

EGFR signaling pathways are major mitogenic pathways, while TGF-β signaling pathways are anti-proliferative in primary human keratinocytes. TGF-β induces protein tyrosine phosphatase-kappa (RPTP-κ), a negative regulator of EGFR function. TGF-β indirectly inhibits EGFR-dependent signaling and proliferation via up-regulation of RPTP-κ.
Abstract
Epidermal growth factor receptor (EGFR) signaling pathways promote human keratinocyte survival and proliferation. In contrast, transforming growth factor-beta (TGF-β) signaling pathways are strongly anti-proliferative. Receptor type protein tyrosine phosphatase-kappa (RPTP-κ) specifically dephosphorylates EGFR, thereby blocking EGFR-dependent signaling, and inhibiting proliferation. We report here that RPTP-κ mediates functional integration of EGFR and TGF-β signaling pathways in human keratinocytes. TGF-β up-regulates RPTP-κ mRNA and protein, in a dose and time dependent manner. Induction of RPTP-κ by TGF-β significantly decreases basal and EGF-stimulated EGFR tyrosine phosphorylation. shRNA-mediated reduction of TGF-β–induced RPTP-κ significantly attenuates the ability of TGF-β to inhibit proliferation. RPTP-κ induction is dependent on activation of transcription factors Smad3 and Smad4. Inhibition of TGF-β receptor kinase completely prevents induction of RPTP-κ. Chromatin immunoprecipitation assays reveal that TGF-β stimulates Smad3 and Smad4 binding to RPTP-κ gene promoter. Smad3/4 binding is localized to an 186-base pair region, which contains a consensus Smad3-binding element. These data describe a novel mechanism of cross-talk between EGFR and TGF-β pathways, in which RPTP-κ functions to integrate growth-promoting and growth-inhibiting signaling pathways.
INTRODUCTION
Epidermal growth factor receptor (EGFR), the prototypical mammalian receptor type protein tyrosine kinase (RTK), regulates fundamental cellular functions such as proliferation, differentiation, migration, and apoptosis (Carpenter, 1987; Schlessinger, 2000; Normanno et al., 2006). Ligand binding to EGFR stabilizes homodimerization, which promotes trans-tyrosine phosphorylation by constitutively active intrinsic tyrosine kinase. Specific phosphorylated tyrosines function as docking sites for a variety of signaling molecules that regulate membrane-proximal steps of signal transduction cascades that ultimately bring about cellular responses to EGFR ligands (Prenzel et al., 2001; Jorissen et al., 2003).
The central role of EGFR in diverse signal transduction pathways dictates that its tyrosine phosphorylation must be strictly regulated by counteracting protein tyrosine phosphatases (PTPs). PTPs are a family of enzymes that catalyze dephosphorylation of protein substrates and usually act to turn off the signals generated by RTKs (Alonso et al., 2004). Dynamic opposition between protein tyrosine kinase and protein tyrosine phosphatase activities is necessary to maintain cellular homeostasis (Tonks, 2006). We have recently reported that receptor type protein tyrosine phosphatase-kappa (RPTP-κ) dephosphorylates EGFR and thereby regulates EGFR tyrosine phosphorylation and subsequent downstream cellular responses, in primary human keratinocytes (Xu et al., 2005).
Transforming growth factor-beta (TGF-β) is a potent regulator of cell proliferation, differentiation, matrix production, migration, angiogenesis, immunity, and apoptosis (Massague, 1998; Derynck et al., 2001; Wang, 2001). The effects of TGF-β are mediated by transmembrane serine/threonine kinase receptors, which directly phosphorylate the Smad proteins, Smad2 and Smad3. Phosphorylated Smad2 and Smad3 separately associate with Smad4 (a CoSmad) and the complexes translocate into the cell nucleus to function as transcription factors (Heldin et al., 1997; Derynck et al., 1998).
Cellular responses to TGF-β are highly cell-type specific. In epithelial cells, TGF-β is a potent inhibitor of cell proliferation (Coffey et al., 1988; Moses et al., 1990). The presence of inactivating mutations in genes encoding TGF-β receptors, Smads, or other downstream effectors in human epithelial cancers, and mouse models of cancer development, demonstrate that the TGF-β signaling pathway is a potent tumor suppressor pathway (Massague et al., 2000; Derynck et al., 2001; Wakefield and Roberts, 2002).
It is well established that EGFR signaling pathways stimulate growth, whereas TGF-β signaling pathways inhibit proliferation, in normal human keratinocytes. However, mechanisms that functionally integrate these two opposing pathways are largely unknown. To address this issue, we have investigated the ability of TGF-β to regulate EGFR activation. We find that TGF-β diminishes EGFR tyrosine phosphorylation and that this inhibition is dependent on Smad3/4-mediated transcriptional induction of RPTP-κ expression. Our findings that TGF-β up-regulates RPTP-κ, and RPTP-κ functions as a negative regulator of EGFR, likely provide a heretofore unknown link between these pivotal pathways, which regulate cellular homeostasis in human skin.
MATERIALS AND METHODS
Adult human primary keratinocytes were purchased from Cascade Biologics (Portland, OR). Human recombinant TGF-β1 was purchased from R&D Systems (Minneapolis, MN). Puromycin and AG1478 were purchased from Calbiochem (Gibbstown, NJ). SB431542 was purchased from Sigma-Aldrich (Cat no. S4317, St. Louis, MO). PD169540 was provided by Dr. David Fry from Pfizer (Ann Arbor, MI). EGFR, Smad2, Smad3, and Smad4 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Phospho-EGFR (Tyr1068) antibody was purchased from Invitrogen (Carlsbad, CA). 293FT cells were obtained from American Type Culture Collection (Manassas, VA). Generation of RPTP-κ antibody has been described previously (Xu et al., 2005).
