

Abstract
Patients with Joubert syndrome 2 (JBTS2) suffer from a neurological disease manifested by psychomotor retardation, hypotonia, ataxia, nystagmus, and oculomotor apraxia and variably associated with dysmorphism, as well as retinal and renal involvement. Brain MRI results show cerebellar vermis hypoplasia and additional anomalies of the fourth ventricle, corpus callosum, and occipital cortex. The disease has previously been mapped to the centromeric region of chromosome 11. Using homozygosity mapping in 13 patients from eight Ashkenazi Jewish families, we identified a homozygous mutation, R12L, in the TMEM216 gene, in all affected individuals. Thirty individuals heterozygous for the mutation were detected among 2766 anonymous Ashkenazi Jews, indicating a carrier rate of 1:92. Given the small size of the TMEM216 gene relative to other JBTS genes, its sequence analysis is warranted in all JBTS patients, especially those who suffer from associated anomalies.
Main Text
Joubert syndrome (JBTS [MIM 213300]) is characterized by a specific midhindbrain malformation, hypotonia, cerebellar ataxia, and developmental delay. Oculomotor apraxia and abnormalities in breathing patterns are frequent findings. Associated anomalies include progressive retinal degeneration,1 renal anomalies (nephronophthisis, NPHP [MIM 256100]),2 both retinal and renal involvement (cerebello-oculo-renal syndromes, CORS [MIM 608091]),3 ocular colobomas and liver abnormalities (COACH [MIM 216360]),4 and both orofacial and digital signs (OFDVI [MIM 277170]).5 So far, Joubert syndrome and related disorders (JSRDs) have been mapped to ten loci, and causative genes have been identified for nine of them.6–10 Most encoded proteins were shown to localize to the cilia or to the centrosome, indicating that ciliary dysfunction is a key factor in the molecular pathogenesis of JSRDs.
We now report on a mutated gene in the tenth locus, 11p12-q13.3, in JSRD patients of Ashkenazi Jewish origin. The subjects were 13 patients from eight Ashkenazi Jewish families. Three of the families belonged to the same clan. Pregnancy was uneventful in all cases, and all the infants in this series were born at term, with normal Apgar scores and appropriate weights for gestational age. In two cases, there was suspicion of posterior fossa abnormalities on the basis of prenatal ultrasound. The initial presentation, evident from birth, consisted of hypotonia, rotatory nystagmus, esotropia, and feeding difficulties due to oral motor dysfunction with associated impaired swallowing and mastication. The physical examination was notable for dolichocephaly and frontal bossing in six patients, metopic craniosynostosis in one patient, and macrocephaly (birth head circumference 41 cm) due to noncommunicating hydrocephalus in one patient. Three patients had postaxial polydactyly in the lower limbs, and two patients had it in their upper limbs as well.
Early psychomotor development was invariably delayed, and the cognitive functioning of all the patients was eventually in the mildly to severely retarded range. Over the first two years of life, the nystagmus improved in all the patients, and visual function, funduscopic examination, and visual evoked potential appeared normal thereafter. Oral motor difficulties were invariably present from birth, resulting in swallowing and chewing difficulties and, in most patients, failure to thrive. Serum transaminases were normal in all the patients. In eight patients, normal renal ultrasound was reported. However, two patients suffered from end-stage renal insufficiency since midadolescence (the clinical data are summarized in Table 1). In brain MRIs, the molar tooth sign (deep interpeduncular fossa; thick, elongated superior cerebellar peduncles and cerebellar vermis hypoplasia) was observed. Associated findings, a complete absence of the vermis with wide space between the cerebellar hemisphere or a dysplastic split vermis without an opening or separation of the cerebellar hemispheres were present in all of the patients (Figure 1). Over the years, the patients underwent extensive metabolic and genetic investigations, including array comparative genomic hybridization, all of which yielded normal results.
Table 1.
