIS proteins required for action potential initiation and the maintenance of neuronal polarity, any functional recovery must include the re-establishment of the AIS cytoskeleton. To determine whether the AIS recovers after injury, or whether 2 h OGD eventually results in widespread cell death, we examined neurons for βIV spectrin immunofluorescence and PI staining at 24 and 72 h after 2 h of OGD (Fig. 7 A,B). By 72 h after OGD, we failed to observe any recovery in the number of cells with clustered AIS proteins despite no significant increase in cell death compared to the 24 h time point.
Figure 7.

Loss of the AIS is irreversible. A, Cultured neurons were treated for 2 h of OGD, followed by recovery for 24 or 72 h. The AIS was labeled with anti-βIV spectrin (green), nuclei by Hoechst (blue), and cell death was assessed with PI (red). B, Quantification of AIS integrity (lines) and cell death (bars) as a percentage of total Hoechst-positive cells (±SEM) in control and 24 or 72 h post OGD treated cells. The AIS was labeled for βIV spectrin. Red asterisks indicate a significant difference in AIS immunoreactivity from control cultures (CTL). C, Live labeling of 10 DIV hippocampal neurons using anti-neurofascin antibodies (A12/18). Arrows indicate the location of axon initial segments. D, Same field of view as in C 8 d after treatment by OGD. Hippocampal neurons were fixed and stained for βIV spectrin immunoreactivity. Arrows indicate the location of the AIS determined 8 d previously. E, The percentage of control and 10 d OGD treated cells that also had βIV spectrin AIS immunoreactivity or were labeled with propidium iodide (PI) Error bars, SD. F, G, 10 DIV hippocampal neuron live-labeled using anti-neurofascin (A12/18; F) and anti-MAP2 (G). The axon is indicated by arrows. Same scale as in I. MAP2 is somatodendritic and excluded from the axon (arrows). H, 10 DIV hippocampal neuron live-labeled using anti-neurofascin (A12/18). The axon is indicated by arrows. Same scale as in I. I, Same neuron as in H 8 d after OGD, labeled for MAP2 immunoreactivity. After OGD the neuron loses polarity and MAP2 invades the axon (arrows). Scale bars: A, 20 μm; I, 5 μm.
Our previous studies have shown that it takes nearly a full week in culture for all hippocampal neurons to develop an AIS (Yang et al., 2007). Therefore, we considered the possibility that >72 h might be needed for the re-establishment of the AIS. Alternatively, it is also possible that disruption of the AIS may result in conversion of dendrites into axons (Gomis-Rüth et al., 2008), and this may require longer recovery times. To test these possibilities, we live-immunolabeled 10 DIV hippocampal neurons using antibodies against neurofascin. We visualized these neurons and their AIS using immunofluorescence microscopy and carefully noted their location using gridded microscope coverglass (Fig. 7 C, arrows). We then exposed these cells to OGD for 2 h and returned them to the incubator for 8–10 d. These cells were then fixed and labeled for both βIV spectrin immunoreactivity and PI staining. Similar to the 72 h time point (Fig. 7 A) we found the loss of initial segments persisted even 8–10 d after OGD (Fig. 7 D,E). Eight days after OGD βIV spectrin immunostaining was very weak and randomly distributed throughout neurons; when we examined the location of the former AIS, we did not detect any recovery of βIV spectrin immunoreactivity (Fig. 7 D, arrows).
We previously showed that dismantling of the AIS by silencing expression of ankG disrupted neuronal polarity and led to axons acquiring the molecular characteristics of dendrites (Hedstrom et al., 2008). To determine whether injury-induced disruption of the AIS cytoskeleton also leads to loss of neuronal polarity, we live-labeled 10 DIV hippocampal neuron AIS using anti-neurofascin antibodies to identify the AIS and axon (Fig. 7 F,H, arrows), and MAP2 to define somatodendritic domains. At this time, MAP2 is excluded from axons (Fig. 7 G). However, after 8 d post OGD treatment, neuronal polarity was disrupted and MAP2 was detected in the axon. Together, these results suggest that proteolysis of the AIS cytoskeleton leads to an irreversible loss of clustered ion channels and neuronal polarity.