Cell Culture
Adult human primary keratinocytes were cultured in Epilife keratinocytes media supplemented with keratinocytes growth supplement (Cascade Biologics) at 37°C under 5% CO2. 293FT cells were culture in DMEM supplemented with 10% fetal bovine serum, nonessential amino acid, l-glutamine, sodium pyruvate, and geneticin at 37°C under 5% CO2.
Real-Time RT-PCR
Total cellular RNA was purified using Miniprep RNA isolation kit according to manufacturer's instruction (Qiagen, Chatsworth, CA). Reverse transcription of all total RNA was carried out using a Taqman Reverse Transcription kit (Applied Biosystems, Foster City, CA). Real-time PCR was performed using a Taqman GEx PCR Master Mix kit (Applied Biosystems) using a 7300 Sequence Detector (Applied Biosystems). Premade primer/probe combinations were purchased from Applied Biosystems. Target gene mRNA levels (number of molecules/10 ng total RNA) were normalized to endogenous 36B4 mRNA levels.
Cell Lysate and Western Blot
Cells were washed twice with ice-cold PBS, scraped from the dishes in WCE buffer (25 mM HEPES, pH 7.2, 75 mM NaCl, 2.5 mM MgCl2, 0.2 mM EDTA, 0.1% Triton X-100, 0.5 mM DTT, 20 mM β-glycerophophate) supplemented with 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitors cocktail (Roche Diagnostics, Indianapolis, IN). Equal amounts of whole cell extract were resolved on a 10% SDS-polyacrylamide gel, and transferred to Immobilon-P filter paper (Millipore, Bedford, MA). Immunoreactive proteins were visualized by enhanced chemifluorescence (ECF) according to manufacturer's protocol (GE Healthcare, Piscataway, NJ). Quantification was performed using a STORM Phosphorimager (Molecular Dynamics, Sunnyvale, CA).
Lentivirus shRNA-mediated Knockdown of Endogenous Proteins
MISSION TurboGFP nontargeting shRNA control vector (Product no. SHC002) and shRNA constructs targeting RPTP-κ (5′-CCGGGCCCAGACTAAGAACATCAATCTCGAGATTGATGTTCTTAGTCTGGGCTTTTT-3′ or 5′-CCGGCCGGCGAGTCAAGTTATCAAACTCGAGTTTGATAACTTGA-CTCGCCGGTTTTT-3′), Smad2 (5′-CCGGCGATTAGATGAGCTTGAGAAACTC-GAGTTTCTCAAGCTCATCTAATCGTTTTTG-3'), Smad3 (5′-CCGGGAGCCTGGT-CAAGAAACTCAACTCGAGTTGAGTTTCTTGACCAGGCTCTTTTT-3′, and Smad4 (5′-CCGGGAGCCTGGTCAAGAAACTCAACTCGAGTTGAGTTTCTTGACCAGGCTCTTTTT-3′) were purchased from Sigma-Aldrich. Lentivirus was produced in 293FT cells after transfection of shRNA construct and helper plasmids using Superfection method as described by the manufacturer (Qiagen). Two days after transfection, supernatant from 293FT cells was used to infect human primary keratinocytes.
Chromatin Immunoprecipitation Experiments
Approximately 6 × 106 human primary keratinocytes were treated with vehicle or with 2.5 ng/ml TGF-β1 for 3 h. Chromatin immunoprecipitation (ChIP) experiments were carried out as described (Orlando et al., 1997) using ChIP grade rabbit anti-Smad3 or Smad4 antibodies (Abcam, Cambridge, MA). After phenol-chloroform extraction and ethanol precipitation, the DNA recovered from the immunoprecipitated chromatin was analyzed by real-time PCR amplification using four sets of primers that covered the 1-kb promoter region immediately before the transcription start site of human RPTP-κ gene. The primer sequences were 5′-GAGTAAATCCAAATACTTGGC-3′ and 5′-GGGCTCGCCAACGACTAACTTTCC-3′ to cover the region 78-327; 5′-TTCAAT-CAGCAACCGCTCGGG-3′ and 5′-GGC-GGCGGTCGGCGACGAGCCT-3′ to cover the region 327-522; 5′-TCCGAGCAG-CGGCTGGCGGCGG-3′ and 5′-AATCAGTTCTCAATGAAACGAG-3′ to cover region 522-706; and 5′-CTCGTTTCATTGAGAACTG-ATT-3′ and 5′-CCTCGTTCTCGCGGTG-TTT3′ to cover region 706-994.
RESULTS
TGF-β Rapidly Induces RPTP-κ, Which Mediates Inhibition of EGFR Activation
We initially examined kinetics and dose dependence of induction of RPTP-κ by TGF-β and its functional consequences on EGFR activation. TGF-β induced RPTP-κ mRNA (Figure 1a) and protein (Figure 1b) levels, in a time-dependent manner. Induction of RPTP-κ by TGF-β was observed at 3 h, was maximal within 24 h, and remained maximally elevated for at least 48 h. Maximal induction of RPTP-κ mRNA (Figure 1c) and protein (Figure 1d) was observed at 1.3 ng/ml TGF-β1. Higher concentrations of TGF-β1 reduced RPTP-κ protein levels, but not mRNA levels, relative to maximal stimulation achieved by 1.3 μg/ml TGF-β1, suggesting posttranslational regulation of RPTP-κ protein expression.
