

Abstract
Ribonucleotide reductases (RNRs) catalyze the conversion of nucleotides to deoxynucleotides in all organisms. The Escherichia coli class Ia RNR is composed of α and β subunits that form an α2β2 active complex. β contains the diferric tyrosyl radical (Y•) cofactor that is essential for the reduction process that occurs on α. [Y•] in vitro is proportional to RNR activity, and its regulation in vivo potentially represents a mechanism for controlling RNR activity. To examine this thesis, N- and C-terminal StrepII-tagged β under the control of an l-arabinose promoter were constructed. Using these constructs and with [l-arabinose] varying from 0 to 0.5 mM in the growth medium, [β] could be varied from 4 to 3300 µM. [Y•] in vivo and on affinity-purified Strep-β in vitro was determined by EPR spectroscopy and Western analysis. In both cases, there was 0.1–0.3 Y• radical per β. To determine if the substoichiometric Y• level was associated with apo β or diferric β, titrations of crude cell extracts from these growths were carried out with reduced YfaE, a 2Fe2S ferredoxin involved in cofactor maintenance and assembly. Each titration, followed by addition of O2 to assemble the cofactor and EPR analysis to quantitate Y•, revealed that β is completely loaded with a diferric cluster even when its concentration in vivo is 244 µM. These titrations, furthermore, resulted in 1 Y• radical per β, the highest levels reported. Whole cell Mössbauer analysis on cells induced with 0.5 mM arabinose supports high iron loading in β. These results suggest that modulation of the level of Y• in vivo in E. coli is a mechanism of regulating RNR activity.
Ribonucleotide reductases (RNRs)1 catalyze the conversion of nucleotides to deoxynucleotides in all organisms, supplying the monomeric precursors required for DNA replication and repair (1–4). The Escherichia coli class I RNR is composed of α and β subunits with an active quaternary structure of α2β2(5). α2 houses the active site for nucleoside diphosphate reduction and additional sites that control the rate and specificity of nucleotide reduction by dNTP and ATP effectors. β contains a diferric tyrosyl radical (Y•) cofactor essential for activity (6,7). The central role of this enzyme in DNA replication and repair and the importance of balanced deoxynucleotide pool sizes for the fidelity of these processes require that RNRs be regulated by many mechanisms. In 1983, Barlow et al. (8) proposed that one mechanism of regulation might involve the control of the concentration of the essential Y•. Studies presented in this paper provide insight into the loading of β with iron and the levels of Y• in vivo, a first step in understanding the mechanism of regulation of RNR activity by modulation of the active metallo-cofactor.
The results of studies by the Reichard and Fontecave laboratories led to the model for diferric Y• radical cofactor assembly and conversion of the diferric cluster of β in which the Y• is reduced (diferric tyrosine or met-β2) to active cofactor (9,10). Our recent discovery of the 2Fe2S cluster ferredoxin, YfaE, in E. coli has resulted in extensive modifications of their original proposal. Our current model is shown in Scheme 1(11) which includes the biosynthetic pathway (A), a maintenance pathway (B), and a regulatory pathway (C).
Scheme 1.

For biosynthesis of the active, diferric Y• cofactor (pathway A, Scheme 1), apo-β2 must be loaded with Fe2+ to generate diferrous β2. The details of this process in vivo, the source of iron, and the control of delivery of the two irons per active site of β without the generation of destructive metabolites of O2 are currently unknown. Once the diferrous β2 is formed, the active cofactor can be assembled by addition of O2 and a reducing equivalent that likely is provided by reduced YfaE (11,12).
The Y• in the active cofactor is inherently unstable [the half-life of the E. coli β2 Y• is several days (13), while that of mouse β2 is 10 min (14)] and is also susceptible to one-electron reduction by small molecules such as hydroxyurea (HU) (13) or potentially a protein. The Y• in crude extracts of E. coli, for example, is rapidly reduced by an unknown mechanism. The inherent instability and reductants both lead to a diferric tyrosine cluster that is inactive in nucleotide reduction (8,10).
Pathway B (Scheme 1) describes the maintenance pathway. In this pathway, activity is regenerated by reduction of the diferric cluster to a diferrous cluster by YfaE and a ferredoxin reductase (potentially Fre) and this cluster is assembled into the active diferric Y• cluster by the biosynthetic pathway (11).
Finally, a regulatory pathway, C in Scheme 1, could have evolved to control the level of Y• as a mechanism of controlling RNR activity. In this pathway, reduction of Y• and its re-formation might be coordinated. For example, outside the S phase of the cell cycle, RNR activity might be turned off by Y• reduction, but once the S phase of the cycle begins, a mechanism for Y• regeneration, which may or may not overlap with the maintenance pathway, may be invoked. To assess the importance of these pathways in vivo, the levels of Y• and the extent of iron loading of β under different growth conditions need to be evaluated.
Our approach to testing this model in E. coli is based on our recent experiments that aimed to determine the Y•/ββ′ ratio in Saccharomyces cerevisiae under different growth conditions (15). In those studies, we constructed a FLAG-tagged β (FLAGβ) which was integrated into the genome of a number of different S. cerevisiae strains and allowed rapid purification to homogeneity of the active protein (ββ′) by affinity chromatography and quantitation of the Y•/ββ′ ratio by EPR spectroscopy. These results were then compared with those from whole cell EPR spectroscopy on the same strains, where the amount of ββ′ in each strain was determined by quantitative Western analysis. We also determined that the amount of Y• was directly correlated with nucleotide reduction activity in crude cell extracts, providing an independent assessment of the concentration of Y•. Under normal growth conditions, the studies together suggested that the cell assembles stoichiometric amounts of Y• and diiron cluster and, consequently, that modulation of the Y• concentration is not involved in regulation of RNR activity (15). However, altered lifetimes of eukaryotic Y• radicals in β, relative to those of E. coli β, suggest that a maintenance pathway might play a very important role in the observed levels of Y•(14). These studies suggest that the regulatory pathway (C, Scheme 1) is unimportant in this organism under the growth conditions thus far examined.
Our studies in S. cerevisiae ββ′ suggested that regulatory mechanisms in play in this organism are different from those suggested to be in play in E. coli. We, therefore, embarked on a similar set of experiments to gain further insight into the role of the Y• concentration in the regulation of RNR activity in E. coli. Instead of using a FLAG-tagged ββ′, we generated an N- or C-terminal StrepII-nrdB and placed the gene behind an l-arabinose (ara) promoter. Levels of β could thus be modulated by varying the concentrations of ara in the growth medium. Affinity chromatography allowed rapid isolation of β2 (<4 h) and the determination of the amount of Y• and iron per β. Whole cell EPR experiments and quantitative Western blots of β in the crude cell extracts of the same cells were analyzed as a compliment to the studies on the isolated StrepII-β. Our recent discovery of [2Fe2S]+-YfaE, a ferredoxin that is involved in maintenance and likely the biosynthesis of the diferric Y• cofactor of E. coli β, has provided us with an additional tool. Titration of met-β2 in crude cell extracts with variable amounts of YfaE in the presence of O2 and monitoring Y• formation allowed us to determine the extent of iron loading of β. Whole cell Mössbauer spectroscopy has provided independent evidence of the high iron loading of β. The studies together suggest that inside the cell, up to 4.4 mM Fe2+ can be directed (redirected) to load β. However, the level of Y• relative to iron loading is always substoichiometric, suggesting that its concentration and consequently RNR activity are regulated by other proteins or small molecules which remain to be identified.