Calpain disrupts the AIS following OGD
Most neurons lose their AIS after 2 h of OGD and die after 3 h of OGD (Fig. 6 A,B). If calpain proteolyses the AIS cytoskeleton, then calpain inhibitors should preserve the AIS cytoskeleton even after 3 h of OGD. To test this, we treated neurons with the calpain inhibitor MDL 28170, and compared this to treatment using the NMDA receptor antagonist MK-801, a well known inhibitor of neuronal excitotoxicity. In the presence of the calpain inhibitor, AIS integrity in vitro was significantly protected in a concentration-dependent manner: at 100 μm MDL 28170, 70.2% of cells had a βIV spectrin-positive AIS. However, cell death was not significantly inhibited since 53.2% of cells were PI+ (Fig. 8 A,B). In contrast to these results, treatment using the cathepsin B inhibitor CA-074 resulted in significant protection from cell death but not preservation of the AIS cytoskeleton (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). Treatment with 10 μm MK-801 resulted in significant protection from cell death (34.4% cells were PI+) (Fig. 8 A,B), but AIS integrity was only modestly protected (46.3% of cells had an identifiable, βIV spectrin labeled AIS). When both MDL 28170 and MK-801 were added to cultures, we observed both protection from cell death and preservation of the AIS (Fig. 8 A,B). Since the improved viability with the combined drug treatment was similar to MK-801 treatment alone, and the amount of AIS preservation was similar to the MDL 28170 treatment alone, we conclude that cell death and disruption of the AIS cytoskeleton are independent events.
Figure 8.

AIS disassembly following OGD is blocked by the calpain inhibitor MDL 28170. A, Cultured cortical neurons (DIV10) were treated during (3 h) and after (24 h) OGD with a calpain inhibitor MDL 28170 (100 μm) and/or an NMDA receptor antagonist MK-801 (10 μm). AIS integrity was assessed with an antibody against βIV spectrin (green) and cell death was assessed with PI (red). Neurons treated with MDL 28170 retained AIS integrity (arrows) whereas protection from cell death was modest. White and red asterisks indicate live and dead neurons with an AIS, respectively. Nuclei are labeled by Hoechst staining. B, Quantification of immunofluorescence as a percentage of Hoechst labeled nuclei for OGD treated cultures in the presence of 0–100 μm MDL 28170, 10 μm MK-801, or 100 μm MDL 28170 + 10 μm MK-801. Asterisks (black corresponds to cell death measurements while red corresponds to AIS measurements) denote percentages that are statistically similar to control, i.e., no OGD treatment (CTL; black bar, red triangle). n = 9 in 3 independent experiments. Error bars, ±SEM. C, Neurons infected with egfp-calpastatin (white bars) or egfp alone (black bars) were deprived of oxygen and glucose for 3 h. AIS integrity was measured by immunostaining with anti-βIV spectrin and counting the number of Hoechst labeled cells with an AIS. After OGD, the AIS was significantly protected in egfp-calpastatin infected cultures (asterisk) compared to control (egfp infected) cultures. D, There was no difference in the percentage of Hoechst labeled nuclei that were also PI-positive between egfp-calpastatin-infected and egfp-infected cultures.
To further confirm the role of calpain in disruption of the AIS cytoskeleton after OGD, we used adenovirus to overexpress enhanced gfp (egfp) or the highly specific endogenous inhibitor of calpain, calpastatin (egfp-clpst) (Fig. 8 C,D). While infection efficiencies were nearly identical (data not shown) and infection itself did not affect the AIS cytoskeleton or cell death (Fig. 8 C,D), overexpression of egfp-clpst significantly protected the AIS cytoskeleton after OGD compared to overexpression of egfp alone (39% and 5% of neurons retain βIV spectrin immunoreactivity, respectively) (Fig. 8 C). Furthermore, neither egfp nor egfp-clpst protected against cell death after OGD (Fig. 8 D). Together, these results suggest that after neuronal injury, increased calpain activity causes cytoskeletal breakdown, which in turn disrupts the molecular organization of the AIS.
Calpain inhibitors protect the AIS after MCAO
The in vitro results described above indicate that calpain disrupts the clustering of AIS ion channels and neuronal polarity by proteolysis of the AIS cytoskeleton. To confirm that calpain mediates the disruption of the AIS in vivo, we subjected rats to 90 min of MCAO followed by reperfusion. Then, at the time of reperfusion, we infused either vehicle or the calpain inhibitor MDL 28170 for 6 h. For comparison, in a separate cohort of animals, we administered MK-801 30 min before MCAO, and then every 3 h beginning at the time of reperfusion for 6 h. We collected brains for immunohistochemical analysis 24 h after MCAO. For both MDL 28170 and MK-801 treated animals, we observed a 40–50% reduction in total infarct size as measured by Fluoro-Jade staining (data not shown), cresyl violet staining (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material), and reduction in NeuN immunoreactivity.
Because cell death and AIS cytoskeleton disruption after OGD are distinct events, we examined the AIS in brain regions that were still clearly infarcted following drug administration in vivo [all quantification and analysis of AIS immunoreactivity was made from infarcted brain regions with at least 50% reduction in NeuN staining (Fig. 9 A, red, C) (Igarashi et al., 2001; Davoli et al., 2002)]). Similar to the in vitro OGD results, calpain inhibition increased the number of immunolabeled AIS in the ipsilateral cortex (Fig. 9 A, green, arrows) by 36.1% (p < 0.01) over vehicle and 24.6% (p < 0.01) over MK-801 treated animals (Fig. 9 B). Within the infarcted region analyzed, NeuN staining did not differ between groups (Fig. 9 C), suggesting that increased AIS immunoreactivity was not due to increased cell survival within the infarcted region. Similar to βIV spectrin, we also found increased levels of AIS Nav channel immunoreactivity in MDL 28170 treated animals (Fig. 9 D, arrows), but not vehicle or MK-801 treated animals. Thus, calpain inhibition protects the molecular organization of the AIS following ischemic injury in vivo.
