

Abstract
Background
The application of high-throughput genomic tools in nutrition research is a widespread practice. However, it is becoming increasingly clear that the outcome of individual expression studies is insufficient for the comprehensive understanding of such a complex field. Currently, the availability of the large amounts of expression data in public repositories has opened up new challenges on microarray data analyses. We have focused on PPARα, a ligand-activated transcription factor functioning as fatty acid sensor controlling the gene expression regulation of a large set of genes in various metabolic organs such as liver, small intestine or heart. The function of PPARα is strictly connected to the function of its target genes and, although many of these have already been identified, major elements of its physiological function remain to be uncovered. To further investigate the function of PPARα, we have applied a cross-species meta-analysis approach to integrate sixteen microarray datasets studying high fat diet and PPARα signal perturbations in different organisms.
Results
We identified 164 genes (MDEGs) that were differentially expressed in a constant way in response to a high fat diet or to perturbations in PPARs signalling. In particular, we found five genes in yeast which were highly conserved and homologous of PPARα targets in mammals, potential candidates to be used as models for the equivalent mammalian genes. Moreover, a screening of the MDEGs for all known transcription factor binding sites and the comparison with a human genome-wide screening of Peroxisome Proliferating Response Elements (PPRE), enabled us to identify, 20 new potential candidate genes that show, both binding site, both change in expression in the condition studied. Lastly, we found a non random localization of the differentially expressed genes in the genome.
Conclusion
The results presented are potentially of great interest to resume the currently available expression data, exploiting the power of in silico analysis filtered by evolutionary conservation. The analysis enabled us to indicate potential gene candidates that could fill in the gaps with regards to the signalling of PPARα and, moreover, the non-random localization of the differentially expressed genes in the genome, suggest that epigenetic mechanisms are of importance in the regulation of the transcription operated by PPARα.
Background
The availability of public gene expression repositories, such as GEO [1] and ArrayExpress [2] has opened up new challenges on microarray data analyses, especially in the field of data integration and meta-analysis [3-6]. Meta-analysis, defined as the analysis of multiple gene expression datasets concerning a common biological problem, is performed to confirm, strengthen and complete the results obtained by single studies and to find common pathways altered in specific physiological or pathological conditions. Pivotal studies of this type have been performed on cancer [4] and have recently demonstrated their capability of retrieving much more relevant information than single experiment datasets [5,6]. In this study, we used a meta-analysis approach to investigate gene regulation and biological processes involved in response to high fat diet and under control of peroxisome-proliferator-activated receptor alpha (PPARα). As demonstrated by Bünger et al. the most of the nutritional science papers usually use transcriptome analysis merely as screening tool in order to focus the study on a single gene or on a pathway. On the one hand this type of approach simplifies the findings and their validations, but in the other hand miss the opportunity to observe the complete picture and infer the mechanism underlying the observations [7,8]. It is becoming increasingly clear that rather than reducing the information the challenge is now to fill gaps integrating information coming from genome-wide data, especially in nutrigenomics field.
Lipids serve as membrane constituents and supply and storage of energy. The related fatty acids are precursors of a wide range of bioactive molecules. They have the ability to regulate a wide variety of cellular processes through the induction of changes in gene expression. Indeed, fatty acids and some oxygenated derivates are ligands able to activate a class of transcription factors, called peroxisome-proliferator-activated receptors (PPARs), key regulators in energy storage and metabolism. PPARs (PPARα, PPARγ and PPARβ/δ) belong to a superfamily of nuclear hormone receptors that share a common action mechanism: the formation of heterodimers with the nuclear receptor RXR and the consequent binding to cis-element of promoter of a target gene.
PPARα is expressed primarily in metabolic tissues (brown adipose tissue, liver, kidney) but elevated levels are also present in the digestive (jejunum, ileum, colon, gall bladder) and cardiopulmonary (aorta, heart) systems, and plays a central role in almost all aspects of fatty acid catabolism in particular in the liver. Recent studies demonstrate that the role of PPARα is not limited only to metabolism but it also acts in many processes like inflammation [9], immunity [10], cardiovascular disease [11] and cancer [12], leading to the concept of an expanded activity of the nuclear receptor on more than one process. Lemay and Hwang in 2006 scanned the whole human genome using a PPRE matrix designed through the analysis of all known PPRE. The research was limited to conserved elements, as in space, evaluating neighbouring nucleotide, as in time, considering the sequence of different species, giving us a list of predicted PPAR targets [13]. Our study complemented the genome-wide analysis conducted to date by adding a meta-analysis performed across species of expression data related to PPARα signaling. Publicly available gene expression studies selected for our meta-analysis included experiments addressing molecular response to high fat diet, PPARα activation by various stimuli and gene expression in PPARα knock-out performed in a number of organisms and different array platforms. The comparison of gene expression across species (Homo sapiens, Mus musculus, Rattus norvegicus and Saccharomyces cerevisiae) is based on the known evolutionary conserved regulatory mechanism responsive to fatty acid-rich diet. In S. cerevisiae the homologous of mammalian PPARα-RXR is Pip2p-Oaf1p [14]. These two proteins form a heterodimer that activates the transcription of genes directly involved in peroxisome proliferation and fatty acid metabolism in response to a nutritional input [15]. Taking advantage of functional evolutionary constraints, comparison across species has potential to improve the comprehension of the biological mechanisms in response to fatty acid-rich diet suggesting novel candidate genes involved in PPARα signaling. Due to the evolutionary conservation, findings resulting from a cross-species analysis are expected to bring forward processes and genes that are most relevant for the studied condition. In addition, the discovery of novel PPARα targets homologous from yeast to human allows characterization of these genes in model organisms and extrapolation of the findings to the human situation.
In this study we provide a list of homolog genes subjected to comparable stimuli, showing significant changes in expression levels after changes in PPARα activity. Thus, taking advantage of all datasets available on PPAR signaling and high fat diet, our study offers a comprehensive overview of the key pathways and cellular processes regulated by PPARα. We identified a series of chromosomal regions in the mouse genome specifically enriched by PPARα related genes suggesting common regulatory mechanisms. Finally we provide a useful method and interesting information to identify new target genes integrating the results of our work on gene expression with those previously obtained by Lemay et Hwang on PPRE sequence [13].
Methods
Data collection
Array Express [16] and GEO [1] databases were used to select expression datasets suitable for the meta-analysis. Only datasets with CEL files for Affymetrix and raw intensity data for the other technologies have been taken into account. Additional datasets were selected to test the results of our work.
The data collection consisted of 16 datasets, that could be divided into three separate groups: (i) experiments on rat, mouse and human hepatocytes, where PPARα signaling is activated treating cells with WY14643, (ii) experiments in which PPARα signaling is completely inhibited using mouse PPARα knockout, (iii) experiments in which the organism under study were fed with high fat diet, comprehensive of the Saccharomyces cerevisiae dataset performed in our laboratory. On the whole, we included in the analysis 202 hybridizations (see Table 1 for details).
Table 1.