Materials and Methods
Materials
E. coli strains used in this study are listed in Table 1. E. coli strains DHB4, RO48, SMG237, and SMG238 were kindly provided by J. Beckwith (Harvard Medical School, Boston, MA). Immun-Blot poly(vinylidene) difluoride (PVDF) membrane (0.2 µm), 10% Tris-HCl SDS−PAGE (1.0 mm) Criterion Precast Gels, a Silver Stain Plus Kit, and AG 1-X8 resin (100–200 Mesh) were from Bio-Rad. Polyclonal rabbit antibodies (Abs) against β were produced by Covance. Secondary Abs (anti-rabbit IgG, horseradish peroxidase from donkey) were from Amersham. Calf intestinal alkaline phosphatase (20 units/µL) and protease inhibitor tablets (Complete Mini, EDTA-free) were from Roche. [5-3H]Cytidine 5′-diphosphate, NH4+ salt (24.0 Ci/mmol) was from GE Healthcare. Bradford reagent and all other chemicals were obtained at the highest available purity from Sigma-Aldrich. The concentration of α was determined using an ϵ280 of 94500 M−1 cm−1. α has a specific activity of 2500 nmol min−1 mg−1. The concentration of β was determined using an ϵ280 of 65500 M−1 cm−1. E. coli thioredoxin (TR) was isolated from overproducing strain SK3981 and had a specific activity of 40 units/mg, and thioredoxin reductase (TRR) had a specific activity of 1800 units/mg (16,17). The amount of iron was determined using the ferrozine assay (18).
Table 1.
Strains Used in This Study
| strain | genotype | ref |
|---|---|---|
| K-12 | F+ | E. coli genetic stock center, CGSC catalog no. 7296 |
| BL21(DE3) | F– ompT gal dcm lon hsdSB(rB− mB−) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) | (48) |
| TOP10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 deoR nupG recA1 araD139 Δ(ara-leu)7697 galU galK rpsL(StrR) endA1 λ− | (49), (50) |
| DHB4 | Δ (ara-leu)7697 araD139ΔlacX74 galE galK rpsL phoRΔ(phoA)PvuII ΔmalF3 thi | (51) |
| RO48 | DHB4 Δ(λattL-lom)::bla nrdA′-′lacZ | (30) |
| SMG237 | RO48 dnaAsup1...MiniTn10Cmr | (30) |
| SMG238 | RO48 dnaNsup...MiniTn10Cmr | (30) |
| pBAD-nrdB | pBAD containing β | this study |
| N-S-nrdB | pBAD containing N-terminal StrepII-β | this study |
| N-C-nrdB | pBAD containing C-terminal StrepII-β | this study |
Construction of N- or C-Terminal StrepII-nrdB (N-S-nrdB or C-S-nrdB, respectively) and pBAD-nrdB
pTB2-R2 was used as a PCR template (19). To generate Ν-S-nrdB, a 5′ primer containing a StrepII tag (WSHPQFEK) and a five-amino acid linker (ALGGH) was used (5′-AAT TAA CCA TGGCGT GGA GCC ACC CGC AGT TCG AAA AAT CTC TGG GCG GCC ATA TGG CAT ATA CCA CCT TTT CAC-3′). The NcoI restriction site including the start codon is in bold, the five-amino acid linker underlined, and the StrepII tag in italics. The 3′ primer was 5′-AAC GCC TCG AGT CAG AGC TGG AAG TTA CTC AAA TCG-3′ [XhoI restriction site (bold) and the last 25 nucleotides of nrdB including the stop codon underlined]. Pfu-Ultra DNA polymerase was used for PCR following the manufacturer’s protocol (Invitrogen). The PCR product and the pBAD-myc-HIS-A vector (Invitrogen) were digested with a 10-fold excess of NcoI and XhoI and ligated with T4 DNA ligase (Promega) in a vector:insert ratio of 1:2.
To make C-S-nrdB, the forward primer, 5′-AGG ACA CAC CCA TGGCAT ATA CCA CCT TTT CAC-3′ which contains a NcoI site (bold) and the first 22 nucleotides of nrdB (underlined), was used. The reverse primer was 5′-AAC GCC TCG AGT CAT TTT TCG AAC TGC GGG TGG CCC AGA GCT GGA AGT TAC TCA AAT C-3′ which contains a XhoI site (bold) followed by a stop codon, a StrepII tag gene (italics), and the last 21 nucleotides of nrdB excluding the stop codon (underlined). The cloning procedures were the same as for N-S-nrdB. To make pBAD-nrdB (with no tag), the forward primer used to make C-S-nrdB and the reverse primer to make N-S-nrdB were used, cut with the appropriate restriction enzymes, and cloned into pBAD-myc-HIS-A. The sequences of all constructs were confirmed by DNA sequencing at the Massachusetts Institute of Technology Biopolymers Laboratory.
Cell Growth
All cell growth was carried out in a 2 L glass bioreactor [BioFlo 2000 from New Brunswick Scientific (Edison, NJ)], an O2 electrode (InPro6110/220), and a pH 2000 control module. The O2 and pH electrodes were purchased from Mettler Toledo. For the expression of N- or C-S-β, a single colony of TOP10/N-S-nrdB or TOP10/C-S-nrdB was inoculated into 5 mL of LB-ampicillin (100 µg/mL) medium and incubated at 37 °C overnight (∼16 h). Four milliliters of the overnight culture were inoculated into 2 L of LB-ampicillin (100 µg/mL) medium in the fermentor at 37 °C and pH 7.0. When A600 reached ∼0.6, ara was added to a final concentration of 0, 0.01, 0.02, 0.05, or 0.5 mM. The cells were grown for an additional 2 h (A600 ∼ 1.9), separated into four batches of 500 mL, and harvested by centrifugation at 7000g for 15 min at 4 °C. Cells were washed twice with 50 mL of buffer A [100 mM Tris, 150 mM NaCl, and 5% glycerol (pH 7.6)], flash-frozen in liquid nitrogen, and stored at –80 °C. Typically, 3.5 g of cell paste per liter of culture was obtained.
Purification of N-S-β and C-S-β.
Cell paste (∼7 g) was washed twice with 50 mL of buffer A and then suspended in 28 mL (4 mL/g of cell paste) of buffer A containing protease inhibitors (Roche) and subjected to two passes through a French press cell (14000 psi). The cell debris was removed at 60000g and 4 °C for 30 min. The supernatant was loaded onto a 5 mL Strep-Tactin Sepharose column (2.5 cm × 2 cm, IBA GmbH, St. Louis, MO). The column was washed with 150 mL of buffer A, and the protein was eluted with buffer A containing 2.5 mM desthiobiotin.
Quantitation of Y•/β in Vivo. (a) Whole Cell EPR Measurements.
The cell paste (∼1.7 g) from a 500 mL culture was suspended in 5 mL of buffer A, and two to four 220 µL aliquots were packed into calibrated EPR tubes (Wilmad). The concentration of cells was 1–3 × 1011 cells/mL determined by cell counting described in the following section. As a control, a cell suspension (500 µL) was incubated with 160 mM hydroxyurea (HU) at 37 °C for 1 h before being packed into an EPR tube. The remaining cell suspension (∼4 mL) was used for Western blot analysis. EPR spectra were recorded using a Bruker ESP-300 X-band (9.4 GHz) spectrometer at 77 K using a liquid nitrogen-cooled finger dewar. Typical parameters used to record the spectra were as follows: 9.38 GHz frequency, 0.05 mW power, 1.5 G modulation amplitude, 2.52 × 103 time constant, and 50−100 scans. The standards for spin quantification were CuSO4(20) and purified E. coli β with 1–882 µM Y•(21). The Y• content was determined by the drop-line correction method (21).
(b) Cell Counting
After the 2 h growth subsequent to ara induction, 1 mL of the cell culture was rapidly cooled on ice and used to count the cells. Serial dilutions of the culture were made and plated on nine LB-Agar plates. The dilutions were made so that there were 100–200 colonies on each plate after growth at 37 °C, overnight. The standard deviation estimated from the cell counting was ~30%.
(c) Data Analysis
The EPR spectra were normalized to the collection parameters and the number of cells. To obtain the EPR spectrum of Y•, the spectrum of the HU-treated cells was subtracted from the spectrum of the untreated cells from the same culture. The total spin was determined by double integration of the difference spectrum and compared to the Y• and CuSO4 standards. The amount of Y• in vivo was calculated from the spin concentration, the cell volume (0.85 fL) (22), and the number of cells per milliliter determined by cell counting. Six to eight 2 L growths were carried out for each set of growth conditions. Typically, the Y• concentration was measured on two to four samples within each growth with a 5–10% standard deviation. Comparison of different growths revealed the in vivo concentration of Y• had a standard deviation of ~30%.
Quantitative Western Blot Analysis of N-S-β.
