

Abstract
Herpes simplex virus type-1 (HSV-1) and type-2 (HSV-2) are highly prevalent human pathogens causing life-long infections. The process of infection begins when the virions bind heparan sulfate moieties present on host cell surfaces. This initial attachment then triggers a cascade of molecular interactions involving multiple viral and host cell proteins and receptors leading to penetration of the viral nucleocapsid and tegument proteins into the cytoplasm. The nucleocapsid is then transported to the nuclear membrane and the viral DNA is released for replication in the nucleus. Recent studies have revealed that HSV entry or penetration into cells may be a highly complex process and the mechanism of entry may demonstrate unique cell-type specificities. While specificities clearly exist, some past and ongoing studies also demonstrate that HSV may share certain common receptors and pathways also used by many other human viruses. This review helps to shed light on recent revelations on HSV entry process.
Introduction and Overview
The herpesvirus family consists of over 100 double-stranded DNA viruses divided into alpha, beta, and gamma subgroups [1]. Only eight herpesviruses are known to commonly infect humans and the rest are animal herpesviruses infecting a wide variety of animal species. All members of the herpesvirus family cause lifelong latent infections and structurally, all have linear, double-stranded DNA genome packaged into an icosahedral capsid (Figure 1) [1]. This capsid, in turn, is enclosed by the tegument, a layer of proteins. The tegument is then covered by a bilayer lipid membrane with embedded proteins and glycoproteins. The protected DNA genome is essential to viral infectivity. Closely related herpes simplex type-1 (HSV-1) and type-2 (HSV-2) viruses are members of the alphaherpesvirus subfamily and are responsible for highly prevalent infections among humans [2], although a number of common experimental animals also demonstrate susceptibility to HSV infections. Symptomatic disease caused by HSV-1 is typically limited to cold sores of the mouth and keratitis in the eyes. HSV-2, in contrast, is mostly responsible for genital lesions. However, both viruses are capable of causing lesions on identical body sites and both can cause life-threatening diseases in immunocompromised individuals including newborns, patients with human immunodeficiency virus (HIV) or patients undergoing immunosuppressive treatment [1–2]. Transmission among humans requires physical contact and often occurs during kissing (HSV-1) or sexual intercourse (HSV-2). The area of the lesions depends on the inoculation site, therefore, sores are most commonly found on the mouth or genital areas. Medical professionals and others not wearing surgical gloves could also acquire lesions on their fingers from the virions shed from their patients’ vaginal mucosa and/or mouth. This is commonly known as finger herpes or herpetic whitlow.
Figure 1.
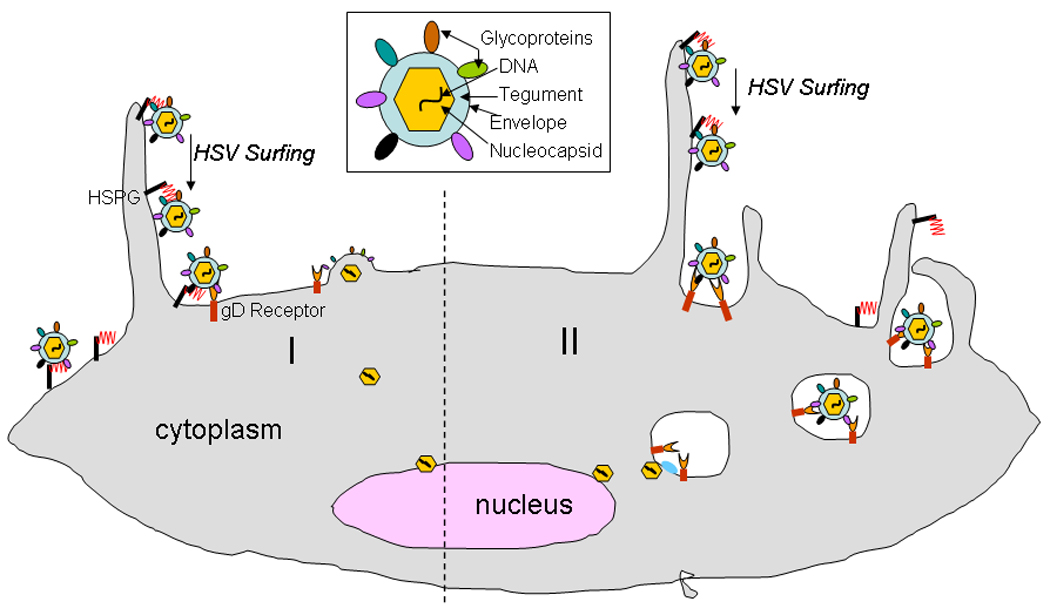
HSV virion and its two major modes of entry into cells. Structural components of a typical HSV virion are shown (box). HSV virions can enter into cells via a pH-independent fusion of viral envelope with the plasma membrane (I) or alternatively, via an endocytic pathway that may be phagocytosis-like (II) in terms of the viral uptake. In both pathways HSV particles may initially associate with filopodia-like membrane protrusions via heparan sulfate proteoglycan (HSPG). Unidirectional transport of extracellular particles bound to filopodia (HSV surfing) then brings the particles closer to the cell body for entry via interactions with the cellular receptors including gD receptor and possibly gB receptor. Fusion at the plasma membrane results in the release of the naked viral nucleocapsid in the cytoplasm for transport to the nucleus. Similarly, endocytosis also requires fusion of the enveloped particles with the vesicular membrane for the release of the viral nucleocapsid proximal to the nucleus.
