

Abstract
Fuchs corneal dystrophy (FCD) is a degenerative genetic disorder of the corneal endothelium that represents one of the most common causes of corneal transplantation in the United States. Despite its high prevalence (4% over the age of 40), the underlying genetic basis of FCD is largely unknown. Here we report missense mutations in TCF8, a transcription factor whose haploinsufficiency causes posterior polymorphous corneal dystrophy (PPCD), in a cohort of late-onset FCD patients. In contrast to PPCD-causing mutations, all of which are null, FCD-associated mutations encode rare missense changes suggested to cause loss of function by an in vivo complementation assay. Importantly, segregation of a recurring p.Q840P mutation in a large, multigenerational FCD pedigree showed this allele to be sufficient but not necessary for pathogenesis. Execution of a genome-wide scan conditioned for the presence of the 840P allele identified an additional late-onset FCD locus on chromosome 9p, whereas haplotype analysis indicated that the presence of the TCF8 allele and the disease haplotype on 9p leads to a severe FCD manifestation with poor prognosis. Our data suggest that PPCD and FCD are allelic variants of the same disease continuum and that genetic interaction between genes that cause corneal dystrophies can modulate the expressivity of the phenotype.
Introduction
FCD represents the most common form of genetic disorders of the corneal endothelium.1,2 The disorder affects as much as 4% of the population over the age of 40 and accounts for a significant fraction of the corneal transplantation performed in the United States every year.2–4 Clinically, FCD is marked by the development of guttae, excrescences of Descemet membrane that appear in the fourth or fifth decade and increase in number over time.5,6 As the disease progresses, visual acuity decreases secondary to corneal edema and endothelial cell loss, with end-stage disease evidenced by the formation of painful epithelial bullae.7
FCD is genetically heterogeneous, exhibiting an autosomal-dominant mode of inheritance with variable penetrance and expressivity. The rare form of early-onset FCD is causally associated with mutations in COL8A2 (MIM 120252), whereas the more common late-onset FCD has been localized to three loci: FCD1, FCD2, and FCD3 on chromosomes 13, 18, and 5, respectively.8–10 Although clinically distinct, corneal endothelial dystrophies share clinical features suggesting that genes implicated in one corneal dystrophy may also harbor mutations liable for other dystrophies. This premise was strengthened when pathogenic mutations in SLC4A11 (MIM 610206), a borate transporter in which loss-of-function mutations cause autosomal-recessive congenital hereditary endothelial dystrophy (CHED2 [MIM 217700]), were identified in sporadic late-onset FCD patients.11 Therefore, we hypothesized that TCF8 (MIM 189909), a transcription factor shown to be causally associated with another corneal dystrophy, PPCD (MIM 609141),12,13 may contribute to the genetic load of late-onset FCD.
Here, we report five missense mutations in TCF8 associated with late-onset FCD, which appear to be causal because of a loss of protein function based on an in vivo complementation assay. One of these mutations, encoding a p.Q840P change, was present in both a sporadic FCD patient and in a large family and was transmitted in a manner consistent with an autosomal-dominant trait. However, the 840P allele could not explain FCD in all patients, suggesting the presence of a second pathogenic allele in this pedigree. A genome-wide scan conditioned to the presence/absence of the TCF8 mutant allele identified a new locus for late-onset FCD, FCD4 on chromosome 9p subsequent genetic and clinical analyses suggested that although mutations in each of TCF8 and FCD4 are sufficient for pathogenesis, genetic interaction between these two loci can lead to a more severe form of the disease. Our data represent the first familial evidence for mutations that cause late-onset FCD, supporting the hypothesis that several corneal dystrophies, although clinically distinct, share the same molecular etiology. Additionally, our data suggest that the quality and quantity of mutations in FCD-associated loci can have a profound impact on the clinical presentation of the phenotype, which in turn will probably impact patient management.
Material and Methods
Recruitment
Family members were recruited through a proband with FCD presenting to our clinics at Johns Hopkins University or Duke University. Extended pedigrees were subsequently developed through interviews and patients were examined in locations proximal to their area of residence. Recruitment, examination, and procedures for DNA sample collection were approved by the Institutional Review Boards for Human Subjects Research at the Johns Hopkins University School of Medicine and Duke University Medical Center, respectively, according to the Declaration of Helsinki. Each study participant provided written consent.
Determination of Phenotype and Disease Severity
Individuals underwent detailed ophthalmic examination including slit-lamp biomicroscopy. Severity was graded on a modified scale according to Krachmer and colleagues with >12 central guttae in either eye (grade 1).5 Progression of confluence was defined by three grades: horizontal width less than 2 mm (grade 2), from 2 to 5 mm (grade 3), and >5 mm (grade 4). The development of stromal and/or epithelial edema elevated this score to grade 5, and grade 6 is disease severe enough to require keratoplasty.
Genotyping
An Affymetrix (Affymetrix, Santa Clara, CA) 5.0 SNP array was used for genome-wide linkage analyses. 500 ng of genomic DNA was used in a multiplexed SNP genotyping assay according to manufacturer's instructions. Arrays were then scanned in a GeneChip Scanner 3000 7G (Affymetrix). The output signal files were checked for quality control (QC) with the Affymetrix BRLMM analysