

Main Text
Next-generation sequencing is a straightforward tool for the identification of disease genes in extended genomic regions. Autozygosity mapping was performed on a five-generation inbred Italian family with three siblings affected with Clericuzio-type poikiloderma with neutropenia (PN [MIM %604173]), a rare autosomal-recessive genodermatosis characterised by poikiloderma, pachyonychia, and chronic neutropenia. The siblings were initially diagnosed as affected with Rothmund-Thomson syndrome (RTS [MIM #268400]), with which PN shows phenotypic overlap. Linkage analysis on all living subjects of the family identified a large 16q region inherited identically by descent (IBD) in all affected family members. Deep sequencing of this 3.4 Mb region previously enriched with array capture revealed a homozygous c.504-2 A>C mismatch in all affected siblings. The mutation destroys the invariant AG acceptor site of intron 4 of the evolutionarily conserved C16orf57 gene. Two distinct deleterious mutations (c.502A>G and c.666_676+1del12) identified in an unrelated PN patient confirmed that the C16orf57 gene is responsible for PN. The function of the predicted C16orf57 gene is unknown, but its product has been shown to be interconnected to RECQL4 protein via SMAD4 proteins. The unravelled clinical and genetic identity of PN allows patients to undergo genetic testing and follow-up.
PN is an autosomal-recessive hereditary poikiloderma, a clinically and genetically heterogeneous group of disorders including RTS. The disorder is characterized by skin manifestations, mainly a papular erythematous rash starting on the limbs and face during the first year of life and evolving into poikiloderma with a pronounced acral involvement, as well as pachyonychia, especially of the toenails. One of the most important extracutaneous symptoms is an increased susceptibility to infections, mainly affecting the respiratory system, primarily due to a chronic neutropenia and to neutrophil functional defects. Bone marrow abnormalities account for neutropenia and may evolve into myelodysplasia associated with the risk of leukemic transformation.1
PN shows phenotypic overlap with RTS, but a few specific phenotypic differences point toward a distinct genetic control. Mutations of RECQL4 (MIM ∗603780), the helicase gene mutated in two thirds of RTS patients, appear to be absent in PN patients.2,3,4
We genotyped a highly consanguineous Italian family consisting of 29 subjects across five generations and including three affected siblings (Figures 1A and 1B). The affected members were initially misdiagnosed as RTS patients as a result of the skin findings that appeared in all three affected siblings before the first year of life, starting from the face and the extensor surface of the arms and then evolving into classical poikiloderma. All siblings also displayed pachyonychia, plantar keratoderma, and dysmorphisms, especially those related to the midfacial hypoplasia (Figure 1A). Severe neutropenia due to myelodysplastic hemopoiesis led to recurrent pulmonary infections, otitis, and sinusitis in two siblings at early infancy. All patients showed growth retardation, mild splenomegaly, and increased level of creatin kinase and lactate dehydrogenase (LDH). Detailed family description, clinical findings, neutrophil count, and testing and evolution of the disease are reported by D.C. (unpublished data). Concomitant acral poikiloderma, pachyonychia, and chronic neutropenia, fairly unusual in RTS patients, were more consistent with the PN diagnosis, which was further supported by the absence of RECQL4 mutations (data not shown). All sampled family members provided written informed consent to participate in the study.
Figure 1.