After initial infection, the virus remains latent in neurons, a key feature of alpha herpesviruses [1]. During this period, hosts are still capable of spreading infection to other humans via asymptomatic shedding of the virions. Reactivation of the virus can be due to a variety of environmental triggers including emotional or physical stress, which subsequently leads to virus replication in epithelial cells and a lifetime of intermittent mucocutaneous lesions [2]. The virus' ability to avoid immune detection and establish latency in significant patient population, up to 80% human adults for HSV-1 and about 40% for HSV-2, is facilitated by its unique ability to productively enter into cells of the epithelia for viral gene expression, replication, and eventually, spread from cell-to-cell to innervating nerves and ultimately to trigeminal (HSV-1) or sacral (HSV-2) ganglia for the establishment of latency. Thus, HSV entry into host cells marks the first and perhaps, the most critical step in viral pathogenesis.
Five viral glycoproteins have been implicated in the viral entry process: gB, gC, gD, gH, and gL [3–4]. All but gC are essential for entry. The initial interaction, or binding to cells, is mediated via interactions of gC and/or gB with heparan sulfate proteoglycans (HSPGs). F-actin-rich membrane protrusions called filopodia may facilitate attachment by providing HSPGs-rich sites for the initial binding (Fig. 1). Although gC is not essential for viral entry, its absence decreases the overall viral binding to cell surfaces [1]. After initial attachment to cells, the process of penetration begins. The latter, depending on the host cell type and the mode of entry [4–5], may require fusion of the virion envelope with the plasma membrane or with the membrane of an intracellular vesicle (Fig. 1) [5]. In either case, the membrane fusion requires essential participation from viral glycoproteins gB, gD, gH and gL. While gD is not considered a fusogen, other essential glycoproteins, more importantly, gB and gH, demonstrate many characteristics of viral fusion proteins [4,6]. Similar to attachment, membrane fusion also requires participation from cellular receptors. A number of unrelated receptors for gD have been discovered. These include nectin-1 and -2, herpesvirus entry mediator (HVEM) and 3-O sulfated heparan sulfate (3-O HS) [3, 7]. The current, widely accepted model for membrane fusion suggests that binding of gD to one of its cognate receptors induces conformational changes in gD that mobilizes a fusion active multi-glycoprotein complex involving gB, gD, gH and gL (Fig. 2) [8]. Fusion of viral envelope with a cellular membrane results in content mixing and eventual release of the viral nucleocapsid and tegument proteins into the host cytoplasm (Fig. 1–2). Thereafter, symbolizing post entry steps, HSV nucleocapsids dissociate with tegument proteins and bind a microtubule (MT)-dependent, minus end- directed motor, dynein [9]. While most of the tegument proteins are required for activation and modulation of viral gene expression and shut-off of host protein synthesis, some may participate in dynein-propelled transport of the nucleocapsids along MT toward the nuclear membrane for uncoating and the release of viral DNA into the nucleus. Transcription, replication of viral DNA, and assembly of progeny capsids take place within the host nucleus. The intricate details of the steps required for entry are discussed below.
Figure 2.
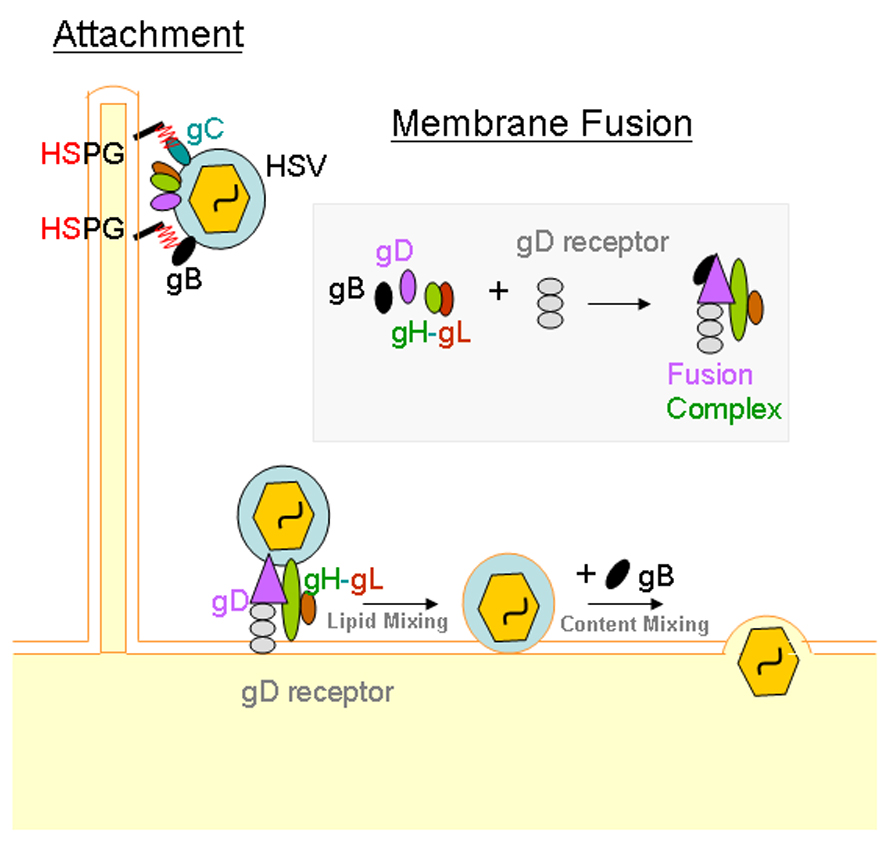
Molecular interactions that facilitate HSV entry. Initial attachment to cells is mediated by interaction between heparan sulfate proteoglycans (HSPGs) with HSV glycoproteins gC and/or gB. Membrane fusion is required for the penetration of viral nucleocapsid and the tegument into the cytoplasm. Interaction between gD, gH-gL and a gD receptor may be sufficient to bring conformational changes within gD to trigger merging of viral and cellular membranes or lipid mixing. However, a fourth glycoprotein, gB, is also required for complete fusion and content mixing, which basically results in the release of the tegument and the nucleocapsid into the cytoplasm. A receptor for gB, PILR-α is also expected to play a role during the fusion process. Its precise role is still emerging and therefore, it is not shown above.