Figure 9.

Calpain inhibition in vivo attenuates AIS disassembly following ischemic injury. A, Rats were subjected to MCAO in the presence or absence (vehicle) of a cell-permeable calpain inhibitor (MDL 28170) or an NMDA antagonist (MK-801). Coronal sections of rat cortex were stained (t = 24 h post-occlusion) with the neuronal marker NeuN (red); the AIS was labeled with anti-βIV spectrin (green) and nuclei were labeled with Hoechst (blue). White arrows indicate βIV spectrin labeled AIS, while black arrows indicate NeuN labeled neurons. B, Quantification of the preservation of the AIS (n = 4 animals for each group). Compared to MK-801 or vehicle treated animals, AIS integrity is significantly increased (asterisk) in the infarcted region of the ipsilateral cortex (white bars) of MDL 28170-treated animals. C, Within the infarcted region of the ipsilateral cortex (white bars), NeuN staining is significantly reduced as compared to the contralateral cortices (black bars) in all treatment groups. D, Within infarcted regions, there was a significant increase in Nav channel/βIV spectrin-labeled (yellow; arrow) AIS in animals treated with the calpain inhibitor (MDL 28170) versus MK-801 or vehicle-treated animals. Scale bars: A, D, 50 μm.
Discussion
An AP is initiated when sufficient excitatory synaptic input depolarizes the AIS membrane to threshold. AP initiation, duration, and modulation also depend on the variety of Nav and Kv channels found at the AIS (Fig. 10 A). The AIS also maintains neuronal polarity by preserving the distinction between axonal and somatodendritic domains (Hedstrom et al., 2008). Given its central role in nervous system function, diseases and injuries that specifically affect the molecular organization and/or integrity of the AIS are expected to have profound neurological effects. Even mild injuries without overt cell death may lead to disruption of the AIS cytoskeleton and modest reductions in ankG at the AIS. This could have significant effects on brain function due to decreased amounts of Nav channels and the corresponding changes in neuronal excitability. Consistent with this idea several AIS proteins, including ankG, have been implicated as susceptibility loci in diverse psychiatric disorders such as autism and bipolar disorder (Alarcón et al., 2008; Ferreira et al., 2008). In the present study we provide evidence that (1) neuronal injury causes rapid and preferential disruption of the AIS, (2) the ankG and βIV spectrin-based AIS cytoskeleton is proteolyzed after injury, (3) proteolysis of the AIS cytoskeleton causes loss of ion channel clusters and neuronal polarity, (4) injury-induced proteolysis of ankG and βIV spectrin depends on the calcium-dependent cysteine protease calpain, (5) calpain inhibitors preserve the molecular organization of the AIS and the integrity of its cytoskeleton after neuronal injury, (6) disruption of the AIS occurs independently of neuron death, and (7) disruption of the AIS is an irreversible event. Together, our results suggest that increased Ca2+ influx resulting from neuronal injury (Fig. 10 B) activates the Ca2+ dependent protease calpain, which in turn preferentially proteolyses ankG and βIV spectrin (Fig. 10 C). Loss of the intact ankG and βIV spectrin based cytoskeleton results in failure to retain the ion channels and CAMs at the AIS leading to their dispersion and/or degradation (Fig. 10 D) (Hedstrom et al., 2008). Although we focused our analyses on the AIS cytoskeleton, both αII and βII spectrin are also proteolyzed after injury. Whether disruption of these cytoskeletal proteins can also contribute to loss of neuronal polarity is an active area of investigation.
Figure 10.