Meta-analysis data collection.
| PPARα signaling | n° | Reference | Dataset Accession Number GEO/AE |
Org | Tissue | Technology |
|---|---|---|---|---|---|---|
| PPARα signaling activated by WY14643 | 1 | [55] | GSE8302/E-GEOD-8302 | Hs | Liver | Affymetrix |
| 2 | [55] | GSE8302/E-GEOD-8302 | Mm | Liver | Affymetrix | |
| 3 | [55] | GSE8302/E-GEOD-8302 | Rn | Liver | Affymetrix | |
| PPARα signaling repressed using PPARα knokout mice | 4 | [55] | GSE8290/E-GEOD-8290 | Mm | Liver | Affymetrix |
| 5 | [55] | GSE8291/E-GEOD-8291 | Mm | Liver | Affymetrix | |
| 6 | [55] | GSE8292/E-GEOD-8292 | Mm | Liver | Affymetrix | |
| 7 | [55] | GSE8295/E-GEOD-8295 | Mm | Liver | Affymetrix | |
| PPARα signaling activated by High fat diet | 8 | [56] | GSE8753/E-GEOD-8753 | Mm | Liver | Affymetrix |
| 9 | [57] | GSE6903/E-GEOD-6853 | Mm | Liver | Affymetrix | |
| 10 | [58] | GSE8524/~ | Mm | Liver | Affymetrix | |
| 11 | [59] | GSE1560/E-GEOD-1560 | Mm | Aorta | Oligo Array | |
| 12 | [60] | GSE8700/E-GEOD-8700 | Rn | Epididymal fat | Affymetrix | |
| 13 | [61] | ~/E-MEXP-893 | Mm | Colon mucosa | Affymetrix | |
| 14 | [62] | ~/E-CBIL-24 | Mm | Liver | Affymetrix | |
| 15 | [63] | GSE432/~ | Mm | Liver | Oligo Array | |
| 16 | Marchi and Cavalieri in preparation | ~/E-TABM-614 | Sc | ~ | Oligo Array | |
Additional datasets were selected to test the results of our work. (see Table 2 for details). The validation set was composed of one dataset of S.cerevisiae with expression data of several knockout yeast for transcription factor involved in oleate response, and two mouse datasets with experiments belonging at three category of experimental design mentioned above.
Table 2.
Validation data sets.
| n° | PPARα signalling | Reference | Dataset Accession Number GEO/AE | Org | Tissue | Technology |
|---|---|---|---|---|---|---|
| 1 | PPARα signaling activated by WY14643 (PPARα WY14643-GSE8396) |
[64] | GSE8396/E-GEOD-8396 | Mm | Liver | Affymetrix |
| 2 | PPARα signaling repressed using PPARα knokout mice (PPARα KO1-GSE8396) |
[64] | GSE8396/E-GEOD-8396 | Mm | Liver | Affymetrix |
| 3 | PPARα signaling repressed using PPARα knokout mice (PPARα KO1-GSE8396) |
[64] | GSE8396/E-GEOD-8396 | Mm | Liver | Affymetrix |
| 4 | PPARα signaling activated by High fat diet (HFD-E-MEXP-1755) |
[65] | ~/E-MEXP-1755 | Mm | Liver | Affymetrix |
| 5 | Oleate response repressed using knokout yeast of a transcription promoter (del_ADR1) |
[50] | GSE5862/~ | Sc | ~ | Oligo Array |
| 6 | Oleate response repressed using knokout yeast of a transcription promoter (del_PIP2) |
[50] | GSE5862/~ | Sc | ~ | Oligo Array |
| 7 | Oleate response repressed using knokout yeast of a transcription promoter (del_OAF1) |
[50] | GSE5862/~ | Sc | ~ | Oligo Array |
| 8 | Oleate response activated using knokout yeast of a transcription repressor (del_OAF3) |
[50] | GSE5862/~ | Sc | ~ | Oligo Array |
| 9 | Oleate response activated by High fat diet (oleate_vs_low_glucose) |
[50] | GSE5862/~ | Sc | ~ | Oligo Array |
Statistical analysis of microarray data
Gene expression of Affymetrix datasets were quantified and separately normalized using rma technique [17] and. EntrezGene Custom CDF file proposed by Dai et al. [18] was used to re-annotate Affymetrix probe sets in order to have an efficient and up-to-date genome annotation of array features. Raw data derived by oligo microarray were normalized using lowess algorithm exploiting MIDAW web tool [19]. In order to identify differentially expressed gene (hereafter DEGs) we performed SAM test [20], a moderated t-test with permutational approach. P-values and then Q-values (false discovery rate, FDR) were used to control test multiplicity, 0.05 was the chosen cut-off. Q-values for each gene were defined as: Q = (p*n)/i, where p is the p-value of the gene, n the total number of genes and i is the number of genes at or better than p. Statistical analyses was performed with R software http://www.r-project.org.
Unfortunately, not all the datasets contained sufficient numbers of biological replicates as required for powerful inference. Fold change cut-off, filtered by variance coefficient, was used to select DEGs in those datasets with less than 3 replicates per gene (GSE8302, GSE9291 and GSE9290).
Pathways analysis on DEGs
Enrichment analysis on metabolic pathways was calculated for each dataset using Fisher exact test based on hypergeometric distribution with a p-value cutoff of 0.1. Similarity structure on metabolic enrichment characteristics across datasets was performed using cluster analysis. A Boolean matrix with pathways in rows and datasets in columns was generated, where matrix cells equal to 1 identified significant enrichment of a given pathway in a given dataset, and 0 otherwise [21,22]. Using TM4 [23] a hierarchical dendrogram based on Euclidean distance, average linkage and with bootstrap support was generated.
Meta-analysis approach
Homologene database [24] was used to match DEG lists across different species. Mouse annotation was used as reference; thus, each gene has been converted to its correspondent Mus musculus HomologeneID.
Given the presence of some datasets without gene p-value (see previous paragraph for details), we decided to adopt as meta-analysis procedure the vote counting approach proposed by Rhodes and colleagues [3]. The vote counting approach allows the identification of a set of genes common to j of the S total number of datasets with j = 2...S. The idea was to compare the observed number of significant genes shared by at least j studies (observed gene enrichment) with the number of significant genes shared by at least j studies obtained by chance (random gene enrichment). The number j was defined through a permutational approach. Permutational steps were the following: i) Q-values of each dataset were randomly permutated so that genes in each signature (list of differentially expressed genes) changed randomly, but the number of genes in each signature remained the same, ii) the number of genes differentially expressed common to at least j datasets was calculated for j ranging from 2 to the total number of datasets, iii) step i) and ii) were repeated 1000 times, iv) average and empirical confidence intervals (at confidence level 95%) of the number of random gene enrichment for each j (across the 1000 simulations) were calculated. Then, we compared the observed number of genes shared by at least j studies with the confidence interval obtained through the permutational approach and choose those js showing a significant difference between observed and random number of gene enrichment. Finally among these js we selected the minimum j such that the ratio between the expected and observed number of genes shared was less than 10%. In our analysis the number of j leading to 4% of false positives was found to be equal to 6 [5]. Finally, meta-analysis approach produced a list of 164 genes, called MDEGs (Meta-analysis Differential Expressed Genes), given by the integration of the DEGs shared by at least 6 datasets (See Additional File 1 and 2 for details).
Pathways analysis on MDEGs
An enrichment analysis, similar to that described in the previous paragraph, was applied on MDEGs. In this case, using hypergeometric distribution, the enrichment test set was represented by the MDEG list, while the reference set should be virtually generated according to the number j identified through the permutational approach described above. The virtual reference set for the hypergeometric distribution was obtained by selecting all the genes common in at least j platforms. In our analysis the reference set contained 15,463 genes. This new general approach, specifically adapted for meta-analysis enrichment analysis, showed several advantages, accurate results, faster and easier execution.
A gene ontology network was drawn and analysed by means of the BINGO plug-in [25] of Cytoscape software version 2.6.0 [26]. Statistical significance was calculated using hypergeometric test with an FDR cut off equal to 0.05.
Gene signature validations
The validation set was divided by organism, we performed one validation with S. cerevisiae and one for mouse data. The differentially expressed genes of each dataset are filtered by the species specific MDEGs and matrix with genes in row and dataset in column were build. Using TM4 [23] a hierarchical dendrogram with bootstrap support was generated.