To ∼4 mL of remaining culture from the whole cell EPR measurements were added protease inhibitors (Roche), and the suspension was passed twice through a French pressure cell (14000 psi). The cell debris was spun down at 60000g and 4 °C for 20 min. The protein concentration of the crude cell extracts was determined by the Bradford assay using BSA as a standard. Part of the crude cell extract was diluted in 2× Laemmli buffer, flash-frozen in liquid nitrogen, and stored at –80 °C. No degradation of β was observed under these storage conditions. To determine the concentration of β in the crude cell extracts, purified N-S-β (5–19 ng) and duplicates of the crude cell extracts (0.025–8 µg) were loaded onto the same 10% SDS gel. Low protein binding tips and Eppendorf tubes (Axygen) were used to increase reproducibility. The proteins were then transferred to PVDF membranes (Bio-Rad) at 200 mA and 4 °C for 1 h in 25 mM Tris, 195 mM glycine, 15% (v/v) methanol, and 0.01% (w/v) SDS. Primary and secondary Abs were used in a 5000-fold dilution. The PVDF membranes were developed using DuraWest chemiluminescence reagents (Pierce). The Western signals were detected using a CCD camera (ChemiDoc XRS, Bio-Rad) and quantified using Bio-Rad’s Quantity One Software. The standard curve was generated from a plot of variable amounts of purified N-S-β, including the control without N-S-β as the background signal. The data were analyzed using a linear fit weighted against the standard deviation of each of the measured band intensities, and the resulting line was used to calculate the unknown concentration of β in the crude cell extracts (Supporting Information, Figure S1). For each extract, duplicates of four different concentrations were examined. The concentration of β inside the cells was calculated from the concentration of β in the crude cell extract, the number of cells determined by cell counting, and the cell volume of 0.85 fL (22).
Specific Activity of Partially Purified Crude Cell Extracts. (a) Purification by Size Exclusion Chromatography
All steps were carried out at 4 °C. Cell paste (∼1 g) was washed twice with 20 mL of 50 mM HEPES, 15 mM MgSO4, 1 mM EDTA, and 3 mM ferrozine (pH 7.6) and suspended in 4 mL of the same buffer containing protease inhibitors. Cell lysis was effected by two passes through a French pressure cell at 14000 psi. Cell debris was removed by centrifugation at 60000g for 20 min. Ammonium sulfate was added to the supernatant to 60% saturation (390 mg/mL). After being stirred for 30 min, the precipitate was collected by centrifugation at 60000g for 20 min, redissolved in 1 mL of buffer A, and loaded onto a Sephadex G-50 column (0.7 cm × 15 cm) pre-equilibrated with 50 mM HEPES, 15 mM MgSO4, and 1 mM EDTA (pH 7.6). Fractions of 500 µL were collected. The fraction with the highest protein concentration determined by the Bradford assay was used immediately for the activity assays. A sample of 200 µL from the same fraction was diluted with 2× Laemmli buffer, frozen in liquid nitrogen, and stored at –80 °C for Western blot analysis.
(b) Measurement of the Activity of β in the Crude Cell Extract
Western blot analysis and Bradford assays described above were used to determine the concentration of β and of total protein in the crude cell extract, respectively. To measure the activity of β, a reaction mixture with a final volume of 170 µL containing different amounts of crude cell extract (0.1–3 mg/mL), 3 mM ATP, 1 mM NADPH, 1 mM [3H]CDP (4290 cpm/nmol), 30 µM TR, 0.5 µM TRR, and 10 µM α (2500 nmol min−1 mg−1) was incubated for 4 min at 25 °C. The reaction was initiated by addition of [3H]CDP. Aliquots of 30 µL (taken every minute from 0 to 4 min) were removed and quenched in boiling water. The production of [3H]dCDP was analyzed using the method of Steeper and Stewart (23).
Quantitation of Iron Loading in β Using YfaE. (a) Sample Preparation
Cell paste (∼1 g) was washed with 25 mL of buffer A and suspended in 2 mL of buffer A containing protease inhibitors. The crude cell extract was prepared as described above. Reduced YfaE was prepared as recently described (11). Reduced YfaE (0–76 µM) was taken from a glovebox (M. Braun, Newburyport, MA) via a gastight syringe (Hamilton) and added to crude cell extracts containing 2–6 µM β in a final volume of 250 µL in buffer A. The samples were incubated at 4 °C for 5 min, and then O2 was blown over the surface of each sample, followed by gentle vortexing. Each sample was then transferred into a calibrated EPR tube to record the spectrum as described above. Four samples using different concentrations of reduced YfaE were prepared for each set of growth conditions.
(b) Data Analysis
The EPR spectrum of a sample from each set of conditions is composed of Y• and other paramagnetic species in the crude cell extracts. To quantitate the Y• in each sample, the Y• standard from purified E. coli β was fit to the low field side of each crude cell extract spectrum. The two spectra were subtracted, and the resulting background signal was compared to background signals from other samples and the background signal from the HU-treated cells described above. The similarity of the background EPR signals is indicative of the reproducibility of the growth conditions and fitting method (Supporting Information, Figure S2).
Titrations of E. coli SMG237 Crude Cell Extract with Reduced YfaE
Sample preparation, Western blot analysis, EPR measurements, and data analysis were performed as described above.
Addition of met-β to TOP10/N-S-nrdB Crude Cell Extracts and Titration with Reduced YfaE
Crude cell extracts from TOP10/N-S-nrdB cells (∼1 g) grown in the absence of ara were prepared as described above. met-β (3.1 µM), reduced YfaE (0–64 µM), and the crude cell extract were mixed in a final volume of 250 µL in buffer A. Cluster reassembly and EPR analysis were performed as in the previous titration experiments (Supporting Information, Figure S3).
Determination of the Stability of Y•. (a) Purified N-S-β
To establish the stability of Y• in N-S-β, 220 µL aliquots of N-S-β (∼20 µM) were packed into EPR tubes immediately after elution from the Strep-Tactin Sepharose column. The EPR spectra were recorded at 77 K. The samples were then thawed and kept on ice for 1 h before being refrozen in liquid nitrogen, and the EPR spectrum was again recorded. This cycle was carried out twice. No loss of radical was detected over 2 h at 4 °C.
(b) In the TOP10/N-S-nrdB Crude Cell Extracts
To determine the stability of Y• during the purification process, cells (∼0.5 g) grown for 2 h with 0.5 mM ara were suspended in buffer A containing protease inhibitors, passed once through the French pressure cell (14000 psi), and spun down at 60000g and 4 °C for 10 min. Aliquots of the crude cell extract (12.4 mg/mL) were assessed by EPR spectroscopy at 77 K. The samples were then thawed and kept on ice for 30 min before being refrozen in liquid nitrogen, and the spectrum was again recorded. The freeze−thaw cycles were repeated five times. The spectra were analyzed as described above for whole cell EPR. A ~20% loss of the Y• signal was found during the period of 3 h and 20 min of the freeze−thaw cycles (Supporting Information, Figure S4).
Preparation of the Whole Cell Mössbauer Sample
A 57Fe3+ stock solution (158 mM) was prepared as previously described (11). TOP10 cells were grown in LB medium which contains ∼12 µM 56Fe determined by the ferrozine assay. One milliliter of an overnight culture of TOP10/pBAD-nrdB was inoculated into 500 mL of LB medium supplemented with 100 µg/mL ampicillin and 50 µM 57Fe3+ in a 2.8 L baffle flask. The cells were grown at 37 °C and 200 rpm until A600 was ∼0.5, at which time l-ara was added to a final concentration of 0.5 mM. The cells were grown for an additional 2 h (A600 ∼ 1.7) before being harvested by centrifugation at 7000g for 20 min at 4 °C. The cells were washed once with 50 mL of buffer A, and the wet cell paste (∼450 µL) was packed directly into Mössbauer cups. A control experiment was carried out under identical growth conditions, but no ara was added.
Mössbauer Spectroscopy
Mössbauer spectra were recorded on a spectrometer from Web Research (Edina, MN), equipped with a SVT-400 cryostat from Janis Research Co. (Wilmington, MA). Spectra were collected in constant acceleration mode in transmission geometry. Isomer shifts are quoted relative to the centroid of α-Fe at room temperature. Spectra were analyzed with WMOSS from Web Research.
