

Abstract
The development of low μM inhibitors of the Mycobacterium tuberculosis phosphatase PtpA is reported. The most potent of these inhibitors (Ki = 1.4 ± 0.3 μM) was found to be selective when tested against a panel of human tyrosine and dual-specificity phosphatases (11-fold vs the highly homologous HCPtpA, and >70-fold vs all others tested). Modeling the inhibitor-PtpA complexes explained the structure–activity relationships observed in vitro and revealed further possibilities for compound development.
Keywords: Mycobacterium tuberculosis, Phosphatase, Inhibitor, PtpA
Tuberculosis (TB) is a chronic infectious disease caused by Mycobacterium tuberculosis (Mtb). Out of over 13 million active cases each year, TB causes nearly 2 million deaths.1 Current treatment of drug-sensitive strains requires 6–9 months to fully eradicate the infection. New Mtb drugs that act on novel targets are needed to shorten treatment and address the emergence of antibiotic resistance.
Mtb encodes two protein tyrosine phosphatases (PTPs), PtpA and PtpB, that are promising new targets for TB drug development.2 These PTPs are secreted by Mtb3 into the cytosol of infected macrophages, obviating the need for inhibitors to enter bacterial cells.4 Although genetic deletion of PtpA or PtpB does not affect Mtb growth in culture,4,5 these deletions severely attenuate growth in sensitive infected macrophages.4 These data suggest that the Mtb PTPs act on macrophage signaling pathways to promote Mtb survival in the infected host. Although not classical drug targets because they are not essential in vitro, targeting the secreted PTPs in the host macrophage circumvents two central resistance mechanisms of Mtb; that is, poor drug permeability due to the Mtb cell wall,6 and pump-mediated drug efflux.7
We previously reported the development of low-molecular weight inhibitors of PtpB8 using a substrate-based, fragment identification and optimization approach termed Substrate Activity Screening (SAS).9 Here, we applied the same method to PtpA to prepare and evaluate a library of inhibitors selective for Mtb PtpA. These studies identified low-micromolar PtpA inhibitors with selectivity versus a panel of human phosphatases. Modeling our compounds bound in the active site of PtpA explained the observed structure–activity relationships (SAR) and highlighted further possibilities for compound development.
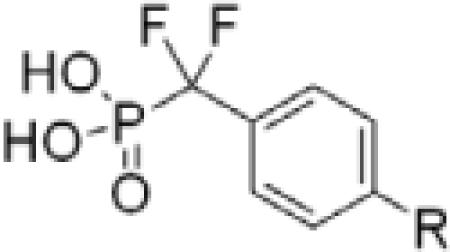
A library of O-aryl phosphate substrate fragments was previously developed to target PtpB.8 Using this library, we identified compounds for further optimization towards PtpA. Due to the ease of synthetic diversification of aryl difluoromethylphosphonic acid (DFMP) inhibitors, we varied DFMP analogs to establish SAR for PtpA inhibition. Although DFMP inhibitors have traditionally exhibited poor cell permeability due to the dianionic nature of this pharmacophore, DFMP inhibitors of the human phosphatase PTP1B, an enzyme involved in insulin signaling, have recently been reported to have cell activity and oral bioavailability in animals.10
A library of phenyl DFMP inhibitors substituted with diverse functionality at the 3- and 4-positions was prepared and tested (Fig. 1). Ki values for each compound were determined using a standard, continuous inhibition assay, with p-nitrophenyl phosphate (pNPP) serving as the chromogenic substrate.11 Triton X-100 detergent (0.004%) was used to prevent nonspecific aggregation commonly observed in inhibition assays.12 Inhibition was also found to be independent of both enzyme (300 and 600 nM) and detergent concentrations (up to 0.01%), and no unusual steepness was observed in the dose–response curves (see Supplementary data). Among this library, the simple benzanilide 20 afforded the most favorable combination of enzyme affinity, solubility, and ease of synthetic diversification.
Figure 1.

Selected members of initial DFMP inhibitor library against PtpA. aBenzimidazole analogs were found not to be soluble at concentrations required for inhibition assays.