Transcription factor binding site search
Over-represented putative transcription factor binding sites have been detected for the lists of differentially expressed genes with oPOSSUM web tool [27]. The default parameters suggested by the Authors have been used to find TFBSs in the genomic flanking regions 2000 bp upstream and downstream the transcription starting site of MDEGs. Two statistical measures (Z-score and Fisher exact one-tail probability) were calculated to determine which TFBS were significantly over-represented in the examined flanking regions. Z-score > 5 and Fisher p-value < 0.05 were used as significant cut-off thresholds.
Chromosomal clustering
Following the approach proposed by Vogel et al. [28] we searched for correlations between chromosome location, regulation and function of genes. In order to find genes located in chromosome clusters along the genome, we used a bioinformatic tool called REEF [29]. The first analysis step used the distribution of MDEGs in the genome, performed with different parameters of "window width" and "window shift". The tool calculate the hypergeometric probability, taking into account the number of studied genes and the number of the genes in the genome contained in each window. Statistical significance was calculated using a cut off equal to 0.05.
Results and discussion
Meta-analysis of gene expression datasets
Our work aims at developing methodological and computational procedures for the study of metabolic pathways and conserved regulatory mechanisms underlying the fundamental biological response to high fat diet, with additional goal to suggest novel candidate genes belonging to PPARα signaling.
We used a cross-species meta-analysis approach for the integration, at the gene level, of sixteen transcriptional datasets from different organisms (human, mouse, rat and yeast) and experimental platforms (Affymetrix, single and double channels spotted oligonucleotides). The datasets included into the analysis were focused on either genetic or dietary perturbations.
First, we evaluated the presence of possible trend in clustering due to organism, platform, tissue and experimental design variability characterising the sixteen datasets selected for the analysis. After inferential and enrichment analysis on each single dataset, functional similarities among studies have been performed through cluster analysis (see Methods for details). We expected that if some biases would be present in our analysis, datasets should be grouped according to the sources of variability. Figure 1 represents the dendrogram resulting from cluster analysis performed on GO and KEGG enriched classes. As shown in Figure 1, despite some differences, all the datasets seem to be highly similar, strictly comparable and not grouped according to the mentioned possible sources of variability. Thus, we proceeded with the meta-analysis approach in order to identify a set of marker genes highly relevant for the response to high fat diet and PPARα signaling. The clustering results support the evidence that dietary conditions modulating PPAR signaling and in general, high fat-low fat diet affect a coherent and evolutionary conserved core of genes, that overcomes the differences in gene expression associated to cell type, tissue and organ.
Figure 1.

Functional enrichment bootstrap support trees of datasets analyzed. Biological Process (BP), KEGG's pathways (KEGG), Cellular Components (CC) and Molecular Functions (MF).
Analyzing the data intra and inter datasets, the meta-analysis approach allows us to find the most frequently deregulated genes in the tested condition.
Comparing the MDEG list with the DEG lists of datasets analyzed individually, we observe that the meta-analysis led us to identify a smaller number of total genes but biologically more strongly correlated to the studied condition, probably reducing the false positive genes and recovering true positive genes eliminated by a strict statistic at a single study level. A cross-species meta-analysis provides an added value to find conserved genes and for this reason is more reliable.
Meta-analysis approach identified 164 differentially expressed genes shared by at least 6 datasets (MDEGs) (for a complete list of MDEG see Additional File 1). The 164 MDEGs are selected with the consensus of at least 6 datasets regardless of the species. All the species are represented in the MDEG list but not all MDEGs are represented in all species excepted for mouse. This is due to the different number of organism specific experiments and the evolutionary distance between the species and the reference species. The Venn diagram represents the contribution of the four organisms in defining the MDEGs (Figure 2). The 100% of MDEGs (164 genes) are found in mice datasets, the 28.6% (47 genes) are found in human, the 25% (41 genes) in yeast and the 55.5% (91 genes) in rat datasets.
Figure 2.
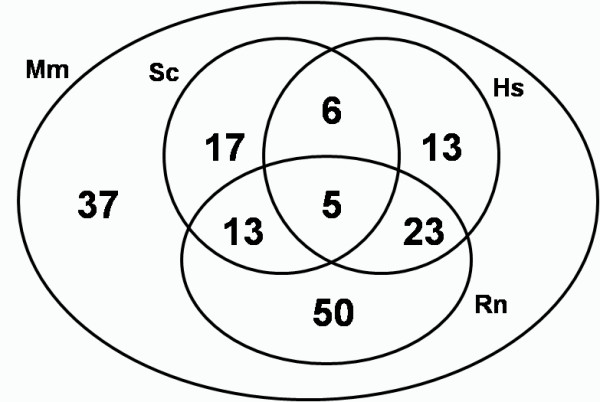
Venn diagram of 164 MDEGs sharing among each organism.
Functional characterization of the MDEGs
To study the identified set of 164 genes, we applied a network-based approach to describe the over-representation of GO categories in our pool of genes. The background organism used for this analysis was mouse, because it best represents all the genes highlighted in this study. This allows assessment of the reliability that our gene set reflects in the response to high fat diet and PPARα signaling as it is known by the literature (Figure 3 &4). For details on number of genes on each category, hypergeometric and FDR correction see Additional File 2.
Figure 3.

Graphical representation of enriched biological process Gene Ontology category in MDEGs. The network represented is a hierarchical network, the arrow from the element A to the element B signifies "the element B is part of the element A". The size of each circle is proportional to the number of gene contained in the category and circles are shaded based on significance. Red indicate P-value close to zero, yellow represent p-value close to cut-off threshold, white are not significant category. Biological Process (GO) category over-represented in MDEGs: A) amino-acid metabolism B) sugar metabolism C) lipid metabolism and transport.
Figure 4.

Graphical representation of enriched cellular component Gene Ontology category in MDEGs. Cellular Component (GO) category over-represented in MDEGs: A) endoplasmic reticulum B) peroxisome C) mitochondrion.
Resulting biological process network (Figure 3) could be divided into three areas: i) group A, representing categories linked to amino acid metabolism, ii) group B, composed by carbohydrate metabolism, specifically related to monosaccharide metabolism and gluconeogenesis, and iii) group C, with categories associated to lipid metabolism and transport. These results give an overview of how PPARα modulates gene expression in order to regulate energy metabolisms.
The main over-represented cluster (cluster C Figure 3) was composed by lipid related categories, biogenesis, catabolism and transport of lipids. Fatty acid metabolism was one of the fundamental category of the network (Hsd17b4, Ehhadh, Dci and Acox1, Acadl and Acadvl), together with biosynthesis of lipids, represented by the enzyme to elongation of fatty acid (Elovl3, Elovl5, Elovl6) and Stearoyl-Coenzyme A desaturase 1 (Scd1) which catalyzes the rate limiting step in biosynthesis from unsaturated to saturated fatty acids. Moreover we found Me1 (Malic enzyme), which catalyzes the generation of NADPH required for fatty acid biosynthesis. Me1 and Scd1 are known as target genes of PPARα responsible for important parts of lipogenesis [30-32]. The activation of PPARα and fatty acid metabolism requires mobilization and transport within the cell and the engagement of various compartments of fatty acids. The categories of fatty acid transport in MDEGs were represented by Cpt1b, Cpt2 and Adfp. Cpt1b is a carnitine palmitoyltransferase enzyme, responsible for the oxidation of fatty acid allowing the translocation across the outer mitochondrial membrane and the starting of fatty acid oxidation. The carnitine palmitoyltransferase II (Cpt2) encodes for an enzyme embedded into inner mitochondrial membrane, that favours the reaction condensating coenzyme A with long-chain fatty acids facilitating the release from mitochondria. Cpt1b and Cpt2 are directly regulated by PPARα [33-38]. The intracellular transport of fatty acids to the nucleus and the nuclear receptor was represented in MDEGs by Fabp1, regulated by PPARα [39-41].
The representation of amino-acid metabolism pathway in GO network could be well explained by the interplay between anabolism and catabolism. In case of caloric restriction, amino acids are precursors for lipids, carbohydrates, and nucleic acids used as co-substrates and co-enzymes in the production of energy. On the contrary in case of dietary surplus, the potentially toxic nitrogen from amino acids has to be eliminated via transamination, deamination, and urea formation. Kersten et al. observed that in fasted mice the simultaneous increase in ketone body concentration and decrease in urea concentration are due to the action of a single factor, PPARα, which regulates the transcription of genes involved in the relative pathways, up-regulating fatty acid oxidation genes and downregulating the ureagenesis and ammonia detoxification.
PPARα regulates amino-acid metabolism through several genes, two of them, Cytosolic aspartate aminotransferase (Got1) and argininesuccinate lyase (Asl) were included in MDEGs [42,43].
Finally, Kersten et al. demonstrated that PPARα regulates carbohydrate metabolisms in particular by acting on gluconeogenesis [42] and governing hepatic glycerol metabolism [44].
All this evidence demonstrates that PPARα, despite its primary role in regulating the metabolism of fatty acids, acts as a master regulator of the rate of utilization of the various energy substrates in relation to food availability, and the identified network of biological process provides full details of all these aspects.
In addition to biological processes network, the cellular component network (Figure 4) highlights that the principal sites of activity during the response to high fat diet and PPARα signaling activation are the endoplasmatic reticulum (cluster A), the peroxisome (cluster B), and the mitochondrion (cluster C). The over-represented cellular components indicate that mitochondrion was the most over-represented scenario (26.8%; 44 genes) of the differential transcription regulation under the studied stimuli.
The results of pathway enrichment analysis are shown in Additional File 2. Several MDEGs belong to pathways related to high fat diet and 16 out of 164 MDEGs (10%) belonged to KEGG's Mouse PPARs signaling pathway (Acadl, Acaa1a, Acox1, Cpt1b, Cpt2, Cyp4a14, Cyp8b1, Fabp1, Hmgcs2, Me1, Scd1, Sorbs1, Acsl5, Angptl4, Ehhadh, Acsl3). Each of these 16 genes could be specifically connected to PPARα signal and not to the others different type of PPARs. Focusing on PPARα target genes in the whole set of 164 MDEGs, we found that the transcription of 42 genes (25.6%) was regulated by this nuclear receptor (Additional File 2). We also observed that only 11 of these 42 genes were characterized by functional PPRE and 5 genes have an in silico predicted PPRE.
Evolutionary conserved markers of high-fat response
Cross-species analysis allows deciphering molecular complexity through evolutionary constrains. As expected mammalian organisms share the largest amount of genes (Figure 2), however, it is interesting to note that 41 yeast ORF (25%) were identified among MDEGs, each of these yeast genes has a homolog in mouse.
Looking at functions and processes linked to the yeast MDEGs, we observed that 7 out 41 genes, (17%; CAT2, FEN1, POT1, FOX2, YAT1, CRC1, SPS19) were directly involved in fatty acid metabolism and transport. CRC1, POT1, YAT1, FOX2 and SPS19 are targets of the transcription induced by Oaf1-Pip2 in yeast [45]. The 41 yeast genes identified by meta-analysis are localized both in mitochondrion (49%; 20 genes) and in peroxisome (12%; 5 genes). This strongly agrees with findings described in literature, stating that S. cerevisiae adapts to oleic acid as a sole carbon source inducing transcriptional modulation of both peroxisomal and mitochondrial function [46]. In addition to lipid metabolism, transcriptional reprogramming induced by oleic acid in yeast, as in the mammalian organisms, deregulates the amino acid metabolism (20%; 8 genes; EHD3, ARG4, CAT2, CYS3, CDC60, YAT1, GLN1, AAT2). Interestingly we discovered 5 yeast genes that are homologous of mammalian genes under control of PPARα. The 5 genes are ADP1 homologous of Abcg2 [47], AAT2 homologous of Got1 [42,43], ARG4 homologous of Asl [42], FOX2 homologous of Hsd17b4 [48,49] and YAT1 homologous of human CPT2 [37,38]. Interestingly only two genes Hsd17b4/FOX2 and CPT2/YAT1 appears to share the same transcriptional regulator PPARa in the mammal and Pip2p-Oaf1p in the yeast. This suggests FOX2 and YAT1 as central and evolutionary conserved response elements for the high fat diet response. Moreover these findings indicate that, either the regulatory structure remains to be completely elucidated, the other three genes could represent valid candidate genes for future investigations and that they could be used as model study for mammals genes exploiting the awesome benefits of yeast genetics.
Gene signature validation
Further validation of our methodology and analyses came from the comparison of our gene list with several external expression dataset. We performed a validation for yeast, that have a gene signature composed of 41 MDEGs and a validation for mouse using all the 164 genes. The validation on yeast was performed using a dataset published in GEO database by Smith et al. [50]. Smith et al. (2007) performed the expression profiles of 4 transcription factor deletion strains (delta_OAF1, delta_PIP2, delta_ADR1 or delta_OAF3) in the presence of oleate and the expression profile of wild type strain in oleate versus low glucose diet. Our 41 yeast genes resulted as a subgroup of the pool of Smith, underlining the consistency of our list of genes in relation to the pathway of signal studied. In order to establish if our list is a useful gene signature to understand the activity of oleate response we selected the expression value of the 41 yeast MDEGs in each experiments of the Smith's dataset and we performed a cluster analysis. This analysis allows us to separate the datasets into two main groups. The first group contains the expression profiles of delta_OAF1, delta_PIP2, delta_ADR1 strains where the oleate-inducible transcription factors are deleted and therefore, the response to oleate diet is repressed. The second group is composed by oleate diet versus low glucose diet dataset, delta_OAF3 strain and our dataset used as a reference. The findings exactly mach to our expectations. As OAF3 is a repressor of oleate-induced transcription, delta_OAF3 strain has a behaviour similar to the activation of transcription induced by oleate. (Tree in Figure 5-B). The same procedure was applied to external expression dataset of M. musculus. The resultant tree of experiments, obtained clustering by similarity of expression the selected 164 mouse MDEGs, perfectly split the 4 experiments in 2 groups. In the first group we find experiment studying activation of PPARα signaling, in the second group experiment in which there is not PPARα. (Tree in Figure 5-A; Complete Matrix with expression values in Additional File 2).
Figure 5.
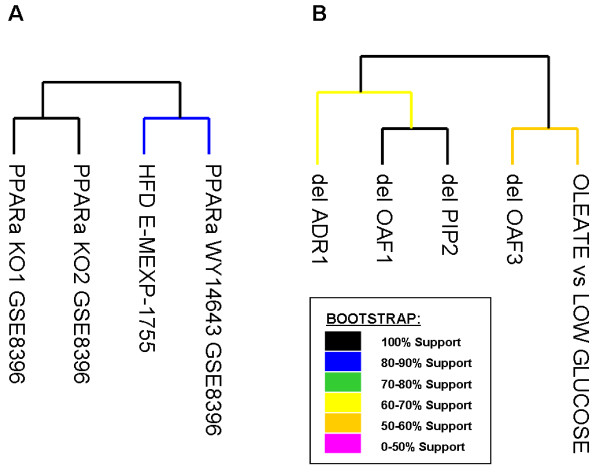
Bootstrap support trees of validation datasets selected for MDEGs. A) Yeast datasets B) Mouse datasets.
The correspondence of the datasets separation to our expectations qualifies the identified genes as putative markers of the biological response to high fat diet.