Results
Determination of the Iron and Y• Content in Purified N-S-β and C-S-β.
The first approach to determining the in vivo concentration of Y• per β and Fe per β was to construct a nrdB fusion with a StrepII tag (WSHPQFEK) at either its N- or C-terminus and to place them in a pBAD vector (24). Each plasmid was transformed into TOP10 cells, and the levels of β were modulated by the concentrations of ara in the growth medium. The goal was to rapidly isolate N-S-β or C-S-β by affinity chromatography and to determine the loading of the diferric Y• cofactor with minimal loss of Y•. This approach has been successful in the rapid purification of S. cerevisiae ββ′ (15). A StrepII tag was chosen for the fusion because in contrast with the more commonly used FLAG and His tags, the StrepII tag has minimal number of side chains that might bind Fe2+ and thus might interfere with iron cluster assembly in vivo.
C-S-β was generated with no linker between β and the StrepII tag as the C-terminal tail of all β subunits is largely responsible for interaction with the α subunit, and thus, any manipulation of this end was expected to result in a construct inactive in nucleotide reduction (25). This construct led to efficient expression of β that was rapidly purified to homogeneity using the Strep-Tactin Sepharose column (Figure 1A, lane 5). The activity assay confirmed that C-S-β is inactive.
Figure 1.

Purification of C-S-β (A) and N-S-β (B) from cells induced with 0.01 mM ara monitored by SDS−PAGE: lane 1, molecular mass standards; lane 2, crude cell extracts; lane 3, flow through from the Strep-Tactin Sepharose column; lane 4, wash from the Strep-Tactin Sepharose column; and lane 5, protein eluted with desthiobiotin from the Strep-Tactin Sepharose column.
Previous N-terminally tagged constructs of β have met with variable success (26,27). Thus, a StrepII tag followed by linkers of variable lengths (GA, ALGGH, and GSGGSG) was examined (data not shown). The construct with the ALGGH linker was chosen as it gave the best binding to the Strep-Tactin Sepharose column. This construct, N-S-β, while binding more weakly to the affinity resin than C-S-β, resulted in its purification to homogeneity (Figure 1B). In this case, the protein is active.
In the case of both constructs, protein purification was complete in 3−4 h, and the expression levels could be induced by addition of ara. Western analysis revealed that the levels of β varied from 4.4 µM in the absence of ara to 3.3 mM in the presence of 0.5 mM ara (Table 2). The TOP10 E. coli strain was chosen to ensure that the level of expression of β in all cells was uniform (24,28).
Table 2.
In Vivo Concentrations of N-S-β and Y• Determined by Whole Cell EPR Spectroscopy and Quantitative Western Analysis
| [ara] (mM) | [β]a (µM) | [Y•] (µM) | Y•/β |
|---|---|---|---|
| 0 | 4.4 ± 0.8 (3) | − | − |
| 0.01 | 16.8 ± 2.8 (3) | − | − |
| 0.02 | 105 ± 7.5 (6) | 25.3 ± 2.3 | 0.24 ± 0.02 |
| 0.05 | 244 ± 48 (5) | 56.6 ± 21 | 0.25 ± 0.09 |
| 0.5 | 3315 ± 960 (8) | 421 ± 153 | 0.13 ± 0.04 |
The number of experiments is given in parentheses.
To analyze the effect of the expression level of β on the level of diferric Y• generated, 2 L growths in an O2/pH-controlled fermentor were carried out. Expression of β was induced with variable amounts of ara; the cells were washed extensively, and the protein was purified by affinity chromatography. For each sample, the amount of Y• was determined by EPR spectroscopy, the protein concentration was determined by ϵ280 (65.5 mM−1 cm−1), and the iron content was determined by the ferrozine assay. For N-C-β, the activity was determined using the spectrophotometric assay. The results are summarized inTable 3.
Table 3.
Iron, Y•, and Specific Activity of Purified N- and C-S-β
| N-S-β |
C-S-βa |
||||
|---|---|---|---|---|---|
| [ara] (mM) | Fe/β | Y•/βc | SA | Fe/β | Y•/βc |
| 0.005 | NDb | NDb | NDb | 2.0 ± 0.05 | 0.35 ± 0.09 (3) |
| 0.01 | 1.75 ± 0.05 | 0.25 ± 0.08 (4) | 3240 ± 547 | 1.8 ± 0.1 | 0.34 ± 0.07 (5) |
| 0.02 | 1.85 ± 0.15 | 0.29 ± 0.09 (5) | 3120 ± 562 | 1.8 ± 0.1 | 0.33 ± 0.08 (5) |
| 0.05 | 1.85 ± 0.15 | 0.26 ± 0.12 (3) | 3116 ± 570 | 1.85 ± 0.2 | 0.25 ± 0.07 (2) |
| 0.5 | 1.7 ± 0.1 | 0.27 ± 0.13 (4) | 3182 ± 530 | 1.8 ± 0.03 | 0.31 ± 0.06 (3) |
C-S-β is inactive.
Not determined.
The number of experiments performed is given in parentheses.
The results revealed that regardless of the levels of β expression, the Y•/β ratio is 0.25−0.35 and the Fe/β ratio is 1.7−1.85. The specific activity of N-S-β also correlates with the amount of Y• per β. Similar amounts of Y• and Fe per β in both constructs suggest that the StrepII tag does not interfere with diferric Y• cofactor assembly. While control experiments revealed that as much as 30% of Y• could be reduced in the crude cell extract if left for ∼3 h at 4 °C, loading onto the affinity column within 30 min at 4 °C minimized this loss. Two results with N-S-β and C-S-β are striking. The first is that the amount of Y• is substoichiometric, suggesting that the level of Y• might be regulating RNR activity as originally proposed by Barlow et al. (8). The second is that the iron content of β, even when β is expressed at 3 mM levels (Table 2), is close to stoichiometric (Table 3). Thus, the majority of the iron in the cells is directed (redirected) toward β, as demonstrated by whole cell Mössbauer experiments (see below).
Whole Cell EPR, Cell Counting, and Western Blot Analysis
Our previous experiments on cofactor assembly and regulation of ββ′ in wt S. cerevisiae showed that whole cell EPR and quantitative Western blots provide an alternative measurement of the Y•/β ratio in vivo. However, previous whole cell EPR experiments and rocket immunoelectrophoresis experiments on wt and thymine-starved E. coli by Sjöberg et al. failed to detect Y• when β was 0.3% (0.9%) of the total protein in crude cell extracts. However, when they analyzed E. coli strain KK546 where β was increased to 2% of the total protein in crude cell extracts, the Y• was readily detected (29). These early studies thus suggested that growth of cells with ara induction would be required to detect Y• in E. coli by whole cell EPR methods.
Initially, we used whole cell EPR methods to examine wt E. coli K-12, BL21-DE3 (Gold), TOP10, DHB4, and RO48 strains (Table 1). No Y• was detected. We also examined several strains of E. coli DHB4 with mutations in dnaA (Ala345Ser, SMG237) and dnaN (Gly157Cys, SMG238) which the Beckwith laboratory reported had 8- and 4-fold higher levels of β relative to the wt strain, respectively (30). Again, no Y• was observed.
We thus turned to the TOP10 cells transformed with N-S-nrdB and carried out cell growth in the presence of variable concentrations of ara under conditions identical to those described above for analysis of purified β. The lowest level of ara induction at which a whole cell EPR signal of Y• could be quantified was 0.02 mM. Signals were also detected at 0.05 and 0.5 mM. Typical results are shown in Figure 2.
Figure 2.