Figure 1.

Time and dose-dependent induction of RPTP-κ by TGF-β in primary human keratinocytes. Keratinocytes were treated with vehicle (0) or TGF-β1 (2.5 ng/ml) for indicated times (a and b), or for 24 h with the indicated concentrations of TGF-β (c and d). (a and c) Total RNA was isolated and RPTP-κ and 36B4 (internal control for normalization) mRNA levels were determined by real-time RT-PCR analyses. n = 3, *p < 0.05. (b and d) Equal amounts of whole cell lysates were analyzed for RPTP-κ and β-actin (internal control for normalization) protein levels by Western blots, which were quantified by chemifluorescence, using a STORM PhosphorImager. Insets, representative Western blots. n = 3, *p < 0.05.
Treatment of keratinocytes with TGF-β suppressed both basal and EGF-stimulated EGFR tyrosine phosphorylation, measured by Western blot (Figure 2a). TGF-β had no effect on total EGFR expression, indicating that TGF-β specifically reduced EGFR tyrosine phosphorylation. To determine the role of RPTP-κ in TGF-β–dependent inhibition of EGF-induced EGFR tyrosine phosphorylation, we used shRNA to knockdown RPTP-κ. Knockdown of RPTP-κ significantly increased basal EGFR tyrosine phosphorylation (Xu and Fisher, 2005). Importantly, RPTP-κ knockdown also significantly attenuated the ability of TGF-β to reduce EGFR phosphorylation (Figure 2b). These data indicate that regulation of RPTP-κ expression by TGF-β mediates cross-talk between the EGFR and TGF-β pathways in human keratinocytes.
Figure 2.

TGF-β reduces EGFR tyrosine phosphorylation in primary human keratinocytes. (a) Keratinocytes were treated with vehicle (−) or TGF-β1 (+, 2.5 ng/ml) for 48 h, before treatment with vehicle (−) or EGF (+, 10 ng/ml) for 30 min. Equal amounts of whole cell lysates were analyzed for phosphorylated (tyrosine 1068, pY-EGFR) and total EGFR protein levels by Western blot, which were quantified by chemifluorescence, using a STORM PhosphorImager. Insets, representative Western blots. n = 3, *p < 0.05. (b) Keratinocytes were infected with nontargeting control (Ctrl shRNA) or RPTP-κ shRNA lentivirus, 24 h before treatment with vehicle (−) or TGF-β1 (+, 2.5 ng/ml), for 48 h. Cells were then treated with EGF (+, 10 ng/ml), for 30 min. Equal amounts of whole cell lysates were analyzed for RPTP-κ, β-actin, phosphorylated (tyrosine 1068, pY-EGFR) and total EGFR protein levels by Western blot, which were quantified by chemifluorescence, using a STORM PhosphorImager. Insets, representative Western blots. n = 3, *p < 0.05.
Induction of RPTP-κ Contributes to Suppression of Human Primary Keratinocyte Proliferation by TGF-β
As shown in Figure 3, inhibition of EGFR function by specific tyrosine kinase inhibitors AG1478 (Fry et al., 1994) or PD169540 (Fry et al., 1998) or addition of TGF-β substantially decreases proliferation of primary adult human keratinocytes. Therefore, we next investigated the role of RPTP-κ in TGF-β–mediated inhibition of human primary keratinocyte growth. Cells were infected with lentivirus carrying either a nontargeting control shRNA or a RPTP-κ–targeting shRNA construct (Figure 4), which also carried a puromycin resistance gene. Cultures were treated with puromycin for 2 d to ensure that the majority of cells were expressing the lentivirus constructs. Puromycin-resistant keratinocyte cultures were treated with vehicle or TGF-β1. TGF-β inhibited control cells proliferation 75%, whereas proliferation of RPTP-κ knockdown cells was reduced only 25% (Figure 4). These data indicate that induction of RPTP-κ mediates, in part, the ability of TGF-β to inhibit human keratinocyte proliferation.
Figure 3.
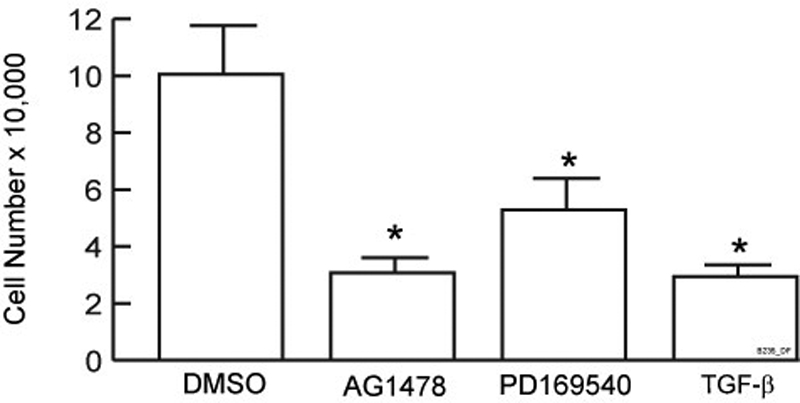
Inhibition of primary human keratinocytes proliferation by TGF-β and specific EGFR kinase inhibitors AG1478 and PD169540. Primary human keratinocytes were seeded at 2500 cell/cm2. The next day, cells were treated with vehicle (DMSO), TGF-β1 (2.5 ng/ml), AG1478 (1 μM), or PD169540 (50 nM). Cell numbers were counted 3 d later using a hemocytometer. Results are means of three independent experiments; error bars, SEM; *p < 0.05 versus DMSO control.