Transcription factor (TF) screening on MDEGs
Mining the literature we can find that Lemay and Hwang [13] have already performed a genome-wide screening of PPRE on human genome. Accordingly we compared the list of MDEG with the list of genes with PPRE provided by Lemay and Hwang, finding that 12 MDEGs have in silico predicted PPRE. We calculated the Fisher exact test p-value to study the overlap. As expected, because of the unbalanced numbers of the comparison, the p-value (p = 0.21) was not significant, although giving an indication of non randomness. However we are aware that the only presence of a PPRE does not necessarily result in a change in gene expression and vice versa many of the changes in gene expression we discover might be due to a not-direct interaction with PPARa or a combined action of more TFs.
With the aim of deeply investigate the presence of other TF binding sites, we accomplished a genome-wide screening of the region flanking the transcription starting site of all mouse MDEGs, searching binding site (TFBS) for all TFs contained in the Jaspar database [51]. The screening was performed using a tool called oPOSSUM [27]. Confirming and strengthening our previous results, the analysis shown that PPRE was the only TFBS over-represented in our pool of genes (see Additional File 2 for details). The target genes with predicted PPRE were 18 (Table 3). As expected the results of the two TF screening on MDEGs were partially different because of two different analysis pipelines [13]. Lemay and Hwang performed the screening using the whole human genome instead of the mouse one, they used a quite different sequence consensus for the PPAR binding site and different methods to assess a score to the findings. However the two analyses were similar for the purpose and, despite of the two different organisms used as reference, each method is developed taking into account species comparison. As consequence the human-mouse conserved elements have to be found and, in our opinion, no method is better than the other providing two complementary results.
Table 3.
Table of MDEG with predicted Peroxisome Proliferator Response Element.
| MDEG with oPOSSUM predicted PPRE in Mus musculus | MDEG with predicted PPRE by Lemay and Hwang in Homo sapiens | ||||||
|---|---|---|---|---|---|---|---|
| Gene Symbol | Entrez Gene ID | TFBS Position |
Homolo Gene ID |
Gene Symbol | Entrez Gene ID | TFBS Position | Reference |
| ACADVL | 37 | chr17:7060717-7060729 | [38] | ||||
| G0S2 | 50486 | chr1:206232580-206232592 | [66] | ||||
| ELOVL3 | 83401 | chr10:103974942-103974954 | [67] | ||||
| TXNIP | 10628 | chr1:142924546-142924558 | [55,68] | ||||
| SLC25A42 | 284439 | chr19:19033468-19033480 | - | ||||
| ACSL3 | 2181 | chr2:223550325-223550337 | - | ||||
| CREB3L3 | 84699 | chr19:4104406-4104418 | - | ||||
| Lpcat3 | 14792 | chr:6 124626752-124630751 | 14678 | LPCAT3 | 10162 | chr12:6996212-6996224 | [69] |
| Hmgcs2 | 15360 | chr:3 98363840-98367839 | 38066 | HMGCS2 | 3158 | chr1:120023667-120023679 | [70-73] |
| Fabp1 | 14080 | chr:6 71127471-71131470 | 1106 | FABP1 | 2168 | chr2:88270538-88270550 | [39-41] |
| Ccnd1 | 12443 | chr:7 144747221-144751220 | 1334 | CCND1 | 595 | chr11:69162605-69162617 | [74] |
| Cpt1b | 12895 | chr:15 89251630-89255629 | 22548 | CPT1B | 1375 | chr22:49311113-49311125 | [34-36,74] |
| Nrp1 | 18186 | chr:8 131243328-131247327 | - | ||||
| Nrp1 | 18186 | chr:8 131241743-131245742 | - | ||||
| Sorbs1 | 20411 | chr:19 40428334-40432333 | - | ||||
| Sorbs1 | 20411 | chr:19 40429124-40433123 | - | ||||
| Sorbs1 | 20411 | chr:19 40434177-40438176 | - | ||||
| Mmd | 67468 | chr:11 90063566-90067565 | - | ||||
| Igfbp2 | 16008 | chr:1 72755699-72759698 | - | ||||
| Stk16 | 20872 | chr:1 75092003-75096002 | - | ||||
| Rtn4 | 68585 | chr:11 29616570-29620569 | - | ||||
| Hadh | 15107 | chr:3 131259199-131263198 | - | ||||
| Cxcl14 | 57266 | chr:13 56304174-56308173 | - | ||||
| St5 | 76954 | chr:7 109391843-109395842 | - | ||||
| Suclg1 | 56451 | chr:6 73176158-73180157 | - | ||||
| Nnmt | 18113 | chr:9 48399563-48403562 | - | ||||
| Etfdh | 66841 | chr:3 79712646-79716645 | [55] | ||||
| Pnpla2 | 66853 | chr:7 141303884-141307883 | |||||
| Pnpla2 | 66853 | chr:7 141304526-141308525 | [55] | ||||
| Pnpla2 | 66853 | chr:7 141306941-141310940 | |||||
We believe that, despite the previous not significant p-value, the overlapping genes might represent an important set of response activators. This belief is strengthened by the fact that the overlap between the results of the two methods is composed by 5 genes (Lpcat3, Hmgcs2, Fabp1, Ccnd1, Cpt1b) all already known as target gene of PPARa (as shown in Table 3). This makes us confident that the remaining 20 genes, 13 of the oPOSSUM list and the 7 of Lemay and Hwang list, could be new candidate targets potentially interesting for further investigations.
Co-localization of MDEGs across the genome
The regulatory mechanism at the basis of PPARα induction of transcription is not well understood. The presence of PPRE in the promoter and the simultaneous expression of the gene with the activation of the nuclear receptor are the best criteria required to confirm the regulation of a gene by PPARα. However, often we do not have both of these evidences to identify a target genes. Some genes without PPRE show fatty acid responsive changes in transcription and they seem to be under control of fatty acid regulation. In literature we find evidences supporting the idea that genes regulated by the same transcription factors and/or that sharing biological functions are co-localized in the genome [28].
Given the validity of our MDEGs, resulting from all the previous findings, for a better comprehension of the molecular mechanism underneath the high fat response, we explored MDEG genome arrangements across a mouse genome.
To increase the power of analysis we added to MDEG list the known PPARα target found in literature and not in our list. The analysis of the arrangement of the genes reveals a non-random chromosomal location, in particular, MDEGs are often co-localized to compose small group consisting of 2 to 5 genes (see Table in Additional File 2). Some of these clusters are very interesting for two reasons: (i) they contained genes that have been already demonstrated to be regulated by PPARα and in some cases the PPRE is known, and (ii) they contained genes with similar function and strongly correlated to the known activity of PPARα.
In our opinion, this finding is a further confirmation that the mechanism of transcription regulation operated by PPARα involve epigenetic processes. Indeed in literature we can find molecular and in silico confirmation of this hypothesis. Lemay and Wang, calculating functional enrichment of genes showing PPRE, have found that one of the most over-represented category was chromatin remodelling. In 1999 Xu et al. demonstrated that the recruitment of transcriptional machinery by nuclear receptors can occur directly or in response to chromatin remodelling, elicited by the dismissing of HDAC (Histone Deacetylase Complex), by ligand and by the recruitment of HAT complex (Histone Acetylase) [52]. Unfortunately this was not demonstrated specifically for PPARα. However, in recent years substantial effort has been invested in studies of chromatin remodelling complexes associated with transcription factors. In particular, Li et al. have shown that SMARCD1 is the molecular link between SWI/SNF chromatin remodelling complex and PPARα transcription factor. The recruitment of SMARCD1 to PPRE, mediated by PGC-1α, leads to a switch in chromatin structure to an active state [53]. In yeast, the connection between diet and chromatin remodelling is well studied. The nutritional status and chromatin state are correlated to health state and replicative life span, by mechanism involving sirtuin activation that regulates mitochondrial biogenesis through changing of the acetylation state of the transcriptional coactivator PGC-1α [54]. The fact that PGC-1α plays important role in epigenetic transcription regulation in both yeast and mammals as a physical link between PPRE bounded by PPARα and chromatin remodelling complex, suggest possible presence of the evolutionary conserved epigenetic regulatory mechanisms.