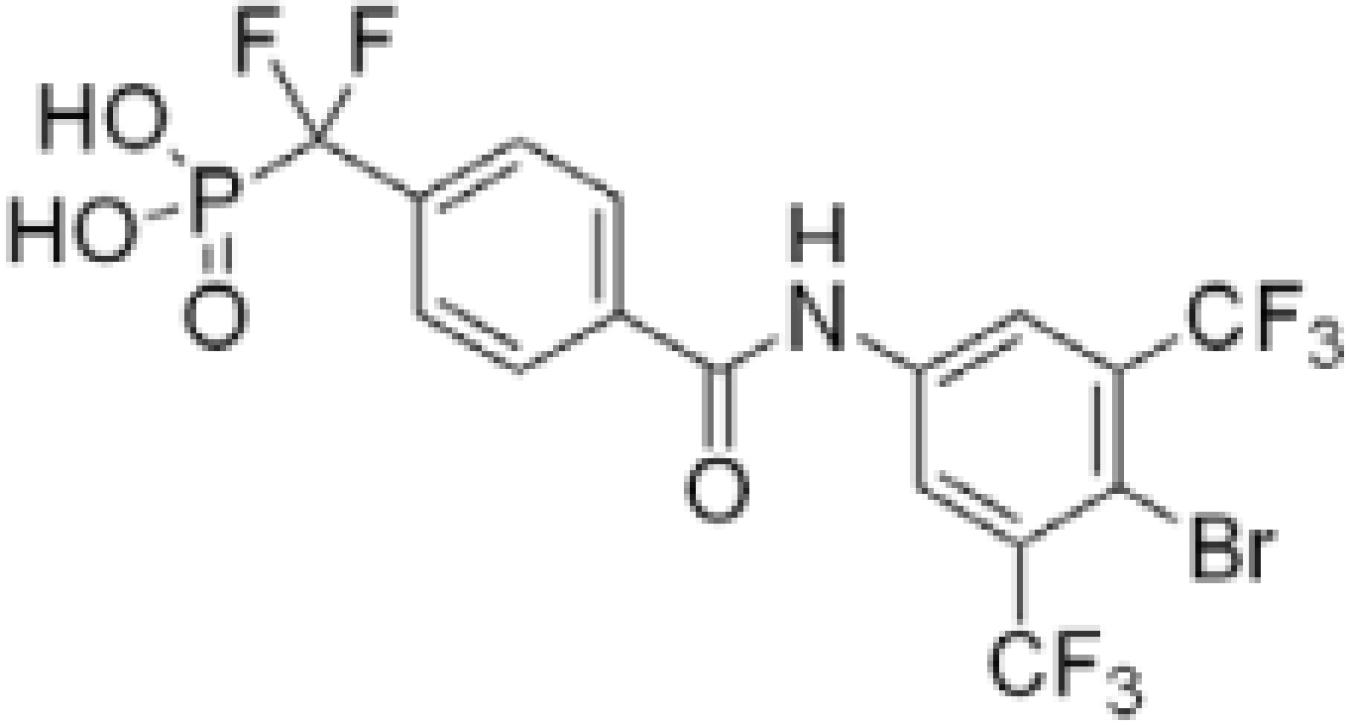
Analogs incorporating electron withdrawing groups provided the most potent compounds, as shown in a subset of our focused library of benzanilide inhibitors (Table 1). Substitution at the meta and para positions (26–33) resulted in a more substantial improvement in affinity than substitution at the ortho position (22–25), with bromine and trifluoromethyl groups resulting in the highest affinity inhibitors. Combining these elements resulted in compound 38, with a Ki of 1.4 ± 0.3 μM, and a Hill coefficient of h = −1.0 ± 0.1.13
Table 1.