Figure 4.

Knockdown of RPTP-κ reduces TGF-β suppression of proliferation in primary human keratinocytes. Keratinocytes were infected with nontargeting control (Ctrl shRNA) or RPTP-κ shRNA lentivirus, coding puromycin resistance. Cells were cultured for 2 d in the presence of puromycin (2 μg/ml), before addition of vehicle (Ctrl) or TGF-β1 (2.5 ng/ml) for 2 d. Cell numbers were obtained by counting the cells using a hemocytometer. Data are means; error bars, SEM; n = 4; *p <0.05 versus Ctrl.
TGF-β Is a Major Regulator of RPTP-κ Expression in Adult Human Keratinocytes
Many cell types, including keratinocytes, constitutively express TGF-β, which then acts as an autocrine or paracrine regulator of cell function. TGF-β primarily initiates cellular responses by binding to its specific cell surface receptor complex. We used the specific TGF-β type I receptor kinase inhibitor SB431542 (Inman et al., 2002) to examine the dependence of RPTP-κ expression on TGF-β receptor signaling. SB431542 substantially inhibited both basal (i.e., in the absence of exogenous TGF-β) and TGF-β–induced RPTP-κ expression. Basal RPTP-κ mRNA (Figure 5a) and protein (Figure 5b) levels were reduced 75%. TGF-β–induced RPTP-κ mRNA (Figure 5a) and protein (Figure 5b) were reduced 95%. These data indicate that TGF-β is a major pathway that regulates RPTP-κ expression in human keratinocytes.
Figure 5.

Induction of RPTP-κ mRNA and protein by TGF-β requires TGF-β receptor function. Primary human keratinocytes were treated with vehicle (−) or SB431542 (+, 10 μM) for 2 h, before addition of TGF-β1 (+, 2.5 ng/ml) for 48 h. (a) Total RNA was isolated and RPTP-κ and 36B4 (internal control for normalization) mRNA levels were determined by real-time RT-PCR analyses. n = 4, *p < 0.05. (b) Equal amounts of whole cell lysates were analyzed for RPTP-κ and β-actin (internal control for normalization) protein levels by Western blots, which were quantified by chemifluorescence, using a STORM PhosphorImager. Data are means; error bars, SEM; n = 3; *p < 0.05. Inset, representative Western blots.
TGF-β Induces RPTP-κ Expression via Smad3 and Smad4, But Not Smad2
The activated TGF-β receptor complex phosphorylates and thereby activates transcription factors Smad2 and Smad3, which together with Smad4 translocate to the nucleus, to regulate target gene transcription. We next investigated the role of Smad proteins in the regulation of RPTP-κ gene expression, using lentivirus mediated shRNA knockdown of endogenous Smad proteins. As shown in Figure 6a, lentivirus shRNA reduced endogenous Smad4 protein by 80%. This reduction resulted in significant reduction of both basal and TGF-β–induced RPTP-κ gene expression. Similarly, knockdown of endogenous Smad3 protein (90%) by lentivirus mediated shRNA substantially reduced TGF-β–induced RPTP-κ gene expression (Figure 6b). In contrast, knockdown of endogenous Smad2 protein (70%) had no significant effect on either basal or TGF-β–induced RPTP-κ expression (Figure 6c). Similar results were obtained by knockdown of Smad2, Smad3, and Smad4 proteins by transfection of siRNA oligonucleotides (data not shown).
Figure 6.

Induction of RPTP-κ by TGF-β requires Smad4 and Smad3, but not Smad2. Primary human keratinocytes were infected with nontargeting control (Ctrl shRNA) shRNA lentivirus, or shRNA lentivirus targeting a) Smad4, b) Smad3, or c) Smad2. Two days after infection cells were treated with vehicle (−) or TGF-β1 (+, 2.5 ng/ml) for 24 h. Total RNA was isolated and RPTP-κ and 36B4 (internal control for normalization) mRNA levels were determined by real-time RT-PCR analyses. Equal amounts of whole cell lysates were analyzed for (a) Smad4, (b) Smad3, or (c) Smad2 and β-actin (internal control for normalization) protein levels by Western blots. Insets, representative Western blots. Data are means; error bars, SEM; n = 4; *p < 0.05.
Smad3 and Smad4 Bind to RPTP-κ Promoter
The data presented above suggest that Smad3/4 may directly regulate RPTP-κ gene transcription. To further substantiate this conclusion, we performed ChIP assay, to determine Smad3/4 binding to the RPTP-κ gene promoter. Human primary keratinocytes were treated with vehicle or TGF-β for 3 h, and cross-linked, sheered chromatin was immunoprecipitated with either Smad3 or Smad4 antibodies. Immunoprecipitated chromatin DNA was amplified using four pairs of primer sets covering the proximal 1-kb region of human RPTP-κ promoter. As shown in Figures 7, a and b, TGF-β specifically induced binding of both Smad3 and Smad4 to a region of the RPTP-κ promoter located between nucleotides 522 and 706. This region of the RPTP-κ promoter contains a GTCT sequence, which closely conforms to the consensus Smad3-binding sequence (Yagi et al., 2002). These data indicate that regulation of RPTP-κ gene expression by TGF-β-activated Smad3/4 (Figure 7) is mediated by direct binding of Smad3/4 to the RPTP-κ promoter.