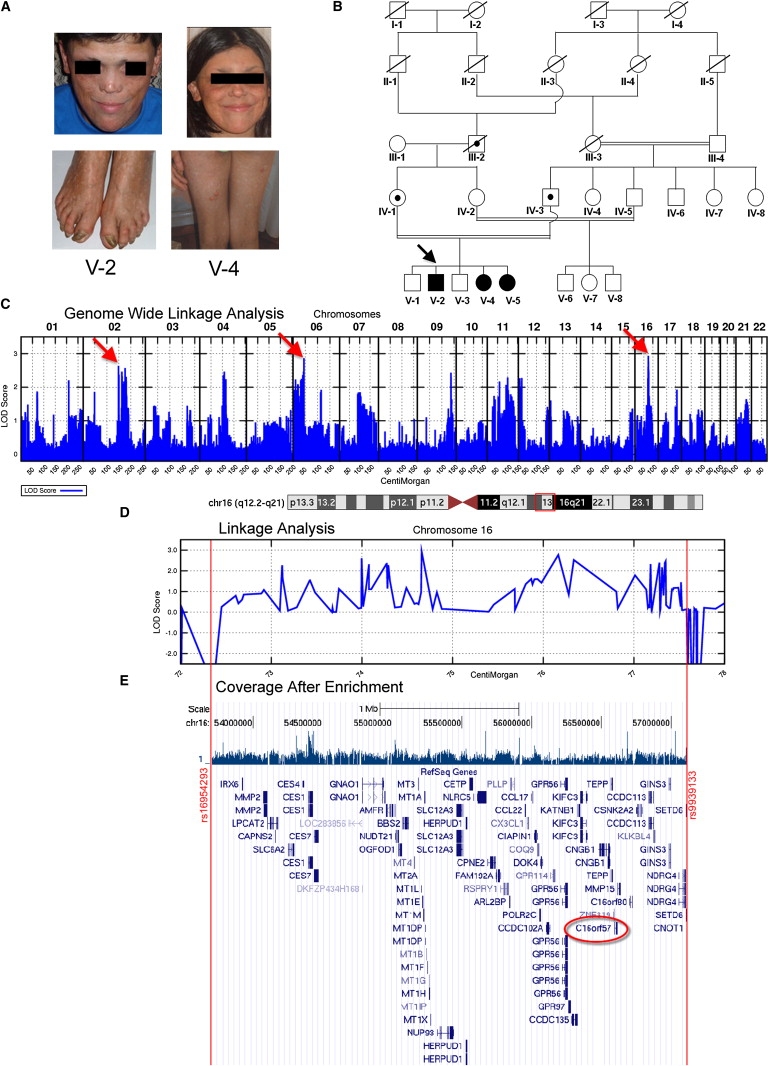
From Clinical Findings to the Candidate Gene
(A) Affected proband (V-2) and sibling (V-4) at ages 22 and 14, respectively, showing facial poikiloderma extending to buttocks and legs and pachyonychia of the toes.
(B) Five-generation pedigree: PN patients are indicated by shaded symbols and obligate carriers by dotted symbols. The inbreeding coefficient of the fifth-generation siblings is 3/64.
(C) Genome-wide parametric linkage analysis revealing LOD score > 2.5 on chromosomes 2, 6, and 16 (arrows). See also Table S1.
(D) LOD score diagram magnification of the 16q region (boxed in the upper ideogram): The region spans from SNP A_1803188 (rs16954293 at 16q12.2) to SNP A_1923765 (rs9939133 at 16q21).
(E) Postcapture array coverage of 16q12.2-q21 from V-2 (calculated enrichment value 241) and UCSC Genome Browser view of all genes mapping to this interval. C16orf57 is circled in red.
All living subjects (n = 18) were genotyped by a genome-wide Affymetrix Genechip Human Mapping 262K NspI SNP Array, and a two-point linkage analysis was performed with SuperLink (v.1.6), assuming an autosomal-recessive trait with 100% penetrance.5,6 SNPs were examined for informative genomic regions that were homozygous in the affected patients but not in any of their healthy siblings and were checked for quality control after removal of ambiguous genotypes and data with a call rate under a 95% threshold.
Linkage analysis identified three regions with a LOD score > 2.5 (Figure 1C and Table S1, available online), among which the 16q12.2-q21 region was selected as the largest genomic interval of which consecutive LOD score values never fall below −2 (Figure 1D). Indeed, the expected length of the IBD-inherited region around the disease locus is a function of the inbreeding coefficient of the proband. The inbreeding coefficient of the last generation is 3/64, predicting a ∼20 cM IBD region.7
The candidate16q region, spanning 3.4 Mb from SNP-A_1803188 (rs16954293) to SNP-A_1923765 (rs9939133), encompasses 276 consecutive SNPs, all consistent with the predicted inheritance pattern, and contains more than 80 known and predicted genes (Figure 1E). Such a list of genes is not manageable in the context of a classical candidate-gene approach, thus prompting us to proceed with array capture-mediated next-generation sequencing (NGS), a strategy enabling an unbiased search for disease-associated mutations in large genomic intervals.8
The adopted stepwise procedure is detailed in Figure S1. In brief, a genomic shotgun library from the 3.4 Mb 16q region of the siblings was prepared with paired-ends adapters in accordance with Illumina guidelines. After quality control, the library was captured by ImaGenes GmbH on a custom repeats-masked 244K solid array (Agilent).