Summary of Clinical Data
| Patient | Sex | Current Age | PMR | Abnormal Eye Movements | Hypotonia | Polydactily | Abnormal Renal Function | Other Clinical Features |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 9 yrs | mild | rotatory nystagmus, esotropia | + | − | − | dolichocephaly, frontal bossing |
| 2 | F | 3 yrs | mod | rotatory nystagmus, esotropia | + | − | − | dolichocephaly, frontal bossing, oromotor difficulties |
| 3 | F | 2 yrs | mod | rotatory nystagmus | + | − | − | dolichocephaly, frontal bossing, oromotor difficulties |
| 4 | M | 2 yrs | mod | rotatory nystagmus | + | − | − | oromotor difficulties |
| 5 | M | 1 yr | dev.delay | rotatory nystagmus, esotropia | + | both feet | − | oromotor difficulties, metopic craniosynostosis |
| 6 | F | 5 yrs | mod | rotatory nystagmus | + | − | − | oromotor difficulties, dolichocephaly, frontal bossing |
| 7 | M | 12 yrs | severe | rotatory nystagmus, esotropia | + | − | − | hydrocephalus |
| 8 | M | 1 yr | dev. delay | rotatory nystagmus, esotropia | + | − | − | oromotor difficulties, dolichocephaly, frontal bossing, hypothyroidism |
| 9 | F | 17 yrs | severe | rotatory nystagmus | + | − | renal insufficiency, renal transplantation | epilepsy, retinopathy |
| 10 | F | 2 yrs | severe | rotatory nystagmus | + | − | − | |
| 11 | M | 26 yrs | severe | − | + | upper and lower limbs | renal insufficiency, peritoneal dialysis | short limbs, dolichocephaly, frontal bossing, oromotor difficulties |
| 12 | M | 16 yrs | severe | nystagmus | + | upper and lower limbs | − | − |
| 13 | M | 2 yrs | mod | rotatory nystagmus | + | − | − | − |
Abbreviations are as follows: m, male; f, female; mod, moderate; dev. delay, developmental delay; PMR, psychomotor retardation. + indicates present, − indicates negative or absent.
Figure 1.
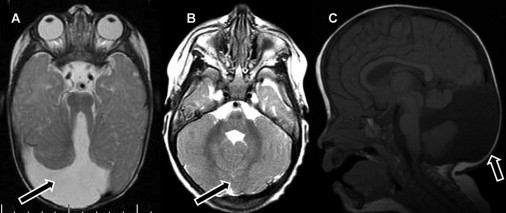
Magnetic Resonance Images of the Patients, Illustrating the Typical Anomalies
(A and B) Axial T2-weighted images showing (A) a completely absent vermis with a wide space between the cerebellar hemispheres (arrow) and (B) a dysplastic split vermis (arrow) without an opening or separation of the cerebellar hemispheres.
(C) Midsagittal T1-weighted image showing cystic dilatation of the fourth ventricle (arrow) and vermian hypoplasia.
Because the clinical and radiological findings were consistent with the diagnosis of JBTS, sequence analysis of known JSRD genes (CEP290 [MIM 610142], TMEM67/MKS3 [MIM 609884], RPGRIP1L [MIM 610937], ARL13B [MIM 608922], AHI1 [MIM 608894], and NPHP1 [MIM 607100]) was performed in three patients but invariably yielded normal results.
In order to localize the mutated gene, we performed homozygosity mapping, using the GeneChip Human Mapping 250K Nsp Array of Affymetrix, as previously described.11 This study was initiated by the Dor Yeshorim organization at the request of families of patients affected with JBTS, and all patients' guardians provided informed consent for participation, in compliance with the research regulations regarding human subjects in Israel. Initially, the DNA samples of three second cousins originating from a single clan were analyzed. Comparing runs of homozygosity longer than 2.4 Mb in each patient, we detected only one homozygous region, spanning 4.46 Mb on chromosome 11 (SNP markers rs11228951–rs11230775, corresponding to 56.73–61.19 Mb; marker positions are provided in accordance with the March 2006 release of the UCSC Human Genome Assembly; build hg18), which was shared by the three patients; the genotypes of the 379 SNP markers that are included in this region were identical. We then analyzed the DNA samples of three additional patients originating from two unrelated families. These patients shared a 2.3 Mb homozygous region on chromosome 11 (SNP markers