Viral Binding to Filopodia
Although the virus binding to cells results from the interaction of gC and/or gB with heparan sulfate (HS), where on the cell this binding can occur the earliest has only recently been elucidated. In human conjunctival epithelial (HCjE) cells, virions were observed attaching to filopodia-like membrane protrusions [10]. It was also observed that virus attachment to filopodia was followed by unilateral movement of the virions towards the cell body. Staining of many natural target cells with anti-HSV receptor antibodies indicated expression of HS but not other entry receptors. Nectin-1 expression, for example, was limited to cell bodies with no detectable expression on filopodia (Oh and Shukla, unpublished results). The phenomenon of extracellular HSV-1 moving unilaterally towards the cell body on filopodia has also been observed in retinal pigment epithelial (RPE) and P19N neural cells [11,12]. A similar phenomenon for the transport of extracellular virus particles, termed “viral surfing”, has been seen with other viruses, such as retroviruses and human papillomavirus type-16 [13, 14]. Surfing is also shared by many additional herpesviruses including cytomegalovirus and human herpesvirus-8 (Tiwari and Shukla, unpublished results).
Exposure of HSV-1 to cells can induce the formation of filopodia [5]. This presumably enhances the efficacy of viral infection by targeted delivery, via surfing, of virus particles to cell bodies for subsequent fusion with plasma membrane or endocytosis [11]. Based on functional analogy with retroviral surfing, it is quite likely that myosin-dependent F-actin retrograde flow is responsible for the HSV movements along filopodia [13]. Some ongoing studies demonstrate that filopodial bridges formed between two cells can help transfer extracellular HSV-1 virions from an infected to an uninfected cell (Tiwari and Shukla, unpublished results). Although viral trafficking on filopodia has not yet been observed or studied in HSV-2, due to strong similarities between HSV-1 and HSV-2 viral entry mechanisms including the use of HS as an attachment receptor, it is quite possible that this phenomenon also plays a role in HSV-2 entry. It is also worth mentioning here that filopodia are not essential for virus attachment, the latter can occur virtually any place on the plasma membrane as long as receptors such as HS are present. However, due to the presence of filopodia at the leading edges of tissue layers, e.g. in vivo during wound healing, they may provide easy “roadways” for HSV to reach cell bodies for infection.
Fusion at cell and vesicular membranes
Fusion at the plasma membrane is a pH-independent process that requires gB, gD, gH and gL (Fig. 2) [3]. Cellular receptors such as nectin-1, HVEM or 3-OS HS are also required. The process is triggered by conformational changes in gD that occur upon receptor binding [15]. Alteration in gD conformation then mobilizes gH and gL, a heterodimer, and gB to initiate the fusion process at the cell surface. Multiprotein complexes involving gD/gB and gD/gH/gL have been detected [9,16] and the entire complex colocalizes at the membrane during fusion [9]. While it is expected that members of fusion active complex undergo additional changes in conformation, no clear information is available due to lack of 3-D structures of any fusion active glycoprotein complexes. It is, however, known that gD bound to its receptor demonstrate significant changes in its conformation [15]. The HSV-induced membrane fusion is accomplished by first bringing the membranes of both the host cell and virus in close contact by receptor/glycoprotein interactions followed by mixing of the membranes or lipids to create an intermediary state, sometimes referred as “hemifusion intermediate” [17]. Subsequently, a fusion pore is formed that allows mixing of the cytoplasmic contents with viral contents, which in case of HSV entry basically implies the delivery of viral tegument proteins and the nucleocapsid into the cytoplasm. While gD and the gH/gL heterodimer may be sufficient to initiate the lipid mixing, a full-scale fusion resulting in the mixing of the contents requires the presence of gB as well (Fig. 2) [17]. While some experts believe that fusion may require a sequential action by the glycoproteins [17], others do not see any pressing evidence for it [16].
Recent studies have implicated gB and gH as having multiple fusogenic domains [4,6,18]. gB is highly conserved among herpesviruses, however, this glycoprotein demonstrates a unique characteristic in HSV-1 and HSV-2 because it remains uncleaved, while many other herpesvirus gBs are post-translationally cleaved. Interestingly, both uncleaved and cleaved forms of gBs share mixed features of class I and class II viral fusion proteins, and thereby, define a new, hybrid class of viral fusogens [6]. Class I proteins contain hairpin trimers with N-terminal hydrophobic membrane penetrating peptides and centrally located α-helical coiled coils. Class II proteins; in contrast, contain β-structures with internally located fusion domains. Similar to class I, gB trimer also contain a central α-helical core but the fusion loops, in similarity with class II, are part of elongated β-hairpins. The gB trimer shows strong resemblance to fusogenic glycoprotein G of vesicular stomatitis virus [6]. Similar to gB, gH was also reported to contain two heptad repeat regions that were found to be necessary for fusion induction [4]. Peptides that corresponded to these regions effectively prevented HSV-1 infection, suggesting the importance of these regions. The exact significance of gL is still unclear but it is likely to play an important role by stabilizing or regulating gH conformations.
The role of gD and its receptor in the fusion process is likely of a catalyst [3, 15]. While gD does not contain any fusion peptides or domains, binding to its receptor is required for the initiation of fusion, unless a poorly understood gD-dependent but gD-receptor-independent pathway is initiated [19]. It is worth noting that gD homologs are rare, and among human herpesviruses, only HSV-1 and HSV-2 seem to express gD [1]. It is unclear why HSV virions have evolved with a gD-based fusion trigger mechanism, whereas many other herpesviruses can do without it. It is conceivable that gD based mechanism may provide some explanation to differences in tissue tropism demonstrated by HSV-1 and HSV-2. It is also possible that involvement of gD, which is capable of interacting with at least three distinct classes of entry receptors, may enhance the host range for HSV since a vast majority of cultured cells of human or animal origin are susceptible to HSV entry. Other human herpesviruses demonstrate more restrictive host ranges in terms of cell lines they infect [1].