The target region, dropped from 3.4 Mb to 1.7 Mb with this procedure, was eventually processed for NGS (Solexa Technology). ELAND (Illumina GAPipeline 1.0) mapping carried out against the full Homo sapiens genome yielded the bed files that were visualized in the UCSC Genome Browser, build March 2006 (Figure 1E). Mapping with the Maq program (v.0.7.1)9 was carried out against the selected 16q region. The best enrichment value (calculated according to the formula in Figure S1) was 241 for patient V-2.
The reads were then aligned to the targeted reference sequence, highlighting a total of 1488 mismatches: 450 were heterozygous mismatches that are unreported in the UCSC database and are accounted for by their location within DNA blocks with high sequence homology; i.e., low copy repeats or duplicons in which chromosome 16 is enriched (UCSC Genome Browser). As regards the homozygous mismatches, 494 have already been reported and 527 lie within intergenic regions; therefore, we focused over the remaining 17 unreported regions located within or very close to genes. We ranked them according to location and evolutionary conservation. As shown in Table S2, the A>C SNP position (56.608.737) appeared to be a strong candidate because it affects the acceptor splice site of intron 4 of the highly conserved C16orf57 gene (NM_024598.2) mapping to 16q13 (Figure 1E and Figure 2A); namely, the c.504-2 A>C mutation destroys the invariant AG dinucleotide splice acceptor (Figure 2B).
Figure 2.
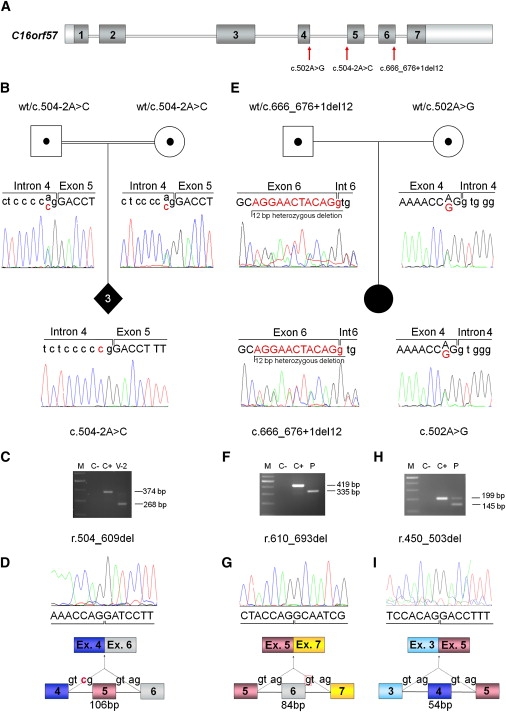
C16orf57 Mutational Analysis
(A) Schematic genomic structure of C16orf57 (16q13) spanning 20 Kb and 7 exons (dark boxes), encoding a 265 aa protein. Red arrows point to the identified mutations (rimers and conditions reported in Table S3).
(B and E) Pedigrees and sequencing of C16orf57 genomic mutations: (B) shows the carrier status of both parents and the homozygous splicing mutation c.504-2A>C of the three affected siblings, and (E) the parental origin and the two mutations c.666_676+1del12 and c.502A>G of the compound-heterozygous sporadic patient. Base changes are indicated with red characters.
(C, F, and H) Agarose gel electrophoresis, showing products of different RT-PCR (primers and conditions reported in Table S4), on lymphoblastoid cell line RNA. Lengths of normal and aberrant products are indicated. The following abbreviations are used: V-2, homozygous patient; P, compound-heterozygous patient; C+, normal control; C−, negative control; M, GeneRuler DNA Ladder Mix (Fermentas).
(D, G, and I) Sequence data of the two PN patients showing aberrant transcripts due to out-of-frame skipping of exon 5, in-frame skipping of exon 6 and exon 4, respectively, and related schematic diagrams of the mutation-associated misspliced cDNAs.