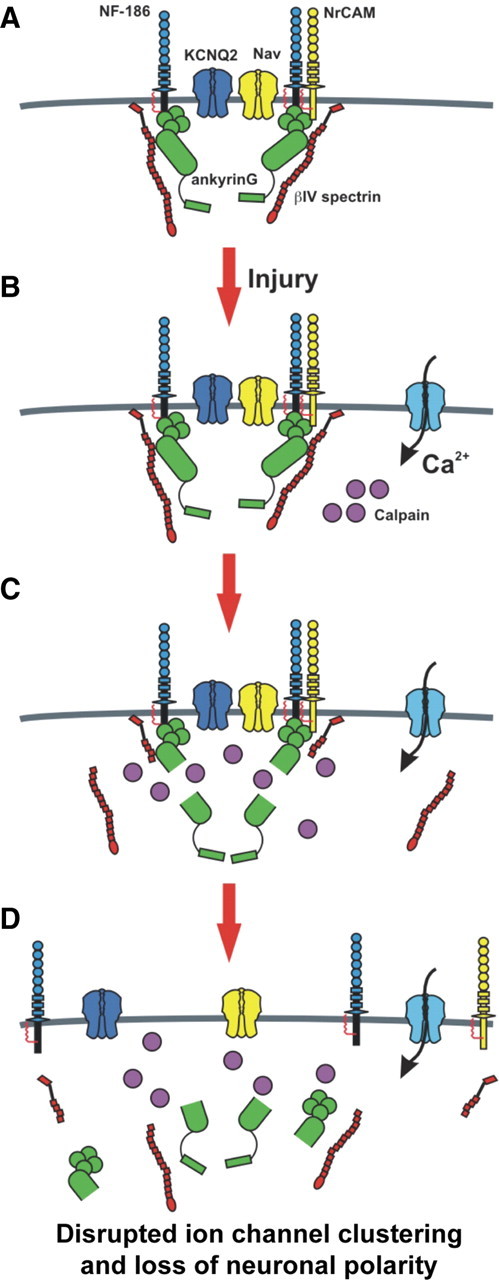
AIS disassembly following acute CNS injury. A, Nav channels, Kv channels, and cell adhesion molecules are clustered at high density at the AIS through interactions with the AIS cytoskeleton comprised of βIV spectrin and ankG. B, Upon injury, calcium enters the cell and activates calpain, a Ca2+ dependent cysteine protease. C, Calpain cleaves the spectrin/ankyrin-based cytoskeleton associated with the AIS. D, Destruction of the AIS cytoskeleton causes loss of high densities of ion channels at the AIS, impaired ability to initiate action potentials, and loss of neuronal polarity.
The AIS also functions to maintain neuronal polarity since its disruption through pharmacological means or through silencing of ankG expression causes disruption in proper protein trafficking and axons acquire the molecular characteristics of dendrites. For example, Winckler et al. (1999) showed that axonal membrane proteins are restricted in their mobility within the AIS membrane when compared to distal axons. Upon pharmacological disruption of the axonal cytoskeleton, this diffusion barrier was lost and membrane proteins were no longer restricted to distinct domains. To further elucidate the molecular mechanisms underlying maintenance of neuronal polarity, Hedstrom et al. (2008) silenced the expression of ankG in fully polarized hippocampal neurons. This dismantled the AIS and caused axons to acquire the molecular characteristics of dendrites including the presence of the somatodendritic protein MAP2. Finally, the ankG-based AIS functions as a filter to exclude dendritic carrier vesicles, and permit entry of axonal carrier vesicles (Song et al., 2009). Together, these experiments demonstrated that ankG is necessary to maintain neuronal polarity and the correct molecular composition of the AIS. Furthermore, these previous studies support our observation that an important consequence of the injury-dependent disruption of the ankG/βIV spectrin-based cytoskeleton is loss of neuronal polarity. We propose that disrupted neuronal polarity represents a previously unappreciated and major consequence of neuronal injury.
It is now well established that in response to axon injury polarity may be plastic such that dendrites can be converted into axons or supernumerary axons can grow from the cell body (Havton and Kellerth, 1987; Hoang et al., 2005). One particularly intriguing experiment showed that in cultured hippocampal neurons this depends on where the axonal transection takes place: transection at sites distal to the cell maintain axon identity, while transection close to the cell body (35 μm from the soma and about the same location as the AIS), causes a fate switch and the conversion of a dendrite into an axon (Gomis-Rüth et al., 2008). Although this change was attributed to altered microtubule stability, an alternative explanation may be the disruption of the AIS cytoskeleton. Our results demonstrating that AIS disruption leads to irreversible dismantling of the molecular organization of the AIS and loss of neuronal polarity supports the latter conclusion. Furthermore, we previously showed that loss of the AIS cytoskeleton converted axons into dendrites (Hedstrom et al., 2008). These differences in dendrite to axon and axon to dendrite conversion may reflect unique aspects of each injury model. In any case, both studies emphasize that preservation of the AIS cytoskeleton after injury may be one important way to reduce inappropriate synaptic connections by preserving the appropriate neuronal polarity and the identity of the axon.