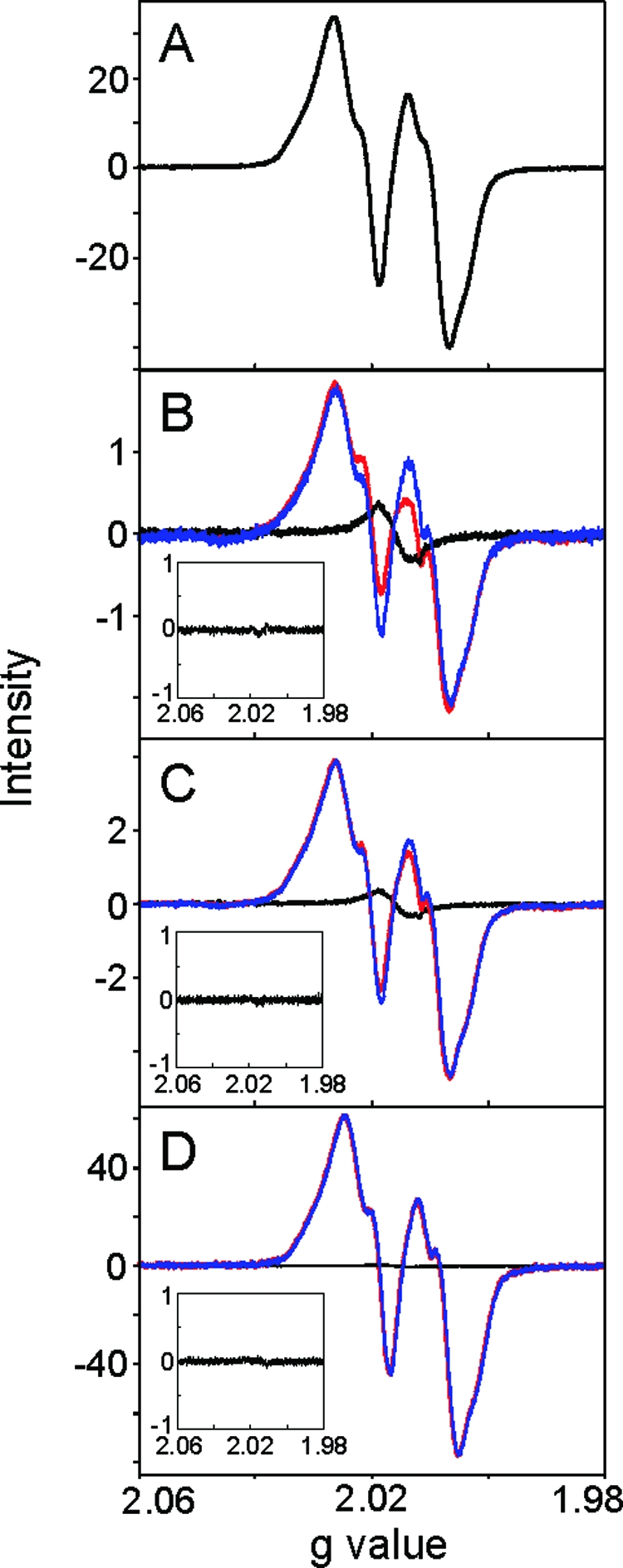
EPR spectrum of purified N-S-β (A) and whole cell EPR spectra of E. coli TOP10/N-S-β cells grown in the presence of 0.02 (B), 0.05 (C), and 0.5 mM l-ara (D). In panels B−D, the EPR spectra of the whole cells (red), the cells treated with HU (black), and subtraction between the two (blue) are shown. All the spectra were normalized to the collection parameters and the cell number. The insets show the subtraction of two background spectra from two independent growths.
As previously observed in whole cell EPR experiments with S. cerevisiae, when the levels of Y• are low, the contributions from unidentified paramagnetic species are readily apparent. To remove the EPR signal associated with these species, part of the cells from the same growth was treated with 160 mM HU for 1 h. Experiments as a function of time revealed that these conditions are sufficient for HU to reduce all Y• and leave behind the background signal. The reproducibility of the background signal between two independent growths is shown as a subtraction of two background spectra (Figure 2 B−D, inset), which reveals the complete disappearance of the signal. To obtain the spectrum of only Y•, the spectra before and after HU treatment were normalized to the collection parameters and the number of cells packed into the EPR tubes (see below). The resulting spectra from the subtractions are shown in Figure 2 B−D (blue). These spectra are very similar to the Y• spectrum from in vitro reconstituted β (Figure 2A). Each spectrum is then integrated and compared to the spectrum of CuSO4 and wt β-Y• standards. At each ara concentration, cell growth was repeated three times (Table 2). Furthermore, the quantification of Y• from each growth was repeated five to eight times (Table 2).
Cell Counting
Calculating the in vivo concentration of Y• requires knowledge of the number of cells in each EPR tube. To obtain the cell concentration, nine dilutions of the original cell culture were plated on LB-agar plates and the colonies were counted after growth at 37 °C for 16 h. The average cell number was used to calculate the Y• concentration in vivo assuming a cell volume of 0.85 fL (22). The calculated concentrations for Y•, the average values from all measurements ± the standard deviation, are summarized in Table 2.
Western Analysis of Crude Cell Extracts of TOP10/N-S-β.
The amount of β in crude cell extracts from each of the growths was determined using Western analysis. Transfer conditions were optimized to ensure complete transfer but not overtransfer from the SDS−PAGE gel (15). The concentration of crude cell extract loaded on the gel was chosen so that the estimated amount of β would fall within the range of the optimized standard curve generated with purified N-S-β (5–19 ng). Duplicates of four different concentrations of each extract were analyzed. Typical results are shown in Figure 3 and Figure S1 of the Supporting Information. The in vivo concentrations of β at different levels generated with different concentrations of ara are summarized in Table 2.
Figure 3.

Quantitative Western blot analysis to determine the in vivo concentration of β in TOP10/N-S-β cells in the presence and absence of l-ara induction. The amount of protein from crude cell extract loaded under each set of growth conditions is indicated above each lane. A representative standard curve of purified N-S-β (5–19 ng) is also shown.
The amount of Y• from the whole cell EPR experiments and the amount of β from the Western blot analysis allowed calculation of the Y•/β ratio (Table 2). Cells induced with 0.02 and 0.05 mM ara gave Y•/β ratios of 0.24 and 0.25, respectively. These values are very similar to the results with purified β (Table 3). The Y•/β ratio in the growth with 0.5 mM ara was 0.13. The standard deviation relative to the other concentrations is larger due to the huge overexpression of β and the larger dilutions required for the Western analysis. The results show that as with purified β, substoichiometric amounts of Y• per β are observed, consistent with a model in which the concentration of Y• might regulate RNR activity.
Activities of N-S-β in Partially Purified Crude Cell Extracts
Our recent experiments with S. cerevisiae showed that the specific activity of ββ′ could be measured in partially purified crude cell extracts and also provided another independent measurement of the amount of Y•(15). Experiments using high-specific activity [14C]CDP, however, under a variety of growth conditions in crude cell extracts showed low activity (152 nmol min−1 mg−1), similar to previous reports (8). When the crude cell extracts were partially purified by (NH4)2SO4 fractionation followed by Sephadex G-50 chromatography, the activity was increased 10-fold but was still lower than that of purified β, suggesting that Y• is reduced during the workup (Table 4). Thus, direct measurement of activity in extracts cannot be used as an independent measure of the concentration of Y•.
Table 4.
Specific Activities of N-S-β in Partially Purified Crude Cell Extracts of TOP10/N-S-β
| [l-ara] (mM) | specific activitya(nmol min−1 mg−1) |
|---|---|
| 0.01 | 1474 ± 408 (5) |
| 0.02 | 1296 ± 128 (3) |
| 0.05 | 920 (1) |
| 0.5 | 928 ± 434 (3) |
The number of experiments is shown in parentheses.
Titrations of Crude Cell Extracts with Reduced YfaE To Determine Iron Loading of β.
The experiments with purified β suggested that despite the low levels of Y•, the protein was almost completely loaded with iron (Table 3). Our recent identification of YfaE, a ferredoxin involved in cofactor maintenance and biosynthesis, provided us with a tool for independently measuring iron loading of β in crude cell extracts (11). In a typical experiment, the level of N-S-β from the crude cell extracts was adjusted to 2.3–5.9 µM, determined by Western blot analysis. The extracts were then titrated with reduced YfaE (0–76 µM) to reduce the diferric cluster to the diferrous cluster and then mixed with O2 to reassemble the active diferric Y• cofactor (Scheme 1). The Y• of each sample was then quantitated by EPR spectroscopy. The results are shown in Figure 4 (red signals, A−D) and summarized in Table 5. Strikingly, cells grown with induction of 0.01, 0.02, or 0.05 mM ara contain 1 Y• per β, indicating that β is fully loaded with iron even when the in vivo concentration of β is as high as 244 µM (Tables 2 and 5). In the case of 0.5 mM ara induction, the number of Y• radicals per β drops to 0.67, suggesting that the cell has reached its limit in terms of delivery of iron to β. However, given the high in vivo concentration of β [∼3.3 mM (Table 2)] under these induction conditions and the stoichiometry of 2 Fe atoms required per Y• generated, up to 4.4 mM iron has been directed or redirected to load β. These results support the observations with purified β. Thus, β appears to be in the met-β state, ready to regenerate the Y• when needed.