Direct capillary sequencing confirmed that the mutation segregates as expected across the last three generations, and it also confirmed the carrier (IV-6, IV-7, V-1, V-3) and noncarrier (III-4, IV-2, IV-4, IV-5, IV-8) status of all the living individuals within the pedigree. Subsequent cDNA analysis on patient V-2 showed an aberrant transcript 106 nucleotides shorter as a result of exon 5 skipping (Figures 2C and 2D). The predicted protein lacks 35 exon 5-encoded aminoacids and, because of frameshift, differs from the original protein sequence in the following 61 residues (p.Thr169IleFsX61) (Figure S2B).
We tested five atypical RTS patients and validated the association between C16orf57 and PN in the only patient reported to have the PN clinical hallmark of neutropenia. Indeed this nonrelated Italian female patient, who was diagnosed with RTS and myelodysplasia10 and tested negative for RECQL4 mutations (data not shown), was found to be a compound heterozygote for C16orf57 mutations. She carries a paternally inherited c.666_676+1del12 mutation in exon 6 and a maternally inherited c.502A>G missense mutation in exon 4, (Figure 2E). The c.502A>G mutation is absent in 175 matched controls and affects a highly conserved Arginine residue mapping within the conserved HVSL domain of C16orf57. The comparative aminoacid sequence analysis in HomoloGene and ClustalW showed complete conservation at position p.R168 in several eukaryotic species (Figure S3). cDNA analysis of the compound heterozygous patient resulted in the identification of two aberrant transcripts (Figures 2F–2I) with the in-frame skipping of exons 6 (paternal allele) and 4 (maternal allele). Mutated C16orf57 proteins lacking 28 (p.D204_Q231del) and 18 (p.F151_R168del) aminoacids are predicted (Figures S2C and S2D).
Little is known about C16orf57 or the functions of its encoded protein, but two independent studies revealed direct interactions between the C16orf57 and SMAD4 proteins,11,12 which are interconnected to RECQL4 through HADAC1, TP53, and/or RAD51 (Figure S4). The phenotypic overlap between RTS and PN can be partially accounted for by SMAD4-mediated signaling of C16orf57 to RECQL4. The fact that C16orf57 is significantly expressed in blood (myeloid lineage) might explain sensitivity to C16orf57 mutations leading to neutropenia and myelodysplastic features, which are distinctive signs of PN patients (1–4 and D.C., unpublished data).
The identification of a gene responsible for PN allows one to test for the C16orf57 mutation in all RECQL4-negative RTS patients fitting the PN clinical presentation, who are probably misdiagnosed, in order to provide adequate onco-hematological surveillance.
The presumptive role of C16orf57 in myeloid cell maturation and function paves the way for investigation of the contribution of this gene to both myelodysplasia and congenital neutropenia syndromes. In addition, the high degree of evolutionary conservation of C16orf57 increases the chance that representative animal models can be developed while such a line of research is pursued.
Acknowledgments
We thank the patients and their relatives for intensive cooperation in the study; A. Renieri and I. Meloni (University of Siena) for providing the lymphoblastoid cell line from the sporadic PN patient; Galliera Genetic Bank for establishing lymphoblastoid cell lines from the affected siblings of the PN family (Telethon project GTB07001); and L. Farinelli from Fasteris SA (CH), who technically and scientifically supported the project steps involving array capture and next-generation sequencing. Bioinformatics support for SNP array data was provided by Roland P. Kuiper (Nijmegen University Centre), and skillful modeling prediction of signalling pathways was performed by Christian Gilissen (Nijmegen University Center). As regards human subjects, we followed the guidelines of the ethical committee of the University of Milan (http://www.unimi.it/cataloghi/comitato_etico/CE_Rec_4_2006_HBMs.pdf). This work was supported by Associazione Italiana per la Ricerca sul Cancro (grant 2008-2009/4217 to L.L.), CARIPLO N.O.B.E.L. (project 2007-2009 to L.L.), and Nando Peretti Foundation (grant 2007-2009/14 to L.V.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BLAST: Basic Local Alignment Search Tool, http://www.ncbi.nlm.nih.gov/blast/Blast.cgi