Besides failure to properly initiate action potentials in the absence of Nav channel clusters (Kole et al., 2008) and maintain neuronal polarity (Hedstrom et al., 2008), what other consequences might result from disruption of the AIS cytoskeleton? In addition to ion channels, the AIS is highly enriched in cell adhesion molecules (CAMs), accessory and signaling proteins, and a unique extracellular matrix (ECM) (Ogawa and Rasband, 2008), all of which depend on an intact AIS cytoskeleton for their localization (Hedstrom et al., 2007). Therefore, loss of the AIS cytoskeleton would be expected to disrupt any molecular or cellular specializations that exist at this site. For example, some data suggest that the AIS CAM neurofascin (NF)-186 and/or ECM directs GABAergic innervation of the AIS (Ango et al., 2004; Szabadics et al., 2006). In addition, AIS recruitment of gephyrin, a postsynaptic scaffolding protein that regulates GABA receptor clustering, also depends on NF-186 (Burkarth et al., 2007). Thus, dispersion of NF-186 away from the AIS through loss of ankG and βIV spectrin might also disrupt GABAergic neurotransmission at this site.
Although we focused our experiments on the AIS, in myelinated axons the node of Ranvier functions to regenerate the AP and has a very similar molecular organization with the same ankG and βIV spectrin-based cytoskeleton (Susuki and Rasband, 2008). Nodes of Ranvier may be a particularly fragile point in the axon since they lack an overlying myelin sheath and could readily contribute to axonal Ca2+ overload. Consistent with this idea it is now appreciated that the permanent functional deficits associated with demyelination are a consequence of axon degeneration, and calpain is upregulated in experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (Bjartmar and Trapp, 2001; Guyton et al., 2005). Indeed, axonal degeneration is attenuated in EAE with the addition of calpain inhibitors (Hassen et al., 2006). However, in our analysis of nodal structure in the striatum after MCAO, although we observed profound loss of initial segments, we failed to observe disruption of nodes of Ranvier even 72 h after injury. These results suggest that proximity of the axonal injury to the cell body may influence proteolysis of the cytoskeleton or that the nodal cytoskeleton is resistant to proteolysis. This may also reflect differences in the amount of calpain found in axonal or somatodendritic cellular compartments. Future experiments focused on white matter injury and responses at nodes should help to clarify these issues.
We have discussed proteolysis of the AIS cytoskeleton as mainly a maladaptive response to neuronal injury. However, it may be that dismantling of the AIS and loss of Nav channels before neuronal death is a neuroprotective response to injury. We demonstrated that 2 h of OGD caused disruption of the AIS, but not cell death. In stroke, waves of depolarization and excitotoxicity are major contributors to the development of the ischemic core and penumbra (the area of the brain bordering the ischemic core where the majority of therapeutic strategies are targeted). Inhibiting neuronal depolarization and a cell's ability to fire APs may protect the cell from increased excitation and spread of an already excitotoxic cellular environment. Consistent with this idea, Nav channel blockers have been shown to be effective neuroprotective agents following ischemic and experimental spinal cord injury (Schwartz and Fehlings, 2001).
In conclusion, we have shown that neuronal injury disrupts the molecular organization of the AIS by calpain-dependent proteolysis of the AIS cytoskeleton. Although we have mainly used ischemia as a model injury, our results in the optic nerve crush model suggest that any injury or disease that causes increases in calpain activity by cytosolic Ca2+ overload will affect the AIS cytoskeleton and neuronal function. Finally, since normal nervous system function requires properly polarized neurons with high densities of ion channels at the AIS, any effort aimed at nervous system protection, repair, or recovery must consider not only strategies to enhance cell survival, but also incorporate ways to preserve or repair the integrity of the AIS.
Footnotes
This research was supported by grants from the National Institutes of Health [NS044916 (M.N.R.), NS050505 (L.D.M.), and NS055215 (L.D.M.)], the Department of Defense [W81XWH-08-2-0145 (M.N.R.)], the Dr. Miriam and Sheldon Adelson Medical Research Foundation, and Mission Connect. D.P.S. was supported in part by National Institutes of Health Training Grant 5T32NS041224. M.N.R. is a Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society. We thank Dr. Sandra Hewett for helpful discussions and the use of her anaerobic chamber. We thank Drs. Michael Stankewich, Stephen Crocker, Elior Peles, and Peter Saggau for helpful discussions. We thank Drs. Yang Yang and Larry Benowitz for providing retinas from animals with optic nerve crush.