Figure 7.

TGF-β promotes binding of Smad3 and Smad4 to the RPTP-κ promoter in primary human keratinocytes. Keratinocytes were treated with vehicle (open bar) or TGF-β1 (closed bar, 2.5 ng/ml) for 3 h. Cross-linked and sheared chromosomal DNA was immunoprecipitated with (a) Smad4 or (b) Smad3 antibody. Immunoprecipitation with IgG was used as control. Immunoprecipitated DNA was amplified by real-time PCR, using four pairs of primers that covered proximal 1-kb region of human RPTP-κ promoter. Data are means; error bars, SEM; n = 3; *p < 0.05.
DISCUSSION
EGFR is a major driving force for cell proliferation in human primary keratinocytes. The intrinsic tyrosine kinase activity of EGFR is kept in check by counteracting dephosphorylation, which is catalyzed by RPTP-κ (Xu et al., 2005). Inhibition of EGFR function either by blocking EGFR kinase activity or by overexpression of RPTP-κ suppresses cell growth (Mendelsohn and Baselga, 2003).
In human skin, keratinocyte proliferation is restricted to the basal (lowest) layer of cells. As keratinocytes mature, they move upward and cease proliferating. Although EGFR is expressed in both proliferating and nonproliferating keratinocytes, RPTP-κ expression is most abundant in nonproliferating cells (Xu et al., 2006a). In addition, RPTP-κ is up-regulated when cultured keratinocytes reach confluency and cease proliferating (Xu et al., 2005). Although the precise mechanism of up-regulation is unknown, we have observed a parallel increase in TGF-β expression as keratinocytes reach confluency (Xu and Fisher, unpublished data). Interestingly, the extracellular region of RPTP-κ is composed of three adhesion molecule-like domains, which can form homophilic intercellular interactions among adjacent cells (Sap et al., 1994; Zondag et al., 1995; McAndrew et al., 1998). These interactions may stabilize RPTP-κ at the cell surface (Gebbink et al., 1995; Fuchs et al., 1996), thereby helping to maintain EGFR in an inactive state. Thus, the level of RPTP-κ expression inversely correlates with keratinocyte proliferation, in human skin in vivo and cultured keratinocytes, consistent with the role of RPTP-κ as a physiological regulator of keratinocyte proliferation.
The TGF-β family of proteins has diverse biological effects, which are cell-type dependent (Rizzino, 1988). For cells of epithelial origin, such as keratinocytes, TGF-β acts as a potent growth inhibitor (Shipley et al., 1986). Induction of cell cycle inhibitors (Moustakas et al., 2002; Seoane et al., 2004; Gomis et al., 2006), suppression of c-myc expression (Coffey et al., 1988), and induction of phosphatase activity (Gruppuso et al., 1991) has been proposed as possible mechanisms for the antiproliferative effect of TGF-β in different types of epithelial cells. In the current study, we found that blocking induction of RPTP-κ allowed human keratinocytes to continue to proliferate in the presence of TGF-β. Thus, induction of RPTP-κ is a significant mechanism by which TGF-β acts to impede keratinocyte growth. However, it is not likely the sole mechanism, but rather RPTP-κ acts in concert with other TGF-β-regulated gene products to block keratinocyte proliferation.
Activated Smad3 and Smad4 bound to a 184-nucleotide base pair region, which contains a consensus Smad3-binding sequence, in the RPTP-κ promoter. Knockdown of Smad3 or Smad4 substantially reduced TGF-β induction of RPTP-κ. These data strongly suggest that TGF-β/Smad3/Smad4 regulates RPTP-κ gene transcription. Future studies employing promoter reporter assays and electrophoretic mobility shift assays will be of interest, to further investigate regulation of RPTP-κ transcription.
Similar to RPTP-κ, cell cycle inhibitors such as p15 (ink4b), p21, and p27 can be induced by TGF-β in human keratinocytes (Xu and Fisher, unpublished data) and are contributing factors in TGF-β–induced cell cycle arrest in epithelial cells (Hannon and Beach, 1994; Reynisdottir et al., 1995). Cell proliferation is regulated by both increased positive input from mitogenic pathways such as EGFR pathway and decreased input from negative regulators such as the above mentioned cell cycle inhibitors. TGF-β can decrease EGFR activity via induction of RPTP-κ and simultaneously induce cell cycle inhibitors in a coordinated manner. The end result is cell cycle arrest. It should be noted that in addition to RPTP-κ, there are several other protein tyrosine phosphatases (such as TCPTP and PTP 1B) and dual specific phosphatases (DUSPs such as Cdc25A), which can also regulate the EGFR signaling pathway (Tiganis et al., 1999; Fawaz et al., 2002; Wang et al., 2002; Caunt et al., 2008). However, the regulation of these phosphatases by TGF-β has not been investigated.