Although fusion at the plasma membrane was originally considered to be the only route of viral entry for HSV, recent studies have supported existence of an alternate entry mechanism utilizing endocytosis [4–5]. This atypical endocytosis resembles phagocytosis in the virus uptake mechanism (Fig. 1) [5]. Under this process, the endocytosed enveloped particles subsequently fuse with a vesicular membrane. Similar interactions between viral gB, gD, gH, gL and host cell receptors are expectedly mirrored within an intracellular vesicle such as endosome to facilitate the fusion. The gD receptor has been colocalized with endosomal markers and electron micrographs show fusion and exit of nuclecapsids from the endosomes [5]. Interestingly, unlike other bacterial and viral entry mechanisms, HSV-1 endocytosis does not appear to be mediated by clathrin-coated pits or caveolae. Also, although endosomes provide an acidic background which may augment viral infectivity in certain cell types, this pH dependency is not required in all cell lines [20–22]. The decision between endocytosis and fusion at the plasma membrane appears to depend on individual cell types. In Vero and Hep2 cells, fusion at the plasma membrane is the mechanism of choice, however, in cell types such as CHO, HeLa, RPE, human epidermal keratinocytes and HCjE, evidence of endocytosis of virions has been observed [11,21–22]. Interestingly, gD was shown to downregulate nectin-1 in cells utilizing endocytosis in HSV-1 entry, such as HeLa cells [23]. In contrast, this downregulation was not seen in cells where HSV fuses at the plasma membrane, such as Vero cells. This suggests that the gD/nectin-1 interaction is a possible factor in cell type specific mode of HSV-1 internalization.
Cellular receptors for gD
The main receptors gD utilizes for cell entry are nectin-1, nectin-2, HVEM, and 3-O HS. HSV-1 and HSV-2 differ in their preference of gD receptor types. HVEM and nectin-1 are utilized by both virus types, however, 3-O HS can only be used by HSV-1. Similarly, nectin-2 has not been shown to allow substantial wild-type HSV-1 entry and it may have a greater effect on HSV-2 entry [3].
Various cell types rely on different gD receptors for HSV-1 entry: T lymphocytes and trabecular meshwork cells utilize HVEM, whereas neuronal and epithelial cells require nectin-1 [3,24]. Dependence of HSV-2 on nectin-1 and HVEM for entry and spread in vivo has been successfully demonstrated using single and double knockout mice [25]. 3-O HS, which is not a receptor for HSV-2, appears to play a major role in HSV-1 entry into primary cultures of corneal fibroblasts [26]. Since 3-O HS is the most recently discovered and perhaps the least understood gD receptor, a relatively detailed analysis of 3-O HS is presented below.
Heparan sulfate (HS) in essence represents a structurally diverse family of polysaccharides sharing a common backbone structure with varying degrees of additional modifications and functions [27]. Demonstrating its structural complexity, HS, which is essentially a polymer of repeating disaccharide units containing a glucosamine and a glucuronic acid residue (Fig.3), is expressed in a variety of chain lengths with varying degrees of additional modifications on cells surfaces and extracellular matrices of almost all cell types [27]. It is an important attachment receptor shared by many pathogenic viruses including all human herpesviruses except Epstein Bar virus [1]. Significance of HS is particularly higher for HSV-1 since in addition to attachment, it can also mediate membrane penetration by HSV-1 virions [7]. While a relatively less modified backbone HS chain can facilitate HSV attachment, a highly modified version, 3-OS HS, is required for interaction with gD and for independently triggering membrane fusion during entry and cell-to-cell spread processes [7, 28–29]. This fusion triggering 3-OS HS is generated after numerous modifications, including 2-O-, 6-O- and 3-O sulfations and epimerization (Fig. 3) [7, 27]. The final step of parent chain sulfation to create 3-O HS is performed by a number of 3-O sulfotransferase (3-OST) isoforms: 3-OST-1, -2, -3A, -3B, -4, -5, -6 [1, 30]. Each isoform may demonstrate cell-type specific expression patterns and may produce a unique 3-OS HS chain with uniquely different functions. 3-OST-1 creates 3-O HS with anti-thrombin binding sites with no gD binding activity, other isoforms such as 3-OST-2, -3, -4, and -6, create forms of 3-O HS that can act as gD receptors [1,7, 30]. Their individual physiological functions are not well known yet.
Figure 3.