References
- Alarcón M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH, Nelson SF, Cantor RM, Geschwind DH. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82:150–159. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ango F, di Cristo G, Higashiyama H, Bennett V, Wu P, Huang ZJ. Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at purkinje axon initial segment. Cell. 2004;119:257–272. doi: 10.1016/j.cell.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Ardelt AA, McCullough LD, Korach KS, Wang MM, Munzenmaier DH, Hurn PD. Estradiol regulates angiopoietin-1 mRNA expression through estrogen receptor-alpha in a rodent experimental stroke model. Stroke. 2005;36:337–341. doi: 10.1161/01.STR.0000153795.38388.72. [DOI] [PubMed] [Google Scholar]
- Benn SC, Woolf CJ. Adult neuron survival strategies–slamming on the brakes. Nat Rev Neurosci. 2004;5:686–700. doi: 10.1038/nrn1477. [DOI] [PubMed] [Google Scholar]
- Berghs S, Aggujaro D, Dirkx R, Jr, Maksimova E, Stabach P, Hermel JM, Zhang JP, Philbrick W, Slepnev V, Ort T, Solimena M. betaIV spectrin, a new spectrin localized at axon initial segments and nodes of ranvier in the central and peripheral nervous system. J Cell Biol. 2000;151:985–1002. doi: 10.1083/jcb.151.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevers MB, Neumar RW. Mechanistic role of calpains in postischemic neurodegeneration. J Cereb Blood Flow Metab. 2008;28:655–673. doi: 10.1038/sj.jcbfm.9600595. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Curr Opin Neurol. 2001;14:271–278. doi: 10.1097/00019052-200106000-00003. [DOI] [PubMed] [Google Scholar]
- Buddle M, Eberhardt E, Ciminello LH, Levin T, Wing R, DiPasquale K, Raley-Susman KM. Microtubule-associated protein 2 (MAP2) associates with the NMDA receptor and is spatially redistributed within rat hippocampal neurons after oxygen-glucose deprivation. Brain Res. 2003;978:38–50. doi: 10.1016/s0006-8993(03)02758-6. [DOI] [PubMed] [Google Scholar]
- Burkarth N, Kriebel M, Kranz EU, Volkmer H. Neurofascin regulates the formation of gephyrin clusters and their subsequent translocation to the axon hillock of hippocampal neurons. Mol Cell Neurosci. 2007;36:59–70. doi: 10.1016/j.mcn.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Cuerrier D, Moldoveanu T, Campbell RL, Kelly J, Yoruk B, Verhelst SH, Greenbaum D, Bogyo M, Davies PL. Development of calpain-specific inactivators by screening of positional scanning epoxide libraries. J Biol Chem. 2007;282:9600–9611. doi: 10.1074/jbc.M610372200. [DOI] [PubMed] [Google Scholar]
- Czogalla A, Sikorski AF. Spectrin and calpain: a ‘target’ and a ‘sniper’ in the pathology of neuronal cells. Cell Mol Life Sci. 2005;62:1913–1924. doi: 10.1007/s00018-005-5097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli MA, Fourtounis J, Tam J, Xanthoudakis S, Nicholson D, Robertson GS, Ng GY, Xu D. Immunohistochemical and biochemical assessment of caspase-3 activation and DNA fragmentation following transient focal ischemia in the rat. Neuroscience. 2002;115:125–136. doi: 10.1016/s0306-4522(02)00376-7. [DOI] [PubMed] [Google Scholar]
- Donkor IO. A survey of calpain inhibitors. Curr Med Chem. 2000;7:1171–1188. doi: 10.2174/0929867003374129. [DOI] [PubMed] [Google Scholar]
- Duckworth EA, Butler TL, De Mesquita D, Collier SN, Collier L, Pennypacker KR. Temporary focal ischemia in the mouse: technical aspects and patterns of Fluoro-Jade evident neurodegeneration. Brain Res. 2005;1042:29–36. doi: 10.1016/j.brainres.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Dzhashiashvili Y, Zhang Y, Galinska J, Lam I, Grumet M, Salzer JL. Nodes of Ranvier and axon initial segments are ankyrin G-dependent domains that assemble by distinct mechanisms. J Cell Biol. 2007;177:857–870. doi: 10.1083/jcb.200612012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MA, O'Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, Smoller JW, Grozeva D, Stone J, Nikolov I, Chambert K, Hamshere ML, Nimgaonkar VL, Moskvina V, Thase ME, Caesar S, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ, ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- Garrido JJ, Giraud P, Carlier E, Fernandes F, Moussif A, Fache MP, Debanne D, Dargent B. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 2003;300:2091–2094. doi: 10.1126/science.1085167. [DOI] [PubMed] [Google Scholar]