Cross-talk between signaling pathways is necessary to fine tune cellular responses to environmental needs. EGFR is a major proproliferative pathway, whereas TGF-β is an antiproliferative pathway in human keratinocytes. The data presented here establish a mechanistic link between these two counteracting pathways in their control of cell proliferation. TGF-β signaling pathway is frequently down-regulated in early tumorigenesis, which enables cancer cells to escape growth suppression caused by TGF-β (Bierie and Moses, 2006; Massague, 2008). Interestingly, we have found a correlation between down-regulation of TGF-β pathway components and reduced RPTP-κ expression, in several squamous cell carcinoma cell lines (Xu, and Fisher, unpublished data). Without sufficient RPTP-κ to maintain EGFR in an inactive state, EGFR can become constitutively active, as seen in many epithelial tumors. Overactivation of EGFR accelerates cancer cells growth and survival, two key properties that give cancer cells advantages over normal cells. Our data suggest that EGFR overactivation via reduced RPTP-κ expression may be a consequence of impairment of TGF-β signaling pathway in epithelial cancer. RPTP-κ mRNA has been shown to be induced by TGF-β treatment in immortalized, nontumorigenic HaCaT keratinocytes (Yang et al., 1996). Recently, it has been shown that restoration of RPTP-κ levels in MCF7 mammary cancer epithelial cells inhibits cell cycle progression (Wang et al., 2005).
RPTP-κ, like all members of the PTP family, contains an active site cysteine that is required for catalytic activity. This cysteine is readily susceptible to oxidation, which inhibits PTP activity (Salmeen et al., 2003; van Montfort et al., 2003). Exposure of human skin to UV irradiation elevates levels of reactive oxygen species, resulting in oxidative inhibition of RPTP-κ (Xu et al., 2006a). This inhibition leads to EGFR activation and consequent stimulation of downstream signal transduction pathways, which mediate multiple responses of keratinocytes to UV irradiation (Xu et al., 2006b). Interestingly, several studies have found that cancer cells generate elevated levels of reactive oxygen species. Increased reactive oxygen species production can result from oncogenic stimulation (Vafa et al., 2002; Hlavata et al., 2003), increased metabolic activity (Behar et al., 2001; Behrend et al., 2003), chronic inflammation (Ernst, 1999; Hussain et al., 2003), or mitochondrial dysfunction (Carew and Huang, 2002; Copeland et al., 2002; Carew et al., 2003; Kumimoto et al., 2004). These data raise the possibility that oxidative inhibition, in addition to reduced responsiveness to TGF-β, may suppress RPTP-κ activity in tumor cells.
The notion that RPTP-κ may function as a tumor suppressor is supported by the fact that expression of RPTP-κ is reduced or undetectable in more than 20% of melanoma cell lines and some melanoma biopsies (Novellino et al., 2003). In addition, the human RPTP-κ gene is localized within a region in chromosome 6 which is frequently deleted in hematological neoplasia, ovary carcinomas, melanomas, and many other types of solid tumors (Nakamura et al., 2003). Future studies in animal models are warranted to verify the tumor suppressor property of RPTP-κ.
ACKNOWLEDGMENTS
We thank Jin Zhou and Laura Gorges for technical assistance and Laura VanGoor and Diane Fiolek for graphic preparation and for administrative assistance. This work is supported by a Dermatology Foundation Research Grant to Y.X. and National Institutes of Health Grant 5R01 ES012920 to G.J.F.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-08-0710) on October 28, 2009.
REFERENCES
- Alonso A., Sasin J., Bottin N., Fried I., Fried I., Osterman A., Godzik A., Hunter T., Dixon J., Mustelin T. Protein tyrosine phosphatases in human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Behar M., Dalyot I., Engelberg D., Levitzki A. Enhanced ROS production in oncogenically transformed cells potentiates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase activation and sensitization to genotoxic stress. Mol. Cell. Biol. 2001;21:6913–6929. doi: 10.1128/MCB.21.20.6913-6926.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrend L., Henderson G., Zwacka R. Reactive oxygen species in oncogenic transformation. Biochem. Sco. Trans. 2003;31:1441–1444. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- Bierie B., Moses H. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Carew J., Huang P. Mitochondrial defeats in cancer. Mol. Cancer. 2002;1:1–9. doi: 10.1186/1476-4598-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carew J., Zhou Y., Albitar M., Carew J., Keating M., Huang P. Mitochondrial DNA mutations on primary leukemia cells after chemotherapy: clinical significance and therapeutic implications. Leukemia. 2003;17:1437–1447. doi: 10.1038/sj.leu.2403043. [DOI] [PubMed] [Google Scholar]
- Carpenter G. Receptors for epidermal growth factor and other polypeptide mitogens. Annu. Rev. Biochem. 1987;56:881–914. doi: 10.1146/annurev.bi.56.070187.004313. [DOI] [PubMed] [Google Scholar]