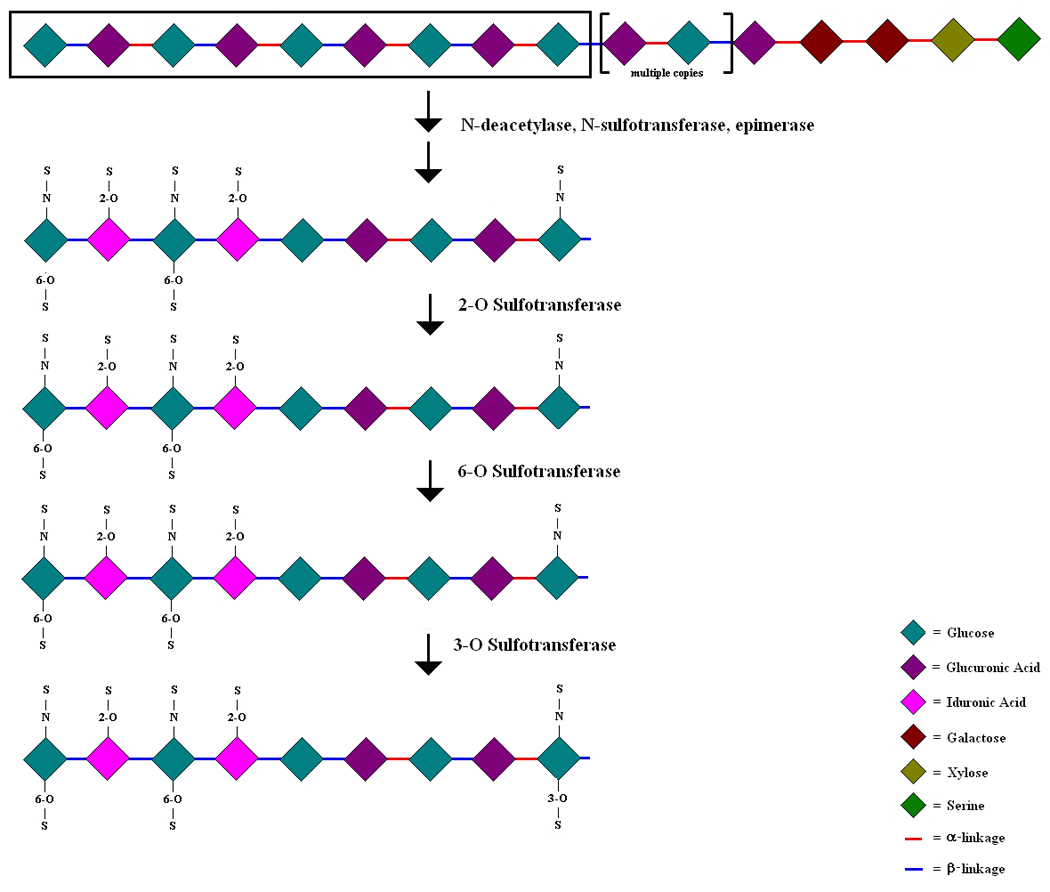
Outline of heparan sulfate (HS) maturation. A number of listed enzymes participate in the modification of the parent chain HS, which is a polymer of repeating disaccharide units containing a glucosamine and a glucuronic acid residue. All possible modifications and their preferred sequences (arrows) are shown.
A 3-dimensional structure for 3-O HS interaction with gD has been suggested [15]. The crystal structure of gD contains a positively charged deep pocket and a flat region with clusters of basic amino acid residues. Mutagenesis studies indicate that the pocket proximal to N-terminus may have a role in 3-OS HS binding since mutations in this region affect 3-OS HS usage by HSV-1 [31]. Interestingly, the same mutations also affect HVEM usage but not nectin-1. Thus, it is quite possible that at least one 3-OS HS binding region of gD may overlap with HVEM binding sites [15, 31]. Efforts have also been made to identify the structural specificity within HS that is needed for gD binding. Apart from a 3-O sulfated glucosamine, an upstream 2-O sulfated iduronic acid appears to be required as well [7]. To define the minimum size requirement for this interaction, a 3-O sulfated octasaccharide was generated [32]. This octasaccharide binds gD and demonstrates an inherent ability to block gD-triggered cell-to-cell fusion. Since the fusion was blocked in cells that may or may not use 3-OS HS as the major receptor for entry, it again suggests that 3-OS HS binding sites may overlap with other receptor (HVEM and possibly nectin-1) binding sites on gD. Another interesting property of 3-OS HS is that HSV-2 gD fails to use it as a receptor for entry [7]. In fact, even for the attachment process, HSV-2 demonstrates many differences in the HS structure recognition than HSV-1 [33]. It remains to be seen whether the inability to use 3-OS HS and additional differences in HS binding may have a role in the tissue tropism shown by the two HSV serotypes.
PILR-α: a new receptor for HSV-1 gB
Although viral attachment to cells shows strong dependence on the interactions of gB and gC with HS, studies have also shown that cells without HS are still susceptible to low-efficiency HSV-1 infection and retain the ability to bind soluble gB [34–35]. This suggestion that there may be other receptors interacting with gB, was upheld by a recent finding of gB’s ability to act as a ligand for PILR-α one of the paired inhibitory receptors found on monocytes, macrophages, and dendritic cells [19]. CHO-K1 cells, naturally resistant to HSV-1 infection, demonstrate susceptibility to infection after transfection with PILRα plasmid. Human cell lines expressing both HVEM and PILR-α experienced decreased viral entry when antibodies to either PILR-α or HVEM were used, supporting a separate need for both receptors.
Additionally, PILR-α contains a tyrosine-based motif in its cytoplasmic domain which delivers inhibitory signals to the host cell. Thus, by interacting with this inhibitory receptor, gB may allow HSV-1 to escape host immune system recognition via suppression while simultaneously exploiting the receptor for viral entry. The most current thought on HSV-induced membrane fusion may include PILR-α as an important and a balancing component of the fusion machinery [36]. Interestingly, HSV-2 entry may not be so strongly dependent on PILR-α as a co-receptor, as shown for retinal pigment epithelial (RPE) cells [37] and further confirmed by additional findings [38].
HSV-1 binding mediated by PILR-α also seems to mediate cell-virus fusion at the plasma membrane. Susceptible CHO cells utilize only endocytosis as an HSV entry mechanism. However, after inhibiting endocytosis and transfecting CHO cells with PILR-α, HSV-1 entry was still possible, presumably at the plasma membrane [38]. Therefore, PILR-α may be responsible for an alternate, but poorly understood, plasma membrane fusion mechanism of entry (Fig. 2). It is likely that PILR-α mediated fusion is a gD-dependent process that may not require a gD receptor as an essential component. Most recent studies seem to indicate that PILR-α plays a role both in binding and fusion; however, more work is needed to better elucidate the importance of this receptor.