Aryl ring optimizationa
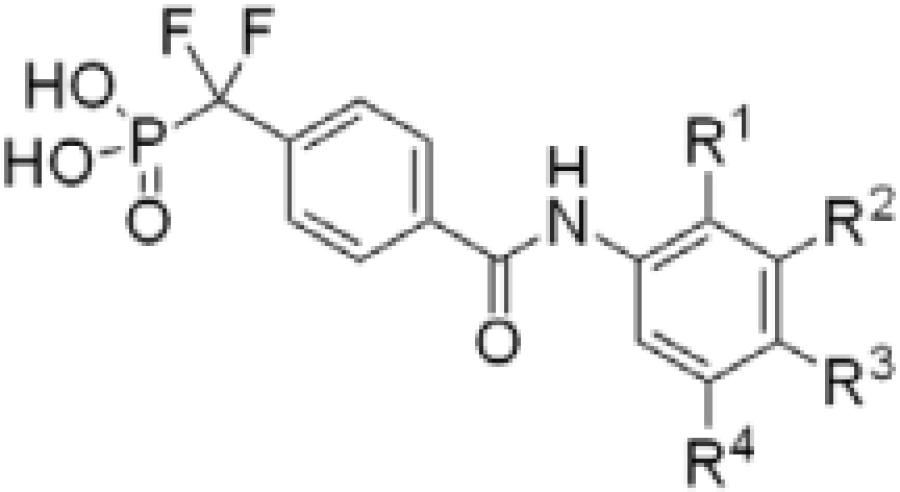 | |||||
|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | Ki (μM) | |
| 20 | H | H | H | H | 24.0 ± 0.9 |
| 22 | F | H | H | H | >100 |
| 23 | Cl | H | H | H | 68.0 ± 6.0 |
| 24 | CF3 | H | H | H | 54.8 ± 14.5 |
| 25 | Br | H | H | H | 44.6 ± 7.4 |
| 26 | H | F | H | H | 41.8 ± 5.8 |
| 27 | H | Cl | H | H | 34.9 ± 3.5 |
| 28 | H | Br | H | H | 18.2 ± 0.9 |
| 29 | H | CF3 | H | H | 10.7 ± 1.2 |
| 30 | H | H | F | H | 22.7 ± 3.2 |
| 31 | H | H | CF3 | H | 18.5 ± 2.5 |
| 32 | H | H | Cl | H | 16.8 ± 7.0 |
| 33 | H | H | Br | H | 10.3 ± 1.0 |
| 34 | H | CF3 | F | H | 11.4 ± 0.3 |
| 35 | H | CF3 | Cl | H | 6.0 ± 0.4 |
| 36 | H | CF3 | Br | H | 4.9 ± 1.7 |
| 37 | H | CF3 | H | CF3 | 3.3 ± 0.6 |
| 38 | H | CF3 | Br | CF3 | 1.4 ± 0.3 |
Ki values were determined using at least four independent measurements.
To investigate the importance of the amide moiety, we synthesized several amide replacement analogs (Table 2). Methylating the nitrogen (39), removing the carbonyl (40), replacing the nitrogen with oxygen in addition to carbonyl removal (41), and replacing the carbonyl with a sulfonyl moiety (42) all resulted in loss of activity. Interestingly, replacing the amide moiety with a urea group resulted in compound 43, with affinity similar to 38.
Table 2.
Amide replacement analogsa
 | ||
|---|---|---|
| Structure | Ki (μM) | |
| 39 | 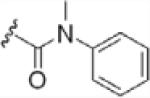 |
>100 |
| 40 | 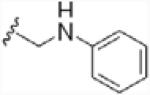 |
>100 |
| 41 | 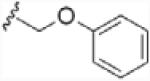 |
>100 |
| 42 | 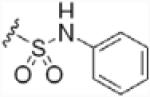 |
>100 |
| 43 | 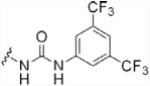 |
3.1 ± 0.4 |
See Table 1 footnote.
To determine the structural features important for affinity, we generated molecular models of our compounds bound in the active site of PtpA. The published crystal structure of apo-PtpA (PDB accession number 1U2P)14 was first relaxed using molecular dynamics in AMBER,15 followed by docking the compounds into the PtpA active site with DOCK 6.4.16 Each of the compounds docked such that the phosphate moiety was in direct contact with the catalytic residues of the protein. The scoring function of the docking program ranked the compounds in the same general order observed experimentally (data not shown). The structural similarity to interactions of DFMP inhibitors with other PTPs, similar rank order of calculated complementarity, and measured binding affinities suggest that our predicted binding modes accurately capture the inhibitor-enzyme interactions.