rs612386–rs7126715, corresponding to 60.71–62.94 Mb) (Figure 2). Notably, the six patients shared identical genotypes within the overlapping homozygous interval and did not share any other genomic region. The overlapping segment (60.71–61.19 Mb on chromosome 11) included 14 open reading frames (PGA3 [MIM 169710], PGA4 [MIM 169720], PGA5 [MIM 169730], VWCE [MIM 611115], DDB1 [MIM 600045], DAK, CYBASC3, TMEM138, TMEM216, CPSF7, C11orf79 [MIM 613019], C11orf66, LRRC10L, and SYT7 [MIM 604146]), and we have determined the sequence of their 117 coding exons and their flanking intronic sequences. This analysis resulted in the identification of a single mutation in the TMEM216 gene, c.35G>T (Figure 2), which is predicted to result in the substitution of arginine at codon 12 by leucine (R12L). In parallel, we took a complementary approach, determining the complete exonic sequence of a mother and an affected daughter by using array-based hybrid selection and deep sequencing.12,13 Genomic DNA samples were fragmented and immortalized by ligation to standard Illumina adapters, followed by PCR-based enrichment. Sequences corresponding to all annotated human exons were enriched by hybridization to an Agilent 1 Million Feature Array.14 Each sample was sequenced in two flow cell lanes on a GAIIx (Illumina), which yielded roughly 30 million paired-end sequence reads per sample. Our analysis did not reveal any potentially relevant insertions or deletions in the targeted region. We called SNPs with SOAPsnp, using a coverage cutoff of at least 5× in the 59–62 Mb region on chromosome 11.15 All SNPs that were not homozygous in the daughter and heterozygous in the mother were discarded. Only 17 SNPs survived this filter (Table S1, available online). Excluding SNPs that were already documented in dbSNP130 reduced the number of candidates to two. One had an obvious effect on a protein, changing amino acid 12 of TMEM216 from arginine to leucine. This was precisely the coding variant identified via combined linkage mapping and candidate resequencing. Thus, not only did these analyses independently support our candidate gene, but they also failed to reveal any similarly compelling potentially causal mutation. The R12L mutation was present in a homozygous state in all 13 patients. Carriers were detected via three different methods. First, direct sequencing was used as described above. Second, restriction fragment length polymorphism (RFLP) analysis was performed, whereby a 176 bp fragment was amplified via mismatch primer amplification (forward primer 5′-CCCAAGTGTGTGGCAGTTC-3′, reverse primer 5′-GGACAACACTTACCAAAAAACTGG-3′), followed by BpmI digestion at 37°C for 2 hrs. In the presence of the mutation, the 176 bp fragment was digested to 136 bp and 40 bp fragments, which were then analyzed on 2% agarose gel. Finally, for mass carrier detection, we used the TaqMan Allelic Discrimination method with the forward primer 5′-GTGATGCTCCTCCTTTATCTTGGAA-3′, reverse primer 5′- GGAGTGGAAATATTCTCTGGACAACA-3′, and the following reporter probes: wild-type, VIC probe 5′-AAGTAATTCGCCTGTTTTT-3′; mutant, FAM probe 5′-TGAAGTAATTCTCCTGTTTTT-3′. The analysis was performed on a 7900HT Real-Time PCR System with SDS version 2.3 software (Applied Biosystems). With these methods used, heterozygosity for the mutation was detected in all of the parents, in 12 of 20 siblings, 23 of 48 first-degree cousins, 58 of 449 more-remote relatives, and 30 of 2766 anonymous individuals of Ashkenazi Jewish origin, indicating a carrier rate of 1:92 for the R12L mutation in this ethnic group.
Figure 2.
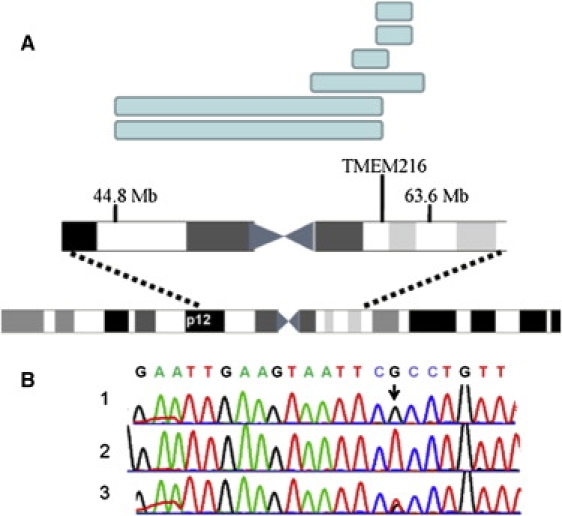
The Homozygous Regions and the TMEM216 Mutation
(A) Homozygous regions of the six SNP-genotyped patients (blue rectangles) within the chromosome 11 centromeric region.