Additional Receptors for HSV entry
A few cell surface molecules have been implicated as putative HSV-1 gH receptors. These include B5 and αvβ3 integrins [39,40]. Expression cloning in porcine kidney cells resistant for HSV-1 entry has led to the discovery of B5, a type-2 membrane protein containing an extracellular heptad repeat potentially capable of forming an α-helix for coiled coils [39]. Such structures may facilitate membrane fusion by interacting with viral proteins containing α-helices. A synthetic peptide identical to the heptad repeat region blocks HSV infection of B5-expressing porcine cells and human HEp-2 cells suggesting a possible role for B5 in HSV-1 entry. Since gH also contains an α-helix, it is speculated that it may be a ligand for B5 [39]. No direct interaction between B5 and gH has been demonstrated thus far. In contrast, in a separate study, a soluble form of gH-gL heterodimer was found to specifically bind cells expressing αvβ3 integrins but effect of this interaction on HSV-1 entry has remain elusive [40]. The binding appears to be highly specific since it can be prevented by mutating a potential integrin-binding motif, Arg-Gly-Asp (RGD), in gH. Again, more work is needed to determine and establish the significance of potential gH receptors.
Cell-to-Cell viral spread
HSV-1 virus is also unique in that the spread of infection is not dependent on a hematogenous or lymphatic route. Cell-to-cell contact is essential in HSV-1 and HSV-2 infection and elucidating the mechanism of cell-to-cell spread is important to fully understand the overall viral infectious process. Viral spread from cell-to-cell depends on the same gD interaction with its receptor(s) as seen when free virions initially infect a host cell [41]. Interestingly, gE and gI are glycoproteins that form a heterodimer required for cell-to-cell spread but are not required for the initial entry of free virus particles [42]. During HSV infection, the gE/gI heterodimers move from the trans-Golgi network (TGN) to epithelial cell junctions along with other viral glycoproteins and virion particles. Removing the gene responsible for the early sorting through the TGN prevents subsequent cell-to-cell spread and virions are observed to travel towards the apical surface instead of the cell-cell junctions. Assortment through the TGN early in infection was found to be necessary for efficient cell-to-cell spread of HSV-1. In addition, gK has also been shown to play an essential role in cell-to-cell spread in corneal and trigeminal ganglia cells, two cell lines that lead to the more devastating consequences of diseases such as blindness and meningitis [43]. Mice infected with mutated virus with gK gene deleted were visualized to show significantly decreased corneal spread and also had decreased clinical signs of infection.
Nectin-1 helps induce HSV-1 cell-to-cell spread. AF6, a multidomain protein that interacts with nectin-1, leads to decreased cell-to-cell HSV-1 spread when knocked down [44]. Similar to nectin-1, this protein is also involved in cell-cell adhesion and is found at cell junctions. Interestingly, AF6 knockdown does not affect the nectin-1 clustering that occurs at junctions for cell-to-cell transmission. Therefore, nectin-1 receptor clustering appears to take place independently of AF6 gene expression and the nectin-1 receptor clustering is not sufficient for cell-to-cell spread.
Although 3-O HS has been shown to act as an gD entry receptor for free virions fusing at the plasma membrane, 3-O HS has also been shown to play a role in cell-to-cell fusion and spread [28]. Previously, HVEM and nectin-1 expression were thought to be required for cell-to-cell spread [41]. However, newer studies imply that 3-O HS can also mediate cell-to-cell fusion, which is required during spread. Cells expressing gB, gD, gH, and gL, and therefore mimicking the essential viral machinery for membrane fusion, were able to fuse with 3-O HS-expressing cells [28]. Importantly, these cells expressed neither nectin-1 nor HVEM. When heparinase was used to eliminate the total HS, cell-to-cell fusion was dramatically decreased. In separate studies it was found that 3-OS HS is crucial for HSV-1-induced cell-to-cell fusion seen with primary cultures of corneal fibroblasts [45]. Therefore, the role of 3-O HS may be more extensive in the process of viral entry and spread than previously thought.
Conclusion
HSV-1 and HSV-2 both utilize similar mechanisms of binding, fusion, and subsequent cell-to-cell spread. Although fewer studies have been performed on HSV-2, the two viruses share 82% of their amino acid sequence and demonstrate high structural similarities [1]. Therefore, many of the concepts developed for HSV-1 are likely to be applicable for HSV-2; although further confirmation is needed. As illustrated, different glycoproteins play unique key roles in the infectious cycle. Binding can be initiated on filopodia with the interaction of gC and gB, however subsequent fusion and penetration utilize gB, gD, gH, and gL. Entry can directly occur at the plasma membrane or via an intracellular vesicle. Fusion appears to be mediated by fusogenic regions of gB and gH triggered by the interaction of gD with its receptor. Finally, cell-to-cell spread utilizes gD receptors, all fusion essential glycoproteins, the gE/gI heterodimer and gK. Clearly, significant advances have been made in our quest to understand the mechanism of HSV entry, although many areas still remain poorly understood. For example, the role of cellular signaling pathways is poorly defined. Activation of Rho signaling has been demonstrated [5, 46] and so is the involvement of focal adhesion kinases [47] but a complete picture is yet to emerge. It is also unclear why the virus undergoes endocytosis when identical receptors can promote fusion at the plasma membrane. A better understanding of how the virus fuses with a vesicular membrane is also needed. For commonly spreading viruses like HSV, such studies would have a high impact. In lieu of a protective vaccine [2], efficient microbicides or prophylactics can be designed by developing better understanding of virus entry mechanisms. Spread of the virus within a host can be contained by antiviral agents that target membrane fusion required for the spread. Similarly, targeting shared entry phenomena such as surfing, can lead to the production of broad-spectrum antiviral agents. In summary, viral entry is an important area of research with strong potential for identifying new strategies to prevent epidemic and pandemic viral diseases caused by HSV, HIV, papilloma, influenza and many additional viruses. Continuing research in this area can lead to a greater understanding of the disease process and new treatments for viral diseases.