- Gerriets T, Stolz E, Walberer M, Kaps M, Bachmann G, Fisher M. Neuroprotective effects of MK-801 in different rat stroke models for permanent middle cerebral artery occlusion: adverse effects of hypothalamic damage and strategies for its avoidance. Stroke. 2003;34:2234–2239. doi: 10.1161/01.STR.0000087171.34637.A9. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Clark BD, Zagha E, Nahmani M, Erisir A, Rudy B. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron. 2008;58:387–400. doi: 10.1016/j.neuron.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis-Rüth S, Wierenga CJ, Bradke F. Plasticity of polarization: changing dendrites into axons in neurons integrated in neuronal circuits. Curr Biol. 2008;18:992–1000. doi: 10.1016/j.cub.2008.06.026. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Wingrave JM, Yallapragada AV, Wilford GG, Sribnick EA, Matzelle DD, Tyor WR, Ray SK, Banik NL. Upregulation of calpain correlates with increased neurodegeneration in acute experimental auto-immune encephalomyelitis. J Neurosci Res. 2005;81:53–61. doi: 10.1002/jnr.20470. [DOI] [PubMed] [Google Scholar]
- Harada K, Fukuda S, Kunimoto M, Yoshida K. Distribution of ankyrin isoforms and their proteolysis after ischemia and reperfusion in rat brain. J Neurochem. 1997;69:371–376. doi: 10.1046/j.1471-4159.1997.69010371.x. [DOI] [PubMed] [Google Scholar]
- Hassen GW, Feliberti J, Kesner L, Stracher A, Mokhtarian F. A novel calpain inhibitor for the treatment of acute experimental autoimmune encephalomyelitis. J Neuroimmunol. 2006;180:135–146. doi: 10.1016/j.jneuroim.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Havton L, Kellerth JO. Regeneration by supernumerary axons with synaptic terminals in spinal motoneurons of cats. Nature. 1987;325:711–714. doi: 10.1038/325711a0. [DOI] [PubMed] [Google Scholar]
- Hedstrom KL, Xu X, Ogawa Y, Frischknecht R, Seidenbecher CI, Shrager P, Rasband MN. Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J Cell Biol. 2007;178:875–886. doi: 10.1083/jcb.200705119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183:635–640. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang TX, Nieto JH, Havton LA. Regenerating supernumerary axons are cholinergic and emerge from both autonomic and motor neurons in the rat spinal cord. Neuroscience. 2005;136:417–423. doi: 10.1016/j.neuroscience.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Igarashi T, Huang TT, Noble LJ. Regional vulnerability after traumatic brain injury: gender differences in mice that overexpress human copper, zinc superoxide dismutase. Exp Neurol. 2001;172:332–341. doi: 10.1006/exnr.2001.7820. [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155:739–746. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM. Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J Neurosci. 2006;26:1935–1944. doi: 10.1523/JNEUROSCI.4664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Ilschner SU, Kampa BM, Williams SR, Ruben PC, Stuart GJ. Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci. 2008;11:178–186. doi: 10.1038/nn2040. [DOI] [PubMed] [Google Scholar]
- Komada M, Soriano P. [Beta]IV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J Cell Biol. 2002;156:337–348. doi: 10.1083/jcb.200110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordeli E, Lambert S, Bennett V. AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem. 1995;270:2352–2359. doi: 10.1074/jbc.270.5.2352. [DOI] [PubMed] [Google Scholar]
- Lacas-Gervais S, Guo J, Strenzke N, Scarfone E, Kolpe M, Jahkel M, De Camilli P, Moser T, Rasband MN, Solimena M. BetaIVSigma1 spectrin stabilizes the nodes of Ranvier and axon initial segments. J Cell Biol. 2004;166:983–990. doi: 10.1083/jcb.200408007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markgraf CG, Velayo NL, Johnson MP, McCarty DR, Medhi S, Koehl JR, Chmielewski PA, Linnik MD. Six-hour window of opportunity for calpain inhibition in focal cerebral ischemia in rats. Stroke. 1998;29:152–158. doi: 10.1161/01.str.29.1.152. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S. Action potential initiation and propagation in CA3 pyramidal axons. J Neurophysiol. 2007;97:3460–3472. doi: 10.1152/jn.01288.2006. [DOI] [PubMed] [Google Scholar]
- Neffe AT, Abell AD. Developments in the design and synthesis of calpain inhibitors. Curr Opin Drug Discov Devel. 2005;8:684–700. [PubMed] [Google Scholar]
- Ogawa Y, Rasband MN. The functional organization and assembly of the axon initial segment. Curr Opin Neurobiol. 2008;18:307–313. doi: 10.1016/j.conb.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Schafer DP, Horresh I, Bar V, Hales K, Yang Y, Susuki K, Peles E, Stankewich MC, Rasband MN. Spectrins and ankyrinB constitute a specialized paranodal cytoskeleton. J Neurosci. 2006;26:5230–5239. doi: 10.1523/JNEUROSCI.0425-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Stuart GJ. Site of action potential initiation in layer 5 pyramidal neurons. J Neurosci. 2006;26:1854–1863. doi: 10.1523/JNEUROSCI.4812-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett V, Scherer SS, Cooper EC. A common ankyrin-G-based mechanism retains KCNQ and Nav channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettigrew LC, Holtz ML, Craddock SD, Minger SL, Hall N, Geddes JW. Microtubular proteolysis in focal cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:1189–1202. doi: 10.1097/00004647-199611000-00013. [DOI] [PubMed] [Google Scholar]