(B) Chromatograms showing the sequence of exon 3, the first coding exon, of the TMEM216 gene. 1, a normal control;. 2, a patient homozygous for the c.35G>T mutation (arrow); 3, an obligatory carrier of the mutation.
JSRDs were heretofore mapped to ten loci, and causative genes have been identified for nine of them. The tenth locus, the centromeric region of chromosome 11 (11p12-q13.3), was mapped in six families, and this form was designated as JBTS2 (MIM 608091). The patients, originating from Sicily, Turkey, Pakistan, United Arab Emirates, and Portugal, suffered from a neurological disease with psychomotor retardation, breathing abnormalities in infancy (alternating brady-tachypnea), hypotonia, truncal ataxia, nystagmus, oculomotor apraxia, and impaired pursuit and saccades. Brain MRI showed the molar tooth sign variably associated with fourth-ventricle cyst, kinked corpus callosum, malformed occipital cortex, and hydrocephalus. Miscarried siblings had occipital encephalocele and enlarged cistern magna. Ophthalmologic abnormalities included microphthalmia, retinal coloboma, retinal dystrophy, and optic atrophy. Renal involvement ranged in severity from a urinary-concentration defect secondary to nephronophthisis to fulminent renal failure due to cystic renal dysplasia. Facial dysmorphism with hypertelorism, low-set ears, and a depressed nasal bridge, as well as postaxial polydactyly, were reported.2,16,17 The critical region on chromosome 11 was initially bordered by D11S1993–D11S1883, corresponding to 43.65–63.13 Mb.2 Mapping of additional families narrowed the region further to 46.12–63.13 Mb (D11S1344–D11S1883).17 Although we did not have the opportunity to analyze the TMEM216 gene sequence in the original JBTS2 patients, the clinical and radiological similarity and the overlapping critical regions suggest that JBTS2 is caused by mutations in the TMEM216 gene.
The three coding exons of the TMEM216 (HSPC244) gene encode a small protein that consists of 87 amino acids, predicted to form two transmembrane domains. The amino acid arginine at codon 12 is conserved in the TMEM216 homologs in Danio rerio and Drosophila melanogaster, and its substitution by leucine is predicted to be deleterious by both the SIFT-BLink software and the PolyPhen software. The segregation of the mutation with the disease state in all family members and the absence of mutations in all other coding exons within the linked region suggest that this mutation is indeed pathogenic. Because all known JSRD genes were shown to localize to the primary cilia or basal body or to directly interact with their components (reviewed in Doherty18), it is tempting to speculate a similar localization for the TMEM216 protein. Ciliary membranes contain receptors and ion-channel proteins mediating mechano- and chemosensations and other types of cell signaling, including Shh, Wnt, and PDGF-alpha signaling pathways. TMEM67/MKS3 (Meckelin), a JSRD protein with seven transmembrane domains, was shown to localize both to the primary cilium and to the plasma membrane in ciliated cell lines and primary cells and to be of importance for centriole migration to the apical membrane and for the consequent formation of the primary cilium.19 The elucidation of the role of the TMEM216 protein in this organelle and in JSRD2 pathogenesis is in progress. Given its small size in comparison to other JSRD genes, sequence analysis of TMEM216 is warranted in every JSRD patient, especially those who suffer from associated anomalies.
Acknowledgments
We are grateful to the patients and their families, as well as to Emily Hodges and Noa Cohen-Itzhaki for dedicated assistance.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
SIFT Blink, http://sift.jcvi.org/www/SIFT_BLink_submit.html
PolyPhen, http://genetics.bwh.harvard.edu/pph/
Note Added in Proof
This version differs from the one originally published online on December 24th, in the following manner: Alan L. Shanske was originally omitted from the author list, and this omission has been corrected in both the online version and the print version of this article.