Figure 4.

EPR spectra of the Y• generated when TOP10/N-S-β crude cell extracts are optimally titrated with reduced YfaE and O2. Cells were induced with (A) 0.01, (B) 0.02, (C) 0.05, and (D) 0.5 mM l-ara. The EPR spectra of the crude cell extracts (red), Y• (blue) optimized to the low field side of the spectrum, and subtraction between the two (black) are shown. All the spectra were normalized to the collection parameters. The insets show the subtraction between two background spectra.
Table 5.
Titrations of TOP10/N-S-β Crude Cell Extracts with Reduced YfaE
| 0.01 mM ara |
0.02 mM ara |
0.05 mM ara |
0.5 mM ara |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| [β]a (µM) | [YfaE] (µM) | Y•/β | [β]a (µM) | [YfaE] (µM) | Y•/β | [β]a (µM) | [YfaE] (µM) | Y•/β | [β]a (µM) | [YfaE] (µM) | Y•/β |
| 2.5 | 0 | 0.25 | 2.3 | 0 | 0.24 | 5.9 | 0 | 0.23 | 5.6 | 0 | 0.20 |
| 2.5 | 7.6 | 0.54 | 2.3 | 7.6 | 0.46 | 5.9 | 25 | 0.59 | 5.6 | 25 | 0.66 |
| 2.5 | 15.3 | 1.00 | 2.3 | 15.3 | 1.05 | 5.9 | 51 | 0.98 | 5.6 | 51 | 0.66 |
| 2.5 | 22.9 | 0.95 | 2.3 | 22.9 | 1.00 | 5.9 | 76 | 1.00 | 5.6 | 76 | 0.67 |
The actual concentration of β in the crude cell extracts at each concentration of ara used in the induction is given in Table 2.
Whole Cell Mössbauer: Detection of Diferric β at 0.5 mM ara
As a further control to show that β generated subsequent to induction with 0.5 mM ara was highly loaded with iron in vivo, we examined by Mössbauer spectroscopy samples of whole cells supplemented with 57Fe. Given the well-described spectrum of the diferric cluster of β, this method was expected to provide an independent verification of iron loading. The spectrum of cells 2 h subsequent to addition of ara to the growth medium is shown in Figure 5A and is dominated by two quadrupole doublets which are assigned to the diferric cluster of β. In addition, there is a small peak at 2.8 mm/s, which is typical of the high-energy line of high-spin Fe(II), and a shoulder at 0.9 mm/s (indicated by an arrow). These features are also observed in the spectrum of the control sample (Figure 5B) in which cells were treated in a fashion identical to that for the experiment except that no ara was added. The observed signals (Figure 5B) are the superposition of the spectral features of all Fe species present in the cell, according to their relative concentrations. Approximately 40% of the iron is high-spin Fe(II) (δ ≈ 1.3 mm/s, ΔEQ ≈ 2.9 mm/s), and the remainder represents several overlapping quadrupole doublets, which may include Fe/S cluster-containing proteins (primarily [4Fe-4S]2+ and [2Fe-2S]2+), low-spin Fe(II) hemes, and possibly fast-relaxing high-spin Fe(III). These features can be approximated as a broad quadrupole doublet with a δapp of 0.46 mm/s and a ΔEQapp of 0.94 mm/s which accounts for ∼60% of the total iron. Mössbauer spectra collected over a wider range of Doppler velocities reveal that the samples contain only a small amount of paramagnetic species (Figure S5, <10% of total iron). Subtraction of the features observed in the spectrum of the control [15% of Fe(II) and 9% of the broad quadrupole doublet] from the spectrum in Figure 5A yields the spectrum of diferric species assumed to be associated with β, which accounts for 76 ± 4% of the total iron (Figure 5C). The spectrum can be simulated with parameters [δ(1) = 0.46 mm/s, ΔEQ(1) = 2.40 mm/s, δ(2) = 0.54 mm/s, and ΔEQ(2) = 1.65 mm/s] that are similar to those reported for the diferric cluster of β (13,31). These experiments demonstrate high iron loading of β and suggest that subsequent to ara induction iron uptake is enhanced. They also suggest that whole cell Mössbauer spectroscopy may be a useful tool for monitoring iron movement in vivo.
Figure 5.

Mössbauer spectra of a sample of whole cells grown on 57Fe-supplemented medium. The production of β was induced with 0.5 mM ara (A), and the cells were grown for an additional 2 h. The control sample (B) is identical to A except that ara was eliminated. The solid lines in panel B represent two quadrupole doublets, one associated with high-spin Fe(II) species and the second with all other Fe species in the cell (see the text for parameters). Removal of the spectral component of these Fe species from the experimental spectrum (A) yields a spectrum of the diferric cluster of β (C), which can be simulated with two quadrupole doublets (solid line) using the parameters given in the text.
Discussion
The genomics revolution has identified through operon organization the importance of biosynthetic pathways for the synthesis of many metallo-cofactors and the redundancy of these processes (32). Identification of genes in the pathway has evoked tremendous interest in understanding the complexity of cofactor assembly. In addition to biosynthetic pathways, it is now clear that many metallo-cofactors can be damaged during catalysis (33–35) or during oxidative stress. Thus, repair or maintenance pathways appear to be universal as well. A recent study of the Pseudomonas aeruginosa class Ia RNR (36) provides one of the most compelling examples of the importance of these pathways for RNR cluster assembly. The Y• of this RNR is very unstable, and its re-formation occurs readily upon addition of O2. This observation is reminiscent of the early studies on the herpes viral RNR (37) and the instability of the mouse RNR (14).
In the case of the class Ia RNRs in E. coli, the biosynthetic and maintenance pathways were proposed many years ago. Only recently, however, has the first protein player directly involved in these pathways been identified, a 2Fe2S ferredoxin, YfaE (11). In vitro studies revealed that met-β2 can be reassembled into the active cofactor when titrated with reduced YfaE followed by addition of O2. To obtain additional support for the maintenance pathway, we have constructed N- and C-S-β, the concentration of which can be regulated by the levels of ara in the growth medium. Use of these constructs has allowed measurement of the amount of Y• per β by two independent methods, whole cell EPR spectroscopy and quantitative Western blots, and by rapid purification of N- or C-S-β via affinity chromatography followed by analysis by EPR spectroscopy and analytical methods (determination of β and Fe concentrations).
Purifications of N-S-β and C-S-β revealed ∼0.25−0.3 Y• radical per β over a broad range of expression levels. Control experiments reveal that this substoichiometric radical is not associated with radical loss during purification or the presence of apo β2. Thus, the explanation that we currently favor is that in the asynchronous cultures, a high Y•:β ratio is present only in the S-phase of the cell cycle, which represents ∼37% of the total population (38). Measurement of the Y•/β ratio by whole cell EPR and Western blot analysis provides an independent verification of this ratio. The substoichiometric Y• levels suggest that there is a mechanism in vivo to reduce this radical, and turn off RNR activity, as well as to regenerate it.
Measurements of N-S-β activity in the crude cell extracts were attempted as a third method of determining the Y•:β ratio. Previous studies have revealed that the activity of E. coli RNR in vitro and the activity of S. cerevisiae RNR in the crude cell extracts is correlated with the amount of Y• per β (15,39). Efforts to measure activity directly in crude cell extracts in E. coli, however, revealed levels substantially lower than those expected on the basis of the measured amount of Y•. These results are similar to those reported by Sjöberg et al. (8,29), which revealed activity in crude cell extracts incompatible with production of sufficient dNTPs for DNA replication. However, when β was partially purified, an increase in activity 10 times relative to that of the crude cell extracts is detected. These data suggest that crude cell extracts contain a small molecule or protein capable of reducing Y•. As noted above, this factor may also play a role in regulation of RNR activity. Thus, the activity assay provides further independent support that Y• is present in substoichiometric amounts relative to β and suggests an important role for a maintenance mechanism in modulating the levels of Y• and, as a consequence, the rates of deoxynucleotide production.