Acknowledgment
The authors thank Dr. Beatrice Yue (UIC) for critical reading of the manuscript. Work described here that was performed in the laboratory of D. Shukla was supported by a NIH RO1 grant AI057860 and a NIH core grant EY01792. D.S. is a recipient of Lew Wasserman Merit award from Research to Prevent Blindness, New York.
References
- 1.Shukla D, Spear PG. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J Clin Invest. 2001;108:503–510. doi: 10.1172/JCI13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitley RJ, Roizman B. Herpes simplex virus infections. Lancet. 2001;357:1513–1518. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- 3.Spear PG. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol. 2004;6:401–410. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- 4.Campadelli-Fiume G, Amasio M, Avitabile E, Cerretani A, Forghieri C, Gianni T, Menotti L. The multipartite system that mediates entry of herpes simplex virus into the cell. Rev Med Virol. 2007;17:313–326. doi: 10.1002/rmv.546. [DOI] [PubMed] [Google Scholar]
- 5.Clement C, Tiwari V, Scanlan PM, Valyi-Nagy T, Yue BY, Shukla D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J Cell Biol. 2006;174:1009–1021. doi: 10.1083/jcb.200509155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 7.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 8.Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc Natl Acad Sci. 2007;104:18718–18723. doi: 10.1073/pnas.0707452104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sodeik B, Ebersold MW, Helenius A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J Cell Biol. 1997;136:1007–1021. doi: 10.1083/jcb.136.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akhtar J, Tiwari V, Oh MJ, Kovacs M, Jani A, Kovacs SK, Valyi-Nagy T, Shukla D. HVEM and nectin-1 are the major mediators of herpes simplex virus 1 (HSV-1) entry into human conjunctival epithelium. Invest Ophthalmol Vis Sci. 2008;49:4026–4035. doi: 10.1167/iovs.08-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiwari V, Oh MJ, Kovacs M, Shukla SY, Valyi-Nagy T, Shukla D. Role for nectin-1 in herpes simplex virus 1 entry and spread in human retinal pigment epithelial cells. FEBS J. 2008;275:5272–5285. doi: 10.1111/j.1742-4658.2008.06655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixit R, Tiwari V, Shukla D. Herpes simplex virus type 1 induces filopodia in differentiated P19 neural cells to facilitate viral spread. Neurosci Lett. 2008;440:113–118. doi: 10.1016/j.neulet.2008.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J Cell Biol. 2005;170:317–325. doi: 10.1083/jcb.200503059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schelhaas M, Ewers H, Rajamaki ML, Day PM, Schiller JT, Helenius A. Human papillomavirus type 16 entry: retrograde cell surface transport along actin-rich protrusions. PLoS Pathog. 2008;4:e1000148. doi: 10.1371/journal.ppat.1000148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol Cell. 2001;8:169–179. doi: 10.1016/s1097-2765(01)00298-2. [DOI] [PubMed] [Google Scholar]
- 16.Gianni T, Amasio M, Campadelli-Fiume G. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL through the C-terminal profusion. J Biol Chem. 2009;284:17370–17382. doi: 10.1074/jbc.M109.005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subramanian RP, Geraghty RJ. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc Natl Acad Sci U S A. 2007;104:2903–2908. doi: 10.1073/pnas.0608374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galdiero S, Vitiello M, D'Isanto M, Falanga A, Cantisani M, Browne H, Pedone C, Galdiero M. The identification and characterization of fusogenic domains in herpes virus glycoprotein B molecules. Chembiochem. 2008;9:758–767. doi: 10.1002/cbic.200700457. [DOI] [PubMed] [Google Scholar]
- 19.Satoh T, Arii J, Suenaga T, Wang J, Kogure A, Uehori J, Arase N, Shiratori I, Tanaka S, Kawaguchi Y, et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell. 2008;132:935–944. doi: 10.1016/j.cell.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gianni T, Campadelli-Fiume G, Menotti L. Entry of herpes simplex virus mediated by chimeric forms of nectin1 retargeted to endosomes or to lipid rafts occurs through acidic endosomes. J Virol. 2004;78:12268–12276. doi: 10.1128/JVI.78.22.12268-12276.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicola AV, McEvoy AM, Straus SE. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol. 2003;77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicola AV, Hou J, Major EO, Straus SE. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J Virol. 2005;79:7609–7616. doi: 10.1128/JVI.79.12.7609-7616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stiles KM, Milne RS, Cohen GH, Eisenberg RJ, Krummenacher C. The herpes simplex virus receptor nectin-1 is down-regulated after trans-interaction with glycoprotein D. Virology. 2008;373:98–111. doi: 10.1016/j.virol.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tiwari V, Clement C, Scanlan PM, Kowlessur D, Yue BY, Shukla D. A role for herpesvirus entry mediator as the receptor for herpes simplex virus 1 entry into primary human trabecular meshwork cells. J Virol. 2005;79:13173–13179. doi: 10.1128/JVI.79.20.13173-13179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor JM, Lin E, Susmarski N, Yoon M, Zago A, Ware CF, Pfeffer K, Miyoshi J, Takai Y, Spear PG. Alternative entry receptors for herpes simplex virus and their roles in disease. Cell Host Microbe. 2007;2:19–28. doi: 10.1016/j.chom.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiwari V, Clement C, Xu D, Valyi-Nagy T, Yue BY, Liu J, Shukla D. Role for 3-O-sulfated heparan sulfate as the receptor for herpes simplex virus type 1 entry into primary human corneal fibroblasts. J Virol. 2006;80:8970–8980. doi: 10.1128/JVI.00296-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindahl U, Kusche-Gullberg M, Kjellen L. Regulated diversity of heparan sulfate. J Biol Chem. 1998;273:24979–24982. doi: 10.1074/jbc.273.39.24979. [DOI] [PubMed] [Google Scholar]