ClustalW software, http://www.hongyu.org/software/clustal.html
ConSeq Server, http://conseq.bioinfo.tau.ac.il
ESE Finder, http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home
Genome-wide Viewer, https://bioinformatics.cancerresearchuk.org/∼cazier01/GWA_View.html
HomoloGene, http://www.ncbi.nlm.nih.gov/homologene
NetGene2 server, http://www.cbs.dtu.dk/services/NetGene2
Illumina, http://illumina.com
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Pathway Studio, http://www.ariadnegenomics.com/products/pathway-studio
PolyPhen, http://genetics.bwh.harvard.edu/pph
PSIPRED: Protein Structure Prediction Server, http://bioinf.cs.ucl.ac.uk/psipred
SIFT, http://sift.jcvi.org
STRING, http://string.embl.de
UCSC Genome Browser, http://genome.ucsc.edu; http://genome.ucsc.edu/cgi-bin/hgTracks
References
- 1.Clericuzio C., Hoyme H.E., Asse J.M. Immune deficient poikiloderma: A new genodermatosis. Am. J. Hum. Genet. 1991;49(Suppl):A661. [Google Scholar]
- 2.Wang L.L., Gannavarapu A., Clericuzio C.L., Erickson R.P., Irvine A.D., Plon S.E. Absence of RECQL4 mutations in poikiloderma with neutropenia in Navajo and non-Navajo patients. J. Med. Genet. 2003;118A:299–301. doi: 10.1002/ajmg.a.10057. [DOI] [PubMed] [Google Scholar]
- 3.Van Hove J.L., Jaeken J., Proesmans M., Boeck K.D., Minner K., Matthijs G., Verbeken E., Demunter A., Boogaerts M. Clericuzio type poikiloderma with neutropenia is distinct from Rothmund-Thomson syndrome. Am. J. Med. Genet. A. 2005;132A:152–158. doi: 10.1002/ajmg.a.30430. [DOI] [PubMed] [Google Scholar]
- 4.Mostefai R., Morice-Picard F., Boralevi F., Sautarel M., Lacombe D., Stasia M.J., McGrath J., Taïeb A. Poikiloderma with neutropenia, Clericuzio type, in a family from Morocco. Am. J. Med. Genet. A. 2008;146A:2762–2769. doi: 10.1002/ajmg.a.32524. [DOI] [PubMed] [Google Scholar]
- 5.Fishelson M., Geiger D. Exact genetic linkage computations for general pedigrees. Bioinformatics. 2002;18(Suppl 1):S189–S198. doi: 10.1093/bioinformatics/18.suppl_1.s189. [DOI] [PubMed] [Google Scholar]
- 6.Silberstein M., Tzemach A., Dovgolevsky N., Fishelson M., Schuster A., Geiger D. Online system for faster multipoint linkage analysis via parallel execution on thousands of personal computers. Am. J. Hum. Genet. 2006;78:922–935. doi: 10.1086/504158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Génin E., Todorov A.A., Clerget-Darpoux F. Optimization of genome search strategies for homozygosity mapping: influence of marker spacing on power and threshold criteria for identification of candidate regions. Ann. Hum. Genet. 1998;62:419–429. doi: 10.1046/j.1469-1809.1998.6250419.x. [DOI] [PubMed] [Google Scholar]
- 8.Brkanac Z., Spencer D., Shendure J., Robertson P.D., Matsushita M., Vu T., Bird T.D., Olson M.V., Raskind W.H. IFRD1 is a candidate gene for SMNA on chromosome 7q22-q23. Am. J. Hum. Genet. 2009;84:692–697. doi: 10.1016/j.ajhg.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pianigiani E., De Aloe G., Andreassi A., Rubegni P., Fimiani M. Rothmund-Thomson syndrome (Thomson-type) and myelodysplasia. Pediatr. Dermatol. 2001;18:422–425. doi: 10.1046/j.1525-1470.2001.01971.x. [DOI] [PubMed] [Google Scholar]
- 11.Rual J.F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G.F., Gibbons F.D., Dreze M., Ayivi-Guedehoussou N. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 12.Colland F., Jacq X., Trouplin V., Mougin C., Groizeleau C., Hamburger A., Meil A., Wojcik J., Legrain P., Gauthier J.M. Functional proteomics mapping of a human signaling pathway. Genome Res. 2004;14:1324–1332. doi: 10.1101/gr.2334104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.