- Caunt C. J., Rivers C. A., Conway-Campbell B. L., Norman M. R., McArdle C. A. Epidermal growth factor receptor and protein kinase C signaling to ERK2, spatiotemporal regulation of ERK2 by dual specificity phosphatases. J. Biol. Chem. 2008;283:6241–6252. doi: 10.1074/jbc.M706624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey R., Bascom C., Sipes N., Graves-Deal R., Weisman B., Moses H. Selective inhibition of growth-related gene expression in murine keratinocytes by transforming growth factor beta. Mol. Cell. Biol. 1988;8:3088–3093. doi: 10.1128/mcb.8.8.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland W., Wachsman J., Johnson F., Penta J. Mitochondrial DNA alterations in cancer. Cancer Invest. 2002;20:557–569. doi: 10.1081/cnv-120002155. [DOI] [PubMed] [Google Scholar]
- Derynck R., Akhurst R., Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- Derynck R., Zhang Y., Feng X. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- Ernst P. Review article: the role of inflammation in the pathogenesis of gastric cancer. Aliment Pharmacol. Ther. 1999;13:13–18. doi: 10.1046/j.1365-2036.1999.00003.x. [DOI] [PubMed] [Google Scholar]
- Fawaz G., Veveer P., Squire A., Neel B., Bastiaens P. Imaging sites of receptor dephosphorylation by PTP1B on the surface of the endoplasmic reticulum. Science. 2002;295:1708–1711. doi: 10.1126/science.1067566. [DOI] [PubMed] [Google Scholar]
- Fry D., et al. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc. Natl. Acad. Sci. USA. 1998;95:12022–12027. doi: 10.1073/pnas.95.20.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry D., Kraker A., McMichael A., Ambroso L., Nelson J., Leopold W., Connors R., Bridges A. A specific inhibitor of the epidermal growth factor receptor tyrosine kinase. Science. 1994;265:1093–1095. doi: 10.1126/science.8066447. [DOI] [PubMed] [Google Scholar]
- Fuchs M., Müller T., Lerch M., Ullrich A. Association of human protein-tyrosine phosphatase κ with members of the armadillo family. J. Biol. Chem. 1996;271:16712–16719. doi: 10.1074/jbc.271.28.16712. [DOI] [PubMed] [Google Scholar]
- Gebbink M., Zondag G., Koningstein G., Feiken E., Wubbolts R., Moolenaar W. Cell surface expression of receptor protein tyrosine phosphatase RPTP-μ is regulated by cell-cell contact. J. Cell Biol. 1995;268:16101–16104. doi: 10.1083/jcb.131.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis R., Alarson C., Nadal C., Van Poznak C., Massague J. C/EBPbeta at the core of the TGFbeta cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell. 2006;10:203–214. doi: 10.1016/j.ccr.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Gruppuso P., Mikumo R., Brautigan D., Braun L. Growth arrest induced by transforming growth factor beta 1 is accompanied by protein phosphatase activation in human keratinocytes. J. Biol. Chem. 1991;266:3444–3448. [PubMed] [Google Scholar]
- Hannon G., Beach D. p15INK4B is potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- Heldin C., Miyazono K., ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Hlavata L., Aguilaniu H., Pichova A., Nystrom T. The oncogenic RAS2(val19) mutation locks respiration, independently of PKA, in a mode prone to generate ROS. EMBO J. 2003;22:3337–3345. doi: 10.1093/emboj/cdg314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S., Hofseth L., Harris C. Radical causes of cancer. Nat. Rev. Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- Inman G., Nicolas F., Callahan J., Harling J., Gaster L., Rieth A., Laping N., Hill C. SB-431542 is a potent and specific inhibitor or transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK 5, and ALK7. Mol. Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- Jorissen R., Walker F., Pouliot N., Gearrett T., Ward C., Burgess A. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp. Cell Res. 2003;284:31–53. doi: 10.1016/s0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- Kumimoto H., Yamane Y., Nishimoto Y., Fukami H., Shinoda M., Hatooka S., Ishizaki K. Frequent somatic mutations of mitochondrial DNA in esophageal squamous cell carcinoma. Int. J. Cancer. 2004;108:228–231. doi: 10.1002/ijc.11564. [DOI] [PubMed] [Google Scholar]
- Massague J. TGF-β signal transduction. Annu. Rev. Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Massague J. TGF-beta in cancer cells. Cell. 2008;134:215–230. [Google Scholar]
- Massague J., Blain S., Lo R. TGF-beta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- McAndrew P., Frostholm A., White R., Rotter A., Burghes A. Identification an characterization of RPTP rho, a novel RPTP mu/kappa-like receptor protein tyrosine phosphatase whose expression is restricted to the central nervous system. Brain Res. Mol. Brain Res. 1998;56:9–21. doi: 10.1016/s0169-328x(98)00014-x. [DOI] [PubMed] [Google Scholar]
- Mendelsohn J., Baselga J. Status of epidermal growth factor receptor receptor antagonists in the biology and treatment of cancer. J. Clin. Oncol. 2003;21:2787–2799. doi: 10.1200/JCO.2003.01.504. [DOI] [PubMed] [Google Scholar]
- Moses H., Yang E., Pientenpol J. TGF-beta stimulation and inhibition of cell proliferation: new mechanistic insights. Cell. 1990;63:245–247. doi: 10.1016/0092-8674(90)90155-8. [DOI] [PubMed] [Google Scholar]
- Moustakas A., Pardali K., Gaal A., Heldin C. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol. Lett. 2002;82:85–91. doi: 10.1016/s0165-2478(02)00023-8. [DOI] [PubMed] [Google Scholar]
- Nakamura M., Kishi M., Sakakim T., Hashimoto H., Nakase H., Sihimada K., Ishida E., Konishi N. Novel tumor suppressor loci on 6q22–23 in primary central nervous system lymphomas. Cancer Res. 2003;63:737–741. [PubMed] [Google Scholar]