All of the modeled compounds exhibited significant hydrogen bonding interactions with PtpA (Fig. 2). Nine hydrogen bonds were found between compound 20 and PtpA active site residues, versus seven for compound 38 and ten for compound 43. The carbonyl of the urea group of 43 is positioned opposite that of 20 and 38, allowing for formation of an additional hydrogen bond with His49 not observed with the other inhibitors.
Figure 2.

Model of (a) parent benzanilide 20, (b) optimized benzanilide 38, and (c) extended urea inhibitor 43 docked in the active site of PtpA (PDB ID 1U2P)14 using DOCK 6.4.16 Hydrogen bonds (green lines) between each inhibitor and active site residues are shown. His49 is emphasized to show H-bonding with compound 43, and Trp48 is emphasized to show pi-stacking interactions with 38 and 43.
Pi-stacking interactions with Trp48 were also predicted in several docked structures (Fig. 2). This binding mode was not unexpected, as Trp pi-stacking has been previously observed in enzyme-inhibitor complexes.17 Compound 20 showed little to no overlap, suggesting that hydrogen bonding interactions are primarily responsible for its affinity, while 38 and 43 have improved access to the aromatic system of Trp48, in addition to electron withdrawing groups that favor more direct pi-stacking. Because amide compounds 20 and 38 hydrogen bond only at the site of the DFMP warhead, pi-stacking appears to be primarily responsible for the observed difference in Ki. Despite the added hydrogen bond observed for 43, the affinity of this compound is lower than that of 38, likely due to less efficient overlap with the aromatic system of Trp48. Taken together, these observations provide a plausible explanation for the observed affinity of 43 for PtpA (Ki = 3.1 ± 0.4 μM, h = −1.1 ± 0.2).
In contrast to 43, the affinity of the other amide replacement analogs (39–42) is greatly reduced. This is likely due to a lack of hydrogen bond acceptors in the correct orientation for interaction with His49. Binding may also be affected by changes in electrostatic interactions or entropic penalties associated with an increased number of rotatable bonds. Modifications to further improve inhibitor potency could include introduction of functionality that takes advantage of hydrogen bonding with His49 while also improving pi-stacking efficiency with Trp48, as well as introduction of functionality off of the pendant anilide ring to extend into an adjacent unfilled enzyme pocket (observed by modeling; see Supplementary data).
Due to the high structural homology of PTP active sites, achieving inhibitor selectivity is a major challenge.18 Compound 38, however, was found to be highly selective (>70-fold) when tested against a panel of tyrosine and dual-specificity phosphatases, including TC-Ptp, an essential human phosphatase (Table 3). This compound was also 11-fold selective for Mtb PtpA versus human low-molecular weight phosphatase, HCPtpA, which shows 38% sequence identity to the Mtb enzyme.19 Compound 38 did not inhibit Mtb PtpB,20 which should enable the use of this inhibitor to dissect the biochemical roles of each of the two Mtb PTPs.
Table 3.
Selectivity of inhibitor 38 against a panel of PTPs
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| PtpA | PtpB | PTP1B | Tc-Ptp | VHR | CD45 | LAR | HCPtpA | |
| Ki (μM) | 1.4 ± 0.3 | >100 | >100 | >100 | >100 | >100 | >100 | 14.8 ± 1.9 |
| Selectivity | – | >70 | >70 | >70 | >70 | >70 | >70 | 11 |
In conclusion, we have identified and developed selective inhibitors for PtpA based on the benzanilide scaffold 20. Our SAR studies resulted in compound 38, which represents the most potent and selective PtpA inhibitor reported in the literature to date.21 Molecular modeling highlighted the importance of pi-stacking with Trp48, and hydrogen bonding with active site residues and His49 for high-affinity binding. Compound 38 is over 70-fold selective for PtpA versus a panel of human phosphatases and 11-fold selective versus the closely related human homologue HCPtpA.