At the light of these findings, in silico analysis suggest that transcription factor induction and chromatin state seem to be the principal factors mediating the response to excess dietary fat. This probably allows PPARα to bind a PPRE and to regulate more than one gene at the same time.
Conclusions
The proposed computational methods contribute towards the advances in integrative analyses of genomic data that still represent a major, and partially unresolved, computational issue. Through the selected strategy we were able to scan the expression data currently available and to suggest directions and new candidates to be investigated. This demonstrates the utility of the undertaken approach to exploit cross-species analysis and define gene signatures of the evolutionary conserved mechanisms as key elements to decipher the complexity of genome-wide data.
Authors' contributions
DC and EC designed the experiment, performed the analyses, developed the method and wrote the paper, CR developed the method supported the analyses and wrote the paper, EM performed the yeast microarray hybridizations, MR discussed the results designed the experiment and corrected the paper, BVO corrected the paper and discussed the results, MM corrected the paper and discussed the results. All authors have read and approved the final manuscript.
Supplementary Material
The MDEGs list.
The file contain the complete output of BINGO, the KEGG enrichment table, the list of MDEGs know as target of PPARα, complete output of oPOSSUM, the table of co-localized genes.
Contributor Information
Duccio Cavalieri, Email: duccio.cavalieri@unifi.it.
Enrica Calura, Email: enrica.calura@unifi.it.
Chiara Romualdi, Email: chiara.romualdi@unipd.it.
Emmanuela Marchi, Email: emanuela.marchi@unifi.it.
Marijana Radonjic, Email: marijana.radonjic@tno.nl.
Ben Van Ommen, Email: ben.vanommen@tno.nl.
Michael Müller, Email: michael.muller@wur.nl.
Acknowledgements
We are grateful to Lisa Rizzetto, Damariz Rivero e Luca Beltrame for helpful discussions. This work was supported by grants EU LSHB-CT-2004-512074, network of Exellence DC-THERA, Dendritic cells for novel immunotherapies http://www.dc-thera.org and by grants UE FOOD- CT-2004-506360 network of excellence NUGO the european nutrigenomics organization http://www.nugo.org.
References
- Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Muertter RN, Edgar R. NCBI GEO: archive for high-throughput functional genomic data. Nucleic acids research. 2009. pp. D885–890. [DOI] [PMC free article] [PubMed]
- Rocca-Serra P, Brazma A, Parkinson H, Sarkans U, Shojatalab M, Contrino S, Vilo J, Abeygunawardena N, Mukherjee G, Holloway E, Kapushesky M, Kemmeren P, Lara GG, Oezcimen A, Sansone SA. ArrayExpress: a public database of gene expression data at EBI. Comptes rendus biologies. 2003;326(10-11):1075–1078. doi: 10.1016/j.crvi.2003.09.026. [DOI] [PubMed] [Google Scholar]
- Rhodes DR, Chinnaiyan AM. Integrative analysis of the cancer transcriptome. Nature genetics. 2005. pp. S31–37. [DOI] [PubMed]
- Rhodes DR, Barrette TR, Rubin MA, Ghosh D, Chinnaiyan AM. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer research. 2002;62(15):4427–4433. [PubMed] [Google Scholar]
- Calura E, Cagnin S, Raffaello A, Laveder P, Lanfranchi G, Romualdi C. Meta-analysis of expression signatures of muscle atrophy: gene interaction networks in early and late stages. BMC genomics. 2008;9:630. doi: 10.1186/1471-2164-9-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romualdi C, De Pitta C, Tombolan L, Bortoluzzi S, Sartori F, Rosolen A, Lanfranchi G. Defining the gene expression signature of rhabdomyosarcoma by meta-analysis. BMC genomics. 2006;7:287. doi: 10.1186/1471-2164-7-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger M, Hooiveld GJ, Kersten S, Muller M. Exploration of PPAR functions by microarray technology--a paradigm for nutrigenomics. Biochimica et biophysica acta. 2007;1771(8):1046–1064. doi: 10.1016/j.bbalip.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Muller M, Kersten S. Nutrigenomics: goals and strategies. Nat Rev Genet. 2003;4(4):315–322. doi: 10.1038/nrg1047. [DOI] [PubMed] [Google Scholar]
- Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454(7203):470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nature reviews. 2006;6(1):44–55. doi: 10.1038/nri1748. [DOI] [PubMed] [Google Scholar]
- Paumelle R, Staels B. Cross-talk between statins and PPARalpha in cardiovascular diseases: clinical evidence and basic mechanisms. Trends in cardiovascular medicine. 2008;18(3):73–78. doi: 10.1016/j.tcm.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Chen YQ, Edwards IJ, Kridel SJ, Thornburg T, Berquin IM. Dietary fat-gene interactions in cancer. Cancer metastasis reviews. 2007;26(3-4):535–551. doi: 10.1007/s10555-007-9075-x. [DOI] [PubMed] [Google Scholar]
- Lemay DG, Hwang DH. Genome-wide identification of peroxisome proliferator response elements using integrated computational genomics. Journal of lipid research. 2006;47(7):1583–1587. doi: 10.1194/jlr.M500504-JLR200. [DOI] [PubMed] [Google Scholar]
- Phelps C, Gburcik V, Suslova E, Dudek P, Forafonov F, Bot N, MacLean M, Fagan RJ, Picard D. Fungi and animals may share a common ancestor to nuclear receptors. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(18):7077–7081. doi: 10.1073/pnas.0510080103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi E, Cavalieri D. Yeast as a model to investigate the mitochondrial role in adaptation to dietary fat and calorie surplus. Genes & nutrition. 2008;3(3-4):159–166. doi: 10.1007/s12263-008-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson H, Kapushesky M, Kolesnikov N, Rustici G, Shojatalab M, Abeygunawardena N, Berube H, Dylag M, Emam I, Farne A, Holloway E, Lukk M, Malone J, Mani R, Pilicheva E, Rayner TF, Rezwan F, Sharma A, Williams E, Bradley XZ, Adamusiak T, Brandizi M, Burdett T, Coulson R, Krestyaninova M, Kurnosov P, Maguire E, Neogi SG, Rocca-Serra P, Sansone SA. ArrayExpress update--from an archive of functional genomics experiments to the atlas of gene expression. Nucleic acids research. 2009. pp. D868–872. [DOI] [PMC free article] [PubMed]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics (Oxford, England) 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ, Meng F. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic acids research. 2005;33(20):e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romualdi C, Vitulo N, Del Favero M, Lanfranchi G. MIDAW: a web tool for statistical analysis of microarray data. Nucleic acids research. 2005. pp. W644–649. [DOI] [PMC free article] [PubMed]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrame L, Rizzetto L, Paola R, Rocca-Serra P, Gambineri L, Battaglia C, Cavalieri D. Using pathway signatures as means of identifying similarities among microarray experiments. PLoS ONE. 2009;4(1):e4128. doi: 10.1371/journal.pone.0004128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalieri D, Castagnini C, Toti S, Maciag K, Kelder T, Gambineri L, Angioli S, Dolara P. Eu. Gene Analyzer a tool for integrating gene expression data with pathway databases. Bioinformatics (Oxford, England) 2007;23(19):2631–2632. doi: 10.1093/bioinformatics/btm333. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open-source system for microarray data management and analysis. BioTechniques. 2003;34(2):374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Ostell J, Pruitt KD, Schuler GD, Shumway M, Sequeira E, Sherry ST, Sirotkin K, Souvorov A, Starchenko G, Tatusov RL. Database resources of the National Center for Biotechnology Information. Nucleic acids research. 2008. pp. D13–21. [DOI] [PMC free article] [PubMed]
- Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics (Oxford, England) 2005;21(16):3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Sui SJ, Mortimer JR, Arenillas DJ, Brumm J, Walsh CJ, Kennedy BP, Wasserman WW. oPOSSUM: identification of over-represented transcription factor binding sites in co-expressed genes. Nucleic acids research. 2005;33(10):3154–3164. doi: 10.1093/nar/gki624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JH, von Heydebreck A, Purmann A, Sperling S. Chromosomal clustering of a human transcriptome reveals regulatory background. BMC bioinformatics. 2005;6:230. doi: 10.1186/1471-2105-6-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe A, Danieli GA, Bortoluzzi S. REEF: searching REgionally Enriched Features in genomes. BMC bioinformatics. 2006;7:453. doi: 10.1186/1471-2105-7-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Fujita T, Usuda N, Cook W, Qi C, Peters JM, Gonzalez FJ, Yeldandi AV, Rao MS, Reddy JK. Peroxisomal and mitochondrial fatty acid beta-oxidation in mice nullizygous for both peroxisome proliferator-activated receptor alpha and peroxisomal fatty acyl-CoA oxidase. Genotype correlation with fatty liver phenotype. The Journal of biological chemistry. 1999;274(27):19228–19236. doi: 10.1074/jbc.274.27.19228. [DOI] [PubMed] [Google Scholar]
- Miller CW, Ntambi JM. Peroxisome proliferators induce mouse liver stearoyl-CoA desaturase 1 gene expression. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(18):9443–9448. doi: 10.1073/pnas.93.18.9443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillou H, Martin P, Jan S, D'Andrea S, Roulet A, Catheline D, Rioux V, Pineau T, Legrand P. Comparative effect of fenofibrate on hepatic desaturases in wild-type and peroxisome proliferator-activated receptor alpha-deficient mice. Lipids. 2002;37(10):981–989. doi: 10.1007/s11745-006-0990-3. [DOI] [PubMed] [Google Scholar]
- Brandt JM, Djouadi F, Kelly DP. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. The Journal of biological chemistry. 1998;273(37):23786–23792. doi: 10.1074/jbc.273.37.23786. [DOI] [PubMed] [Google Scholar]
- Mascaro C, Acosta E, Ortiz JA, Marrero PF, Hegardt FG, Haro D. Control of human muscle-type carnitine palmitoyltransferase I gene transcription by peroxisome proliferator-activated receptor. The Journal of biological chemistry. 1998;273(15):8560–8563. doi: 10.1074/jbc.273.15.8560. [DOI] [PubMed] [Google Scholar]
- Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. The Journal of clinical investigation. 1999;103(11):1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(13):7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrero MJ, Camarero N, Marrero PF, Haro D. Control of human carnitine palmitoyltransferase II gene transcription by peroxisome proliferator-activated receptor through a partially conserved peroxisome proliferator-responsive element. The Biochemical journal. 2003;369(Pt 3):721–729. doi: 10.1042/BJ20020851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha) The Journal of biological chemistry. 1998;273(10):5678–5684. doi: 10.1074/jbc.273.10.5678. [DOI] [PubMed] [Google Scholar]
- Poirier H, Niot I, Monnot MC, Braissant O, Meunier-Durmort C, Costet P, Pineau T, Wahli W, Willson TM, Besnard P. Differential involvement of peroxisome-proliferator-activated receptors alpha and delta in fibrate and fatty-acid-mediated inductions of the gene encoding liver fatty-acid-binding protein in the liver and the small intestine. The Biochemical journal. 2001;355(Pt 2):481–488. doi: 10.1042/0264-6021:3550481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfrum C, Borrmann CM, Borchers T, Spener F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha- and gamma-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(5):2323–2328. doi: 10.1073/pnas.051619898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Aoyama T, Burns AM, Gonzalez FJ. Bezafibrate is a dual ligand for PPARalpha and PPARbeta: studies using null mice. Biochimica et biophysica acta. 2003;1632(1-3):80–89. doi: 10.1016/s1388-1981(03)00065-9. [DOI] [PubMed] [Google Scholar]
- Kersten S, Mandard S, Escher P, Gonzalez FJ, Tafuri S, Desvergne B, Wahli W. The peroxisome proliferator-activated receptor alpha regulates amino acid metabolism. Faseb J. 2001;15(11):1971–1978. doi: 10.1096/fj.01-0147com. [DOI] [PubMed] [Google Scholar]
- Edgar AD, Tomkiewicz C, Costet P, Legendre C, Aggerbeck M, Bouguet J, Staels B, Guyomard C, Pineau T, Barouki R. Fenofibrate modifies transaminase gene expression via a peroxisome proliferator activated receptor alpha-dependent pathway. Toxicology letters. 1998;98(1-2):13–23. doi: 10.1016/S0378-4274(98)00042-3. [DOI] [PubMed] [Google Scholar]
- Patsouris D, Mandard S, Voshol PJ, Escher P, Tan NS, Havekes LM, Koenig W, Marz W, Tafuri S, Wahli W, Muller M, Kersten S. PPARalpha governs glycerol metabolism. The Journal of clinical investigation. 2004;114(1):94–103. doi: 10.1172/JCI20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpichev IV, Small GM. Global regulatory functions of Oaf1p and Pip2p (Oaf2p), transcription factors that regulate genes encoding peroxisomal proteins in Saccharomyces cerevisiae. Molecular and cellular biology. 1998;18(11):6560–6570. doi: 10.1128/mcb.18.11.6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter PJ. The genetics of fatty acid metabolism in Saccharomyces cerevisiae. Annual review of nutrition. 2001;21:97–119. doi: 10.1146/annurev.nutr.21.1.97. [DOI] [PubMed] [Google Scholar]
- Hirai T, Fukui Y, Motojima K. PPARalpha agonists positively and negatively regulate the expression of several nutrient/drug transporters in mouse small intestine. Biological & pharmaceutical bulletin. 2007;30(11):2185–2190. doi: 10.1248/bpb.30.2185. [DOI] [PubMed] [Google Scholar]
- Corton JC, Bocos C, Moreno ES, Merritt A, Marsman DS, Sausen PJ, Cattley RC, Gustafsson JA. Rat 17 beta-hydroxysteroid dehydrogenase type IV is a novel peroxisome proliferator-inducible gene. Molecular pharmacology. 1996;50(5):1157–1166. [PubMed] [Google Scholar]
- Fan LQ, Cattley RC, Corton JC. Tissue-specific induction of 17 beta-hydroxysteroid dehydrogenase type IV by peroxisome proliferator chemicals is dependent on the peroxisome proliferator-activated receptor alpha. The Journal of endocrinology. 1998;158(2):237–246. doi: 10.1677/joe.0.1580237. [DOI] [PubMed] [Google Scholar]
- Smith JJ, Ramsey SA, Marelli M, Marzolf B, Hwang D, Saleem RA, Rachubinski RA, Aitchison JD. Transcriptional responses to fatty acid are coordinated by combinatorial control. Molecular systems biology. 2007;3:115. doi: 10.1038/msb4100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic acids research. 2008. pp. D102–106. [DOI] [PMC free article] [PubMed]