- Normanno N., De Luca A., Bianco C., Strizzi L., Mancino M., Maiello M., Carotenuto A., De Fei G., Caponigro F., Salomon D. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- Novellino L., et al. Identification of a mutated receptor-like protein tyrosine phosphatase kappa as a novel, class II HLA-restricted melanoma antigen. J. Immunol. 2003;170:6363–6370. doi: 10.4049/jimmunol.170.12.6363. [DOI] [PubMed] [Google Scholar]
- Orlando V., Strutt H., Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997;11:205–214. doi: 10.1006/meth.1996.0407. [DOI] [PubMed] [Google Scholar]
- Prenzel N., Fischer O., Streit S., Hart S., Ullrich A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr. Cancer. 2001;8:11–31. doi: 10.1677/erc.0.0080011. [DOI] [PubMed] [Google Scholar]
- Reynisdottir I., Polyak K., Iavarone A., Massague J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Gene Dev. 1995;9:1831–1845. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- Rizzino A. Transforming growth factor-beta: Multiple effects on cell differentiation and extracellular matrices. Dev. Biol. 1988;130:411–422. doi: 10.1016/0012-1606(88)90337-5. [DOI] [PubMed] [Google Scholar]
- Salmeen A., Andersen J., Myers M., Meng T., Hinks J., Tonks N., Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- Sap J., Jiang J.-P., Friedlander D., Grument M., Schlessinger J. Receptor tyrosine phosphate R-PTP-κ mediates homophilic binding. Mol. Cell. Biol. 1994;14:1–9. doi: 10.1128/mcb.14.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Seoane J., Le H., Anderson S., Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and gliblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- Shipley G., Pittelkow M., Wille J., Jr, Scott R., Moses H. Reversible inhibition of normal human prokeratinocyte proliferation by type beta transforming growth factor-growth inhibitor in serum-free medium. Cancer Res. 1986;46:2068–2071. [PubMed] [Google Scholar]
- Tiganis T., Kemp B., Tonks N. The protein-tyrosine phosphatase TCPTP regulates epidermal growth factor receptor-mediated and phosphatidylinositol 3-kinase-dependent signaling. J. Biol. Chem. 1999;274:27768–27775. doi: 10.1074/jbc.274.39.27768. [DOI] [PubMed] [Google Scholar]
- Tonks N. Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- Vafa O., Wade M., Kern S., Beeche M., Pandita T., Hampton G., Wahl G. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol. Cell. 2002;9:1031–1144. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- van Montfort R., Congreve M., Tisi D., Carr R., Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- Wakefield L., Roberts A. TGF-beta signaling: positive and negative effects on tumorigenesis. Curr. Opin. Genet. Dev. 2002;12:22–29. doi: 10.1016/s0959-437x(01)00259-3. [DOI] [PubMed] [Google Scholar]
- Wang S., Wu F., Shin I., Qu S., Arteaga C. Transforming growth factor β (TGF-β)-Smad target gene protein tyrosine phosphatase receptor type kappa is required for TGF-β function. Mol. Cell. Biol. 2005;25:4703–4715. doi: 10.1128/MCB.25.11.4703-4715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Role of TGF-beta signaling in skin carcinogenesis. Microsc Res. Tech. 2001;52:420–429. doi: 10.1002/1097-0029(20010215)52:4<420::AID-JEMT1027>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Wang Z., Wang M., Lazo J., Carr B. Identification of epidermal growth factor receptor as a target of Cdc25A protein phosphatase. J. Biol. Chem. 2002;277:19470–19475. doi: 10.1074/jbc.M201097200. [DOI] [PubMed] [Google Scholar]
- Xu Y., Fisher G. Ultraviolet (UV) light irradiation induced signal transduction in skin photoaging. J. Dermatol. Sci. Suppl. 2005;1:S1–S8. [Google Scholar]
- Xu Y., Shao Y., Voorhees J., Fisher G. Oxidative inhibition of receptor-type protein-tyrosine phosphatase κ by ultraviolet irradiation activates epidermal growth factor receptor in human keratinocytes. J. Biol. Chem. 2006a;291:27389–27397. doi: 10.1074/jbc.M602355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Tan L., Grachtchouk V., Voorhees J., Fisher G. Receptor-type protein-tyrosine phosphatase-κ regulates epidermal growth factor receptor function. J. Biol. Chem. 2005;280:42694–42700. doi: 10.1074/jbc.M507722200. [DOI] [PubMed] [Google Scholar]
- Xu Y., Voorhees J., Fisher G. Epidermal growth factor receptor is a critical mediator of ultraviolet B irradiation-induced signal transduction in immortalized human keratinocyte HaCaT cells. Am. J. Pathol. 2006b;169:823–830. doi: 10.2353/ajpath.2006.050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi K., Furuhashi M., Aoki H., Goto D., Kuwani H., Sugamura K., Miyazono K., Kato M. c-myc is a downstream target of the Samd pathway. J. Biol. Chem. 2002;277:854–861. doi: 10.1074/jbc.M104170200. [DOI] [PubMed] [Google Scholar]
- Yang Y., Gil M., Byun S., Choi I., Pyun K., Ha H. Transforming growth factor-beta1 inhibits human keratinocyte proliferation by upregulation of a receptor-type tyrosine phosphatase R-PTP-kappa gene expression. Biochem. Biophys. Res. Comm. 1996;228:807–812. doi: 10.1006/bbrc.1996.1736. [DOI] [PubMed] [Google Scholar]
- Zondag G., Koningstein G., Jiang J.-P., Sap J., Moolenaar W., Gebbink M. Homophilic interactions mediated by receptor tyrosine phosphatases μ and κ. A critical role for the novel extracellular MAM domain. J. Biol. Chem. 1995;270:14247–14250. doi: 10.1074/jbc.270.24.14247. [DOI] [PubMed] [Google Scholar]