Our recent discovery that YfaE in crude cell extracts results in highly efficient cofactor assembly has provided an important new tool, a method for measuring iron loading. Our titration of crude cell extracts of cells in which expression levels of N-S-β were induced with 0.01, 0.02, or 0.05 mM ara revealed 1 Y• per β and 2 Fe atoms per β! This is the first time that stoichiometric loading of β in any organism has been observed. The most amazing result is that even when the β concentration is ∼250 µM it is fully loaded with active cofactor. Our YfaE titration studies and whole cell Mössbauer data further indicate that high levels of iron loading have occurred even at millimolar β concentrations. This result suggests that the cell is able to take up iron from the growth medium and direct (redirect) iron into β within the cell at the expense of other iron-requiring or -binding proteins.
Our studies provide the foundation for experiments aimed at addressing a major unresolved issue, movement of iron within the cell. The availability of a wide range of deletion mutants related to iron uptake and storage (40,41) and the choice of appropriate growth conditions should allow us to generate a moderately homogeneous source of iron and, subsequent to ara induction of β, to monitor iron movement by whole cell Mössbauer spectroscopy. Finally, our unexpected observation of 1 Y• per β suggests that now might be an appropriate time to revisit spectroscopically the structure of intermediate X in diferric cluster assembly (42–46). In all previous studies, the spectroscopy was complicated by the observation that only 0.5 X per β was observed despite the detection of one cluster per β at the end of the assembly process. Thus, 0.5 equivalent of an unidentified precursor to the final diferric cluster is unaccounted for. If 1.0 X per β can now be generated, the 17O ENDOR experiments might prove to be informative (47).
Supporting Information Available
A typical standard curve of purified N-S-β used for determining the level of β in the crude cell extracts of TOP10/N-S-β by Western blot analysis (Figure S1), comparison of the EPR spectrum from the HU-treated cells and the background spectra from the titrations of the crude cell extracts of TOP10/N-S-β with reduced YfaE (Figure S2), titrations of met-β2 in the presence of TOP10/N-S-β crude cell extracts with reduced YfaE (Figure S3), stability of Y• of N-S-β in the crude cell extracts of TOP10/N-S-β (Figure S4), and 4.2 K, 53 mT whole cell Mössbauer spectra collected over a wide range of Doppler velocities (Figure S5). This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: α, ribonucleotide reductase large subunit; ara, l-arabinose; β, ribonucleotide reductase small subunit; apo-β2, iron-free form of β2; met-β2, radical-free β2 that contains a diiron(III/III) cluster; RNR, ribonucleotide reductase; wt, wild type; Y•, tyrosyl radical; YfaE, 2Fe2S ferredoxin; Abs, antibodies; HU, hydroxyurea; buffer A, 100 mM Tris, 150 mM NaCl, and 5% glycerol (pH 7.6); N-S-β, N-terminal StrepII-tagged β; C-S-β, C-terminal StrepII-tagged β; PVDF, poly(vinylidene) difluoride; TR, thioredoxin; TRR, thioredoxin reductase.
Supplementary Material
References
- Nordlund P.; Reichard P. (2006) Ribonucleotide reductases. Annu. Rev. Biochem. 75, 681–706. [DOI] [PubMed] [Google Scholar]
- Jordan A.; Reichard P. (1998) Ribonucleotide reductases. Annu. Rev. Biochem. 67, 71–98. [DOI] [PubMed] [Google Scholar]
- Licht S., and Stubbe J. (1999) Mechanistic investigations of ribonucleotide reductases, in Comprehensive Natural Products Chemistry (Poulter C. D., Ed.) pp 163–203, Elsevier Science, New York. [Google Scholar]
- Kolberg M.; Strand K. R.; Graff P.; Andersson K. K. (2004) Structure, function, and mechanism of ribonucleotide reductases. Biochim. Biophys. Acta 1699, 1–34. [DOI] [PubMed] [Google Scholar]
- Wang J.; Lohman G.; Stubbe J. (2007) Enhanced subunit interactions with gemcitabine-5′-diphosphate inhibit ribonucleotide reductases. Proc. Natl. Acad. Sci. U.S.A. 104, 14324–14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöberg B. M.; Reichard P. (1977) Nature of the free radical in ribonucleotide reductase from Escherichia coli. J. Biol. Chem. 252, 536–541. [PubMed] [Google Scholar]
- Sjöberg B. M.; Reichard P.; Gräslund A.; Ehrenberg A. (1978) The tyrosine free radical in ribonucleotide reductase from Escherichia coli. J. Biol. Chem. 253, 6863–6865. [PubMed] [Google Scholar]
- Barlow T.; Eliasson R.; Platz A.; Reichard P.; Sjöberg B. M. (1983) Enzymic modification of a tyrosine residue to a stable free radical in ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A. 80, 1492–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontecave M.; Eliasson R.; Reichard P. (1987) NAD(P)H:flavin oxidoreductase of Escherichia coli. A ferric iron reductase participating in the generation of the free radical of ribonucleotide reductase. J. Biol. Chem. 262, 12325–12331. [PubMed] [Google Scholar]
- Fontecave M.; Eliasson R.; Reichard P. (1989) Enzymatic regulation of the radical content of the small subunit of Escherichia coli ribonucleotide reductase involving reduction of its redox centers. J. Biol. Chem. 264, 9164–9170. [PubMed] [Google Scholar]
- Wu C. H.; Jiang W.; Krebs C.; Stubbe J. (2007) YfaE, a ferredoxin involved in diferric-tyrosyl radical maintenance in Escherichia coli ribonucleotide reductase. Biochemistry 46, 11577–11588. [DOI] [PubMed] [Google Scholar]
- Stubbe J.; Riggs-Gelasco P. (1998) Harnessing free radicals: Formation and function of the tyrosyl radical in ribonucleotide reductase. Trends Biochem. Sci. 23, 438–443. [DOI] [PubMed] [Google Scholar]
- Atkin C. L.; Thelander L.; Reichard P.; Lang G. (1973) Iron and free radical in ribonucleotide reductase. Exchange of iron and Mössbauer spectroscopy of the protein B2 subunit of the Escherichia coli enzyme. J. Biol. Chem. 248, 7464–7472. [PubMed] [Google Scholar]
- Thelander L.; Gräslund A.; Thelander M. (1983) Continual presence of oxygen and iron required for mammalian ribonucleotide reduction: Possible regulation mechanism. Biochem. Biophys. Res. Commun. 110, 859–865. [DOI] [PubMed] [Google Scholar]
- Ortigosa A. D.; Hristova D.; Perlstein D. L.; Zhang Z.; Huang M.; Stubbe J. (2006) Determination of the in vivo stoichiometry of tyrosyl radical per ββ′ in Saccharomyces cerevisiae ribonucleotide reductase. Biochemistry 45, 12282–12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russel M.; Model P. (1985) Direct cloning of the trxB gene that encodes thioredoxin reductase. J. Bacteriol. 163, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn C. A.; Kathju S.; Wallace B. J.; Kushner S. R.; Pigiet V. (1984) Amplification and purification of plasmid-encoded thioredoxin from Escherichia coli K12. J. Biol. Chem. 259, 10469–10474. [PubMed] [Google Scholar]
- Fish W. W. (1988) Rapid colorimetric micromethod for the quantitation of complexed iron in biological samples. Methods Enzymol. 158, 357–364. [DOI] [PubMed] [Google Scholar]
- Salowe S. P.; Stubbe J. (1986) Cloning, overproduction, and purification of the B2 subunit of ribonucleoside-diphosphate reductase. J. Bacteriol. 165, 363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmström B. G.; Reinhammar B.; Vänngard T. (1970) The state of copper in stellacyanin and laccase from the lacquer tree Rhus vernicifera. Biochim. Biophys. Acta 205, 48–57. [DOI] [PubMed] [Google Scholar]