- Xu L, Glass CK, Rosenfeld MG. Coactivator and corepressor complexes in nuclear receptor function. Current opinion in genetics & development. 1999;9(2):140–147. doi: 10.1016/S0959-437X(99)80021-5. [DOI] [PubMed] [Google Scholar]
- Li S, Liu C, Li N, Hao T, Han T, Hill DE, Vidal M, Lin JD. Genome-wide coactivation analysis of PGC-1alpha identifies BAF60a as a regulator of hepatic lipid metabolism. Cell metabolism. 2008;8(2):105–117. doi: 10.1016/j.cmet.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J, Maslov AY, Suh Y. Aging: a sirtuin shake-up? Cell. 2008;135(5):797–798. doi: 10.1016/j.cell.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Rakhshandehroo M, Sanderson LM, Matilainen M, Stienstra R, Carlberg C, de Groot PJ, Muller M, Kersten S. Comprehensive Analysis of PPARalpha-Dependent Regulation of Hepatic Lipid Metabolism by Expression Profiling. PPAR research. 2007;2007:26839. doi: 10.1155/2007/26839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MY, Zhao Y, Heng CK. Sequential responses to high-fat and high-calorie feeding in an obese mouse model. Obesity (Silver Spring, Md) 2008;16(5):972–978. doi: 10.1038/oby.2008.32. [DOI] [PubMed] [Google Scholar]
- Lo JC, Wang Y, Tumanov AV, Bamji M, Yao Z, Reardon CA, Getz GS, Fu YX. Lymphotoxin beta receptor-dependent control of lipid homeostasis. Science (New York, NY) 2007;316(5822):285–288. doi: 10.1126/science.1137221. [DOI] [PubMed] [Google Scholar]
- de Wilde J, Mohren R, Berg S van den, Boekschoten M, Dijk KW, de Groot P, Muller M, Mariman E, Smit E. Short-term high fat-feeding results in morphological and metabolic adaptations in the skeletal muscle of C57BL/6J mice. Physiological genomics. 2008;32(3):360–369. doi: 10.1152/physiolgenomics.00219.2007. [DOI] [PubMed] [Google Scholar]
- Tabibiazar R, Wagner RA, Spin JM, Ashley EA, Narasimhan B, Rubin EM, Efron B, Tsao PS, Tibshirani R, Quertermous T. Mouse strain-specific differences in vascular wall gene expression and their relationship to vascular disease. Arteriosclerosis, thrombosis, and vascular biology. 2005;25(2):302–308. doi: 10.1161/01.ATV.0000151372.86863.a5. [DOI] [PubMed] [Google Scholar]
- Li S, Zhang HY, Hu CC, Lawrence F, Gallagher KE, Surapaneni A, Estrem ST, Calley JN, Varga G, Dow ER, Chen Y. Assessment of diet-induced obese rats as an obesity model by comparative functional genomics. Obesity (Silver Spring, Md) 2008;16(4):811–818. doi: 10.1038/oby.2007.116. [DOI] [PubMed] [Google Scholar]
- Toye AA, Dumas ME, Blancher C, Rothwell AR, Fearnside JF, Wilder SP, Bihoreau MT, Cloarec O, Azzouzi I, Young S, Barton RH, Holmes E, McCarthy MI, Tatoud R, Nicholson JK, Scott J, Gauguier D. Subtle metabolic and liver gene transcriptional changes underlie diet-induced fatty liver susceptibility in insulin-resistant mice. Diabetologia. 2007;50(9):1867–1879. doi: 10.1007/s00125-007-0738-5. [DOI] [PubMed] [Google Scholar]
- Biddinger SB, Almind K, Miyazaki M, Kokkotou E, Ntambi JM, Kahn CR. Effects of diet and genetic background on sterol regulatory element-binding protein-1c, stearoyl-CoA desaturase 1, and the development of the metabolic syndrome. Diabetes. 2005;54(5):1314–1323. doi: 10.2337/diabetes.54.5.1314. [DOI] [PubMed] [Google Scholar]
- Raab RM, Bullen J, Kelleher J, Mantzoros C, Stephanopoulos G. Regulation of mouse hepatic genes in response to diet induced obesity, insulin resistance and fasting induced weight reduction. Nutrition & metabolism. 2005;2:15. doi: 10.1186/1743-7075-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson LM, de Groot PJ, Hooiveld GJ, Koppen A, Kalkhoven E, Muller M, Kersten S. Effect of synthetic dietary triglycerides: a novel research paradigm for nutrigenomics. PLoS ONE. 2008;3(2):e1681. doi: 10.1371/journal.pone.0001681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder SP, Kaisaki PJ, Argoud K, Salhan A, Ragoussis J, Bihoreau MT, Gauguier D. Comparative analysis of methods for gene transcription profiling data derived from different microarray technologies in rat and mouse models of diabetes. BMC genomics. 2009;10:63. doi: 10.1186/1471-2164-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandbergen F, Mandard S, Escher P, Tan NS, Patsouris D, Jatkoe T, Rojas-Caro S, Madore S, Wahli W, Tafuri S, Muller M, Kersten S. The G0/G1 switch gene 2 is a novel PPAR target gene. The Biochemical journal. 2005;392(Pt 2):313–324. doi: 10.1042/BJ20050636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson A, Jorgensen JA, Jacobsson A. Differential regulation of fatty acid elongation enzymes in brown adipocytes implies a unique role for Elovl3 during increased fatty acid oxidation. American journal of physiology. 2005;289(4):E517–526. doi: 10.1152/ajpendo.00045.2005. [DOI] [PubMed] [Google Scholar]
- Billiet L, Furman C, Cuaz-Perolin C, Paumelle R, Raymondjean M, Simmet T, Rouis M. Thioredoxin-1 and its natural inhibitor, vitamin D3 up-regulated protein 1, are differentially regulated by PPARalpha in human macrophages. Journal of molecular biology. 2008;384(3):564–576. doi: 10.1016/j.jmb.2008.09.061. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Chen YQ, Bonacci TM, Bredt DS, Li S, Bensch WR, Moller DE, Kowala M, Konrad RJ, Cao G. Identification and characterization of a major liver lysophosphatidylcholine acyltransferase. The Journal of biological chemistry. 2008;283(13):8258–8265. doi: 10.1074/jbc.M710422200. [DOI] [PubMed] [Google Scholar]
- Le May C, Pineau T, Bigot K, Kohl C, Girard J, Pegorier JP. Reduced hepatic fatty acid oxidation in fasting PPARalpha null mice is due to impaired mitochondrial hydroxymethylglutaryl-CoA synthase gene expression. FEBS letters. 2000;475(3):163–166. doi: 10.1016/S0014-5793(00)01648-3. [DOI] [PubMed] [Google Scholar]
- Kok T, Bloks VW, Wolters H, Havinga R, Jansen PL, Staels B, Kuipers F. Peroxisome proliferator-activated receptor alpha (PPARalpha)-mediated regulation of multidrug resistance 2 (Mdr2) expression and function in mice. The Biochemical journal. 2003;369(Pt 3):539–547. doi: 10.1042/BJ20020981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JC, Gil-Gomez G, Hegardt FG, Haro D. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by fatty acids. The Journal of biological chemistry. 1994;269(29):18767–18772. [PubMed] [Google Scholar]
- Hegardt FG. Transcriptional regulation of mitochondrial HMG-CoA synthase in the control of ketogenesis. Biochimie. 1998;80(10):803–806. doi: 10.1016/S0300-9084(00)88874-4. [DOI] [PubMed] [Google Scholar]
- Anderson SP, Yoon L, Richard EB, Dunn CS, Cattley RC, Corton JC. Delayed liver regeneration in peroxisome proliferator-activated receptor-alpha-null mice. Hepatology (Baltimore, Md) 2002;36(3):544–554. doi: 10.1053/jhep.2002.35276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The MDEGs list.
The file contain the complete output of BINGO, the KEGG enrichment table, the list of MDEGs know as target of PPARα, complete output of oPOSSUM, the table of co-localized genes.