Supplementary Material
Acknowledgments
We thank the Bertozzi and M. Chang groups for use of equipment, and Cyrus Maher for help with the initial modeling studies. J.A.E. acknowledges the support of the NIH (GM054051). K.A.R. was supported by an ACS Division of Medicinal Chemistry Pre-doctoral fellowship, sponsored by Eli Lilly. P.T.L., T.A. and C.G. acknowledge the support of the TB Structural Genomics Consortium (NIH P01 AI68135).
Footnotes
Supplementary data
Experimental details include enzyme assay protocols, synthesis procedures, analytical characterization of inhibitors, and additional modeling figures referenced in the text. Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2009.10.090.
References and notes
- 1.World Health Organization Global Tuberculosis Report 2009. http://www.who.int/tb/publications/global_report/2009/pdf/full_report.pdf.
- 2.Cowley SC, Babakaiff R, Av-Gay Y. Res. Microbiol. 2002;153:233. doi: 10.1016/s0923-2508(02)01309-8. [DOI] [PubMed] [Google Scholar]
- 3.Koul A, Choidas A, Treder M, Tyagi AK, Drlica K, Singh Y, Ullrich A. J. Bacteriol. 2000;182:5425. doi: 10.1128/jb.182.19.5425-5432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bach H, Papavinasasundaram KG, Wong D, Hmama Z, Av-Gay Y. Cell Host Microbe. 2008;3:316. doi: 10.1016/j.chom.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 5.Grundner C, Cox JS, Alber T. FEMS Microbiol. Lett. 2008;287:181. doi: 10.1111/j.1574-6968.2008.01309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brennan PJ, Nikaido H. Annu. Rev. Biochem. 1995;64:29. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 7.Louw GE, Warren RM, Gey van Pittius NC, McEvoy CRE, Van Helden PD, Victor TC. Antimicrob. Agents Chemother. 2009;53:3181. doi: 10.1128/AAC.01577-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soellner MB, Rawls KA, Grundner C, Alber T, Ellman JA. J. Am. Chem. Soc. 2007;129 doi: 10.1021/ja0727520. [DOI] [PubMed] [Google Scholar]
- 9.(a) Wood WJL, Patterson AW, Tsuruoka H, Jain RK, Ellman JA. J. Am. Chem. Soc. 2005;127:15521. doi: 10.1021/ja0547230. [DOI] [PubMed] [Google Scholar]; (b) Patterson AW, Wood WJL, Hornsby M, Lesley S, Spraggon G, Ellman JA. J. Med. Chem. 2006;49:6298. doi: 10.1021/jm060701s. [DOI] [PubMed] [Google Scholar]; (c) Salisbury CM, Ellman JA. ChemBioChem. 2006;7:1034. doi: 10.1002/cbic.200600081. [DOI] [PubMed] [Google Scholar]; (d) Inagaki H, Tsuruoka H, Hornsby M, Lesley SA, Spraggon G, Ellman JA. J. Med. Chem. 2007;50:2693. doi: 10.1021/jm070111+. [DOI] [PubMed] [Google Scholar]; (e) Brak K, Doyle PS, McKerrow JH, Ellman JA. J. Am. Chem. Soc. 2008;130:6404. doi: 10.1021/ja710254m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han Y, Belley M, Bayly CI, Colucci J, Dufresne C, Giroux A, Lau CK, Leblanc Y, McKay D, Therien M, Wilson M-C, Skorey K, Chan C-C, Scapin G, Kennedy BP. Bioorg. Med. Chem. Lett. 2008;18:3200. doi: 10.1016/j.bmcl.2008.04.064. [DOI] [PubMed] [Google Scholar]
- 11.Montalibet J, Skorey KI, Kennedy BP. Methods. 2005;35:2. doi: 10.1016/j.ymeth.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 12.For reviews and general references, see: McGovern SL, Caselli E, Grigorieff N, Shoichet BK. J. Med. Chem. 2002;45:1712. doi: 10.1021/jm010533y. McGovern SL, Helfand BT, Feng B, Shoichet BK. J. Med. Chem. 2003;46:4265. doi: 10.1021/jm030266r. Shoichet BK. Drug Discovery Today. 2006;11:607. doi: 10.1016/j.drudis.2006.05.014. Shoichet BK, Feng BY, Coan KED. Comput. Struct. Approaches Drug Discovery. 2008:223.