References
- 1.King M.D., Dudgeon J., Stephenson J.B. Joubert's syndrome with retinal dysplasia: neonatal tachypnoea as the clue to a genetic brain-eye malformation. Arch. Dis. Child. 1984;59:709–718. doi: 10.1136/adc.59.8.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valente E.M., Salpietro D.C., Brancati F., Bertini E., Galluccio T., Tortorella G., Briuglia S., Dallapiccola B. Description, nomenclature, and mapping of a novel cerebello-renal syndrome with the molar tooth malformation. Am. J. Hum. Genet. 2003;73:663–670. doi: 10.1086/378241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Satran D., Pierpont M.E., Dobyns W.B. Cerebello-oculo-renal syndromes including Arima, Senior-Löken and COACH syndromes: more than just variants of Joubert syndrome. Am. J. Med. Genet. 1999;86:459–469. [PubMed] [Google Scholar]
- 4.Verloes A., Lambotte C. Further delineation of a syndrome of cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, coloboma, and hepatic fibrosis. Am. J. Med. Genet. 1989;32:227–232. doi: 10.1002/ajmg.1320320217. [DOI] [PubMed] [Google Scholar]
- 5.Haug K., Khan S., Fuchs S., König R. OFD II, OFD VI, and Joubert syndrome manifestations in 2 sibs. Am. J. Med. Genet. 2000;91:135–137. doi: 10.1002/(sici)1096-8628(20000313)91:2<135::aid-ajmg11>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 6.Valente E.M., Brancati F., Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur. J. Med. Genet. 2008;51:1–23. doi: 10.1016/j.ejmg.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Cantagrel V., Silhavy J.L., Bielas S.L., Swistun D., Marsh S.E., Bertrand J.Y., Audollent S., Attié-Bitach T., Holden K.R., Dobyns W.B., International Joubert Syndrome Related Disorders Study Group Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am. J. Hum. Genet. 2008;83:170–179. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coene K.L., Roepman R., Doherty D., Afroze B., Kroes H.Y., Letteboer S.J., Ngu L.H., Budny B., van Wijk E., Gorden N.T. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am. J. Hum. Genet. 2009;85:465–481. doi: 10.1016/j.ajhg.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacoby M., Cox J.J., Gayral S., Hampshire D.J., Ayub M., Blockmans M., Pernot E., Kisseleva M.V., Compère P., Schiffmann S.N. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 2009;41:1027–1031. doi: 10.1038/ng.427. [DOI] [PubMed] [Google Scholar]
- 10.Gorden N.T., Arts H.H., Parisi M.A., Coene K.L., Letteboer S.J., van Beersum S.E., Mans D.A., Hikida A., Eckert M., Knutzen D. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am. J. Hum. Genet. 2008;83:559–571. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodges E., Xuan Z., Balija V., Kramer M., Molla M.N., Smith S.W., Middle C.M., Rodesch M.J., Albert T.J., Hannon G.J., McCombie W.R. Genome-wide in situ exon capture for selective resequencing. Nat. Genet. 2007;39:1522–1527. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- 13.Hodges E., Rooks M., Xuan Z., Bhattacharjee A., Benjamin Gordon D., Brizuela L., Richard McCombie W., Hannon G.J. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nat. Protoc. 2009;4:960–974. doi: 10.1038/nprot.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li R., Li Y., Fang X., Yang H., Wang J., Kristiansen K., Wang J. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19:1124–1132. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keeler L.C., Marsh S.E., Leeflang E.P., Woods C.G., Sztriha L., Al-Gazali L., Gururaj A., Gleeson J.G. Linkage analysis in families with Joubert syndrome plus oculo-renal involvement identifies the CORS2 locus on chromosome 11p12-q13.3. Am. J. Hum. Genet. 2003;73:656–662. doi: 10.1086/378206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valente E.M., Marsh S.E., Castori M., Dixon-Salazar T., Bertini E., Al-Gazali L., Messer J., Barbot C., Woods C.G., Boltshauser E. Distinguishing the four genetic causes of Jouberts syndrome-related disorders. Ann. Neurol. 2005;57:513–519. doi: 10.1002/ana.20422. [DOI] [PubMed] [Google Scholar]
- 18.Doherty D. Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin. Pediatr. Neurol. 2009;16:143–154. doi: 10.1016/j.spen.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dawe H.R., Smith U.M., Cullinane A.R., Gerrelli D., Cox P., Badano J.L., Blair-Reid S., Sriram N., Katsanis N., Attie-Bitach T. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum. Mol. Genet. 2007;16:173–186. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.