- 28.Tiwari V, Clement C, Duncan MB, Chen J, Liu J, Shukla D. A role for 3-O-sulfated heparan sulfate in cell fusion induced by herpes simplex virus type 1. J Gen Virol. 2004;85:805–809. doi: 10.1099/vir.0.19641-0. [DOI] [PubMed] [Google Scholar]
- 29.O'Donnell CD, Tiwari V, Oh MJ, Shukla D. A role for heparan sulfate 3-O-sulfotransferase isoform 2 in herpes simplex virus type 1 entry and spread. Virology. 2006;346:452–459. doi: 10.1016/j.virol.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 30.Xu D, Tiwari V, Xia G, Clement C, Shukla D, Liu J. Characterization of heparan sulphate 3-O-sulphotransferase isoform 6 and its role in assisting the entry of herpes simplex virus type 1. Biochem J. 2005;385:451–459. doi: 10.1042/BJ20040908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoon M, Zago A, Shukla D, Spear PG. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J Virol. 2003;77:9221–9231. doi: 10.1128/JVI.77.17.9221-9231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Copeland R, Balasubramaniam A, Tiwari V, Zhang F, Bridges A, Linhardt RJ, Shukla D, Liu J. Using a 3-O-sulfated heparin octasaccharide to inhibit the entry of herpes simplex virus type 1. Biochemistry. 2008;47:5774–5783. doi: 10.1021/bi800205t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herold BC, Gerber SI, Belval BJ, Siston AM, Shulman N. Differences in the susceptibility of herpes simplex virus types 1 and 2 to modified heparin compounds suggest serotype differences in viral entry. J Virol. 1996;70:3461–3469. doi: 10.1128/jvi.70.6.3461-3469.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banfield BW, Leduc Y, Esford L, Schubert K, Tufaro F. Sequential isolation of proteoglycan synthesis mutants by using herpes simplex virus as a selective agent: evidence for a proteoglycan-independent virus entry pathway. J Virol. 1995;69:3290–3298. doi: 10.1128/jvi.69.6.3290-3298.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bender FC, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J Virol. 2005;79:11588–11597. doi: 10.1128/JVI.79.18.11588-11597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan Q, Lin E, Satoh T, Arase H, Spear PG. Differential effects on cell fusion activity of mutations in herpes simplex virus 1 gB dependent on whether a gD receptor or a gB receptor is over-expressed. J Virol. 2009 doi: 10.1128/JVI.00087-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shukla SY, Singh YK, Shukla D. Role of Nectin-1, HVEM, and PILR-alpha in HSV-2 Entry into Human Retinal Pigment Epithelial Cells. Invest Ophthalmol Vis Sci. 2009;50:2878–2887. doi: 10.1167/iovs.08-2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arii J, Uema M, Morimoto T, Sagara H, Akashi H, Ono E, Arase H, Kawaguchi Y. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor {alpha} J Virol. 2009 doi: 10.1128/JVI.02601-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perez A, Li QX, Perez-Romero P, Delassus G, Lopez SR, Sutter S, McLaren N, Fuller AO. A new class of receptor for herpes simplex virus has heptad repeat motifs that are common to membrane fusion proteins. J Virol. 2005;79:7419–7430. doi: 10.1128/JVI.79.12.7419-7430.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parry C, Bell S, Minson T, Browne H. Herpes simplex virus type 1 glycoprotein H binds to alphavbeta3 integrins. J Gen Virol. 2005;86:7–10. doi: 10.1099/vir.0.80567-0. [DOI] [PubMed] [Google Scholar]
- 41.Pertel PE, Fridberg A, Parish ML, Spear PG. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology. 2001;279:313–324. doi: 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- 42.Farnsworth A, Johnson DC. Herpes simplex virus gE/gI must accumulate in the trans-Golgi network at early times and then redistribute to cell junctions to promote cell-cell spread. J Virol. 2006;80:3167–3179. doi: 10.1128/JVI.80.7.3167-3179.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.David AT, Baghian A, Foster TP, Chouljenko VN, Kousoulas KG. The herpes simplex virus type 1 (HSV-1) glycoprotein K(gK) is essential for viral corneal spread and neuroinvasiveness. Curr Eye Res. 2008;33:455–467. doi: 10.1080/02713680802130362. [DOI] [PubMed] [Google Scholar]
- 44.Keyser J, Lorger M, Pavlovic J, Radziwill G, Moelling K. Role of AF6 protein in cell-to-cell spread of Herpes simplex virus 1. FEBS Lett. 2007;581:5349–5354. doi: 10.1016/j.febslet.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 45.Tiwari V, ten Dam GB, Yue BY, van Kuppevelt TH, Shukla D. Role of 3-O-sulfated heparan sulfate in virus-induced polykaryocyte formation. FEBS Lett. 2007;581:4468–4472. doi: 10.1016/j.febslet.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Donnell CD, Shukla D. A novel function of heparan sulfate in the regulation of cell-cell fusion. J Biol Chem. 2009 Sep 2; doi: 10.1074/jbc.M109.037960. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheshenko N, Liu W, Satlin LM, Herold BC. Focal adhesion kinase plays a pivotal role in herpes simplex virus entry. J Biol Chem. 2005;280:31116–31125. doi: 10.1074/jbc.M503518200. [DOI] [PubMed] [Google Scholar]