- 13.Hill coefficients were calculated using GraphPad prism version 5.00 for Windows, GraphPad Software, San Diego California USA, www.graphpad.com, using variable slope parameters, with the equation Y = Bottom + (Top – Bottom)/(1 + 10((Log IC50–X)*HillSlope)), where X is the log of inhibitor concentration. Values are negative because dose–response curves are used, where values are plotted from high to low inhibitor concentrations. A Hill coefficient of −1 indicates completely independent binding.
- 14.Madhurantakam C, Rajakumara E, Mazumdar PA, Saha B, Mitra D, Wiker HG, Sankaranarayanan R, Das AK. J. Bacteriol. 2005;187:2175. doi: 10.1128/JB.187.6.2175-2181.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The ff03 force field was designed by and is available from: Case DA, Darden TA, Cheatham TE, III, Simmerling C, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker R, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews DH, Schafmeister C, Ross WS, Kollman P. AMBER. Vol. 9. University of California; San Francisco: 2006.
- 16.Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID. RNA-Publ. RNA Soc. 2009;15:1219. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Kryger G, Silman I, Sussman J. L. Structure. 1999;7:297. doi: 10.1016/s0969-2126(99)80040-9. [DOI] [PubMed] [Google Scholar]; (b) Rao FV, Andersen OA, Vora KA, DeMartino JA, Van Aalten DMF. Chem. Biol. 2005;12:973. doi: 10.1016/j.chembiol.2005.07.009. [DOI] [PubMed] [Google Scholar]; (c) Schuettelkopf AW, Andersen OA, Rao FV, Allwood M, Lloyd C, Eggleston IM, van Aalten DMF. J. Biol. Chem. 2006;281:27278. doi: 10.1074/jbc.M604048200. [DOI] [PubMed] [Google Scholar]; (d) Zsila F, Matsunaga H, Bikadi Z, Haginaka J. Biochim. Biophys. Acta, Gen. Subj. 2006;1760:1248. doi: 10.1016/j.bbagen.2006.04.006. [DOI] [PubMed] [Google Scholar]; (e) Zsila F, Iwao Y. Biochim. Biophys. Acta, Gen. Subj. 2007;1770:797. doi: 10.1016/j.bbagen.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 18.For reviews on PTP inhibitor development, see: Moller NPH, Andersen HS, Jeppesen CB, Iversen LF. Handbook Exp. Pharmacol. 2005;167:215. Lee S, Wang Q. Med. Res. Rev. 2007;27:553. doi: 10.1002/med.20079. Zhang S, Zhang Z-Y. Drug Discovery Today. 2007;12:373. doi: 10.1016/j.drudis.2007.03.011. Vintonyak VV, Antonchick AP, Rauh D, Waldmann H. Curr. Opin. Chem. Biol. 2009;13:272. doi: 10.1016/j.cbpa.2009.03.021.
- 19.See Supplementary data for a structural overlay of PtpA and HCPtpA, focusing on the PTP active site and variable loops.
- 20.This result was not surprising given the large structural differences in the variable loops of PtpA and PtpB. See Supplementary data for a structural overlay of these enzymes, focusing on the PTP active site and variable loops.
- 21.For other PtpA inhibitor efforts, see: Manger M, Scheck M, Prinz H, von Kries JP, Langer T, Saxena K, Schwalbe H, Fuerstner A, Rademann J, Waldmann H. ChemBioChem. 2005;6:1749. doi: 10.1002/cbic.200500171. Chiaradia LD, Mascarello A, Purificacao M, Vernal J, Cordeiro MNS, Zenteno ME, Villarino A, Nunes RJ, Yunes RA, Terenzi H. Bioorg. Med. Chem. Lett. 2008;18:6227. doi: 10.1016/j.bmcl.2008.09.105.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.