- Bollinger J. M. Jr.; Tong W. H.; Ravi N.; Huynh B. H.; Edmondson D. E.; Stubbe J. A. (1995) Use of rapid kinetics methods to study the assembly of the diferric-tyrosyl radical cofactor of Escherichia coli ribonucleotide reductase. Methods Enzymol. 258, 278–303. [DOI] [PubMed] [Google Scholar]
- Neidhardt F. C., and Ingraham J. L. (1987) Escherichia coli and Salmonella typhimurium: Cellular and molecular biology, American Society for Microbiology, Washington, DC. [Google Scholar]
- Steeper J. R.; Steuart C. D. (1970) A rapid assay for CDP reductase activity in mammalian cell extracts. Anal. Biochem. 34, 123–130. [DOI] [PubMed] [Google Scholar]
- Guzman L. M.; Belin D.; Carson M. J.; Beckwith J. (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose pBAD promoter. J. Bacteriol. 177, 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Climent I.; Sjöberg B. M.; Huang C. Y. (1991) Carboxyl-terminal peptides as probes for Escherichia coli ribonucleotide reductase subunit interaction: Kinetic analysis of inhibition studies. Biochemistry 30, 5164–5171. [DOI] [PubMed] [Google Scholar]
- Kasrayan A.; Birgander P. L.; Pappalardo L.; Regnström K.; Westman M.; Slaby A.; Gordon E.; Sjöberg B. M. (2004) Enhancement by effectors and substrate nucleotides of R1-R2 interactions in Escherichia coli class Ia ribonucleotide reductase. J. Biol. Chem. 279, 31050–31057. [DOI] [PubMed] [Google Scholar]
- Yee C. S.; Seyedsayamdost M. R.; Chang M. C.; Nocera D. G.; Stubbe J. (2003) Generation of the R2 subunit of ribonucleotide reductase by intein chemistry: Insertion of 3-nitrotyrosine at residue 356 as a probe of the radical initiation process. Biochemistry 42, 14541–14552. [DOI] [PubMed] [Google Scholar]
- Morgan-Kiss R. M.; Wadler C.; Cronan J. E. Jr. (2002) Long-term and homogeneous regulation of the Escherichia coli araBAD promoter by use of a lactose transporter of relaxed specificity. Proc. Natl. Acad. Sci. U.S.A. 99, 7373–7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson S.; Sjöberg B. M.; Hahne S. (1977) Ribonucleoside diphosphate reductase from Escherichia coli. An immunological assay and a novel purification from an overproducing strain lysogenic for phage lambda dnrd. J. Biol. Chem. 252, 6132–6138. [PubMed] [Google Scholar]
- Ortenberg R.; Gon S.; Porat A.; Beckwith J. (2004) Interactions of glutaredoxins, ribonucleotide reductase, and components of the DNA replication system of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 101, 7439–7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch J. B.; Juarez-Garcia C.; Münck E.; Que L. Jr. (1989) Mössbauer and EPR studies of the binuclear iron center in ribonucleotide reductase from Escherichia coli. A new iron-to-protein stoichiometry. J. Biol. Chem. 264, 8091–8096. [PubMed] [Google Scholar]
- Johnson D. C.; Dean D. R.; Smith A. D.; Johnson M. K. (2005) Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 74, 247–281. [DOI] [PubMed] [Google Scholar]
- Djaman O.; Outten F. W.; Imlay J. A. (2004) Repair of oxidized iron-sulfur clusters in Escherichia coli. J. Biol. Chem. 279, 44590–44599. [DOI] [PubMed] [Google Scholar]
- Toraya T. (2000) Radical catalysis of B12 enzymes: Structure, mechanism, inactivation, and reactivation of diol and glycerol dehydratases. Cell. Mol. Life Sci. 57, 106–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K.; Gravel R. A.; Toraya T.; Matthews R. G. (2006) Human methionine synthase reductase is a molecular chaperone for human methionine synthase. Proc. Natl. Acad. Sci. U.S.A. 103, 9476–9481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrents E.; Westman M.; Sahlin M.; Sjöberg B. M. (2006) Ribonucleotide reductase modularity: Atypical duplication of the ATP-cone domain in Pseudomonas aeruginosa. J. Biol. Chem. 281, 25287–25296. [DOI] [PubMed] [Google Scholar]
- Atta M.; Lamarche N.; Battioni J. P.; Massie B.; Langelier Y.; Mansuy D.; Fontecave M. (1993) Escherichia coli and herpes-simplex-virus ribonucleotide reductase R2 subunit. Compared reactivities of the redox centres. Biochem. J. 290, Part 3807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D.; Epstein J.; Boye E.; Fahrner K.; Berg H.; Kleckner N. (2005) The Escherichia coli baby cell column: A novel cell synchronization method provides new insight into the bacterial cell cycle. Mol. Microbiol. 57, 380–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenberg A.; Reichard P. (1972) Electron spin resonance of the iron-containing protein B2 from ribonucleotide reductase. J. Biol. Chem. 247, 3485–3488. [PubMed] [Google Scholar]
- Abdul-Tehrani H.; Hudson A. J.; Chang Y. S.; Timms A. R.; Hawkins C.; Williams J. M.; Harrison P. M.; Guest J. R.; Andrews S. C. (1999) Ferritin mutants of Escherichia coli are iron deficient and growth impaired, and fur mutants are iron deficient. J. Bacteriol. 181, 1415–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grass G.; Franke S.; Taudte N.; Nies D. H.; Kucharski L. M.; Maguire M. E.; Rensing C. (2005) The metal permease ZupT from Escherichia coli is a transporter with a broad substrate spectrum. J. Bacteriol. 187, 1604–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgeon B. E.; Burdi D.; Chen S. X.; Huynh B. H.; Edmondson D. E.; Stubbe J.; Hoffman B. M. (1996) Reconsideration of X, the diiron intermediate formed during cofactor assembly in Escherichia coli ribonucleotide reductase. J. Am. Chem. Soc. 118, 7551–7557. [Google Scholar]
- Riggs-Gelasco P. J.; Shu L. J.; Chen S. X.; Burdi D.; Huynh B. H.; Que L.; Stubbe J. (1998) EXAFS characterization of the intermediate X generated during the assembly of the Escherichia coli ribonucleotide reductase R2 diferric tyrosyl radical cofactor. J. Am. Chem. Soc. 120, 849–860. [Google Scholar]
- Mitic N.; Saleh L.; Schenk G.; Bollinger J. M. Jr.; Solomon E. I. (2003) Rapid-freeze-quench magnetic circular dichroism of intermediate X in ribonucleotide reductase: New structural insight. J. Am. Chem. Soc. 125, 11200–11201. [DOI] [PubMed] [Google Scholar]
- Han W. G.; Liu T.; Lovell T.; Noodleman L. (2006) Density functional theory study of Fe(IV) d-d optical transitions in active-site models of class I ribonucleotide reductase intermediate X with vertical self-consistent reaction field methods. Inorg. Chem. 45, 8533–8542. [DOI] [PubMed] [Google Scholar]
- Mitic N.; Clay M. D.; Saleh L.; Bollinger J. M. Jr.; Solomon E. I. (2007) Spectroscopic and electronic structure studies of intermediate X in ribonucleotide reductase R2 and two variants: A description of the FeIV-Oxo bond in the FeIII-O-FeIV dimer. J. Am. Chem. Soc. 129, 9049–9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdi D.; Willems J.-P.; Riggs-Gelasco P.; Antholine W. E.; Stubbe J.; Hoffman B. M. (1998) The core stucture of X generated in the assembly of the diiron cluster of ribonucleotide reductase: 17O2 and H217O ENDOR. J. Am. Chem. Soc. 120, 12910–12919. [Google Scholar]
- Studier F. W.; Moffatt B. A. (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130. [DOI] [PubMed] [Google Scholar]
- Casadaban M. J.; Cohen S. N. (1980) Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J. Mol. Biol. 138, 179–207. [DOI] [PubMed] [Google Scholar]
- Grant S. G.; Jessee J.; Bloom F. R.; Hanahan D. (1990) Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. U.S.A. 87, 4645–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd D.; Manoil C.; Beckwith J. (1987) Determinants of membrane protein topology. Proc. Natl. Acad. Sci. U.S.A. 84, 8525–8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.