- Pike BR, Flint J, Dave JR, Lu XC, Wang KK, Tortella FC, Hayes RL. Accumulation of calpain and caspase-3 proteolytic fragments of brain-derived alphaII-spectrin in cerebral spinal fluid after middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 2004;24:98–106. doi: 10.1097/01.WCB.0000098520.11962.37. [DOI] [PubMed] [Google Scholar]
- Pineda JA, Lewis SB, Valadka AB, Papa L, Hannay HJ, Heaton SC, Demery JA, Liu MC, Aikman JM, Akle V, Brophy GM, Tepas JJ, Wang KK, Robertson CS, Hayes RL. Clinical significance of alphaII-spectrin breakdown products in cerebrospinal fluid after severe traumatic brain injury. J Neurotrauma. 2007;24:354–366. doi: 10.1089/neu.2006.003789. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Bansal R, Hedstrom KL, Pfeiffer SE, Rasband MN. Does paranode formation and maintenance require partitioning of neurofascin 155 into lipid rafts? J Neurosci. 2004;24:3176–3185. doi: 10.1523/JNEUROSCI.5427-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz G, Fehlings MG. Evaluation of the neuroprotective effects of sodium channel blockers after spinal cord injury: improved behavioral and neuroanatomical recovery with riluzole. J Neurosurg. 2001;94:245–256. doi: 10.3171/spi.2001.94.2.0245. [DOI] [PubMed] [Google Scholar]
- Sedarous M, Keramaris E, O'Hare M, Melloni E, Slack RS, Elce JS, Greer PA, Park DS. Calpains mediate p53 activation and neuronal death evoked by DNA damage. J Biol Chem. 2003;278:26031–26038. doi: 10.1074/jbc.M302833200. [DOI] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Baudry M, Lynch G. Brain fodrin: substrate for calpain I, an endogenous calcium-activated protease. Proc Natl Acad Sci U S A. 81:3572–3576. doi: 10.1073/pnas.81.11.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song AH, Wang D, Chen G, Li Y, Luo J, Duan S, Poo MM. A selective filter for cytoplasmic transport at the axon initial segment. Cell. 2009;136:1148–1160. doi: 10.1016/j.cell.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Susuki K, Rasband MN. Spectrin and ankyrin-based cytoskeletons at polarized domains in myelinated axons. Exp Biol Med (Maywood) 2008;233:394–400. doi: 10.3181/0709-MR-243. [DOI] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Molnár G, Oláh S, Barzó P, Tamás G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- Türeyen K, Vemuganti R, Sailor KA, Dempsey RJ. Infarct volume quantification in mouse focal cerebral ischemia: a comparison of triphenyltetrazolium chloride and cresyl violet staining techniques. J Neurosci Methods. 2004;139:203–207. doi: 10.1016/j.jneumeth.2004.04.029. [DOI] [PubMed] [Google Scholar]
- Uemoto Y, Suzuki S, Terada N, Ohno N, Ohno S, Yamanaka S, Komada M. Specific role of the truncated betaIV-spectrin Sigma6 in sodium channel clustering at axon initial segments and nodes of ranvier. J Biol Chem. 2007;282:6548–6555. doi: 10.1074/jbc.M609223200. [DOI] [PubMed] [Google Scholar]
- Uliasz TF, Hewett SJ. A microtiter trypan blue absorbance assay for the quantitative determination of excitotoxic neuronal injury in cell culture. J Neurosci Methods. 2000;100:157–163. doi: 10.1016/s0165-0270(00)00248-x. [DOI] [PubMed] [Google Scholar]
- Wang KK, Yuen PW. Development and therapeutic potential of calpain inhibitors. Adv Pharmacol. 1997;37:117–152. doi: 10.1016/s1054-3589(08)60949-7. [DOI] [PubMed] [Google Scholar]
- White BC, Sullivan JM, DeGracia DJ, O'Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci. 2000;179:1–33. doi: 10.1016/s0022-510x(00)00386-5. [DOI] [PubMed] [Google Scholar]
- Winckler B, Forscher P, Mellman I. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 1999;397:698–701. doi: 10.1038/17806. [DOI] [PubMed] [Google Scholar]
- Yang Y, Lacas-Gervais S, Morest DK, Solimena M, Rasband MN. BetaIV spectrins are essential for membrane stability and the molecular organization of nodes of Ranvier. J Neurosci. 2004;24:7230–7240. doi: 10.1523/JNEUROSCI.2125-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ogawa Y, Hedstrom KL, Rasband MN. {beta}IV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J Cell Biol. 2007;176:509–519. doi: 10.1083/jcb.200610128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu G, He Z. Signaling mechanisms of the myelin inhibitors of axon regeneration. Curr Opin Neurobiol. 2003;13:545–551. doi: 10.1016/j.conb.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Harada K. Proteolysis of erythrocyte-type and brain-type ankyrins in rat heart after postischemic reperfusion. J Biochem. 1997;122:279–285. doi: 10.1093/oxfordjournals.jbchem.a021750. [DOI] [PubMed] [Google Scholar]
- Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143:1295–1304. doi: 10.1083/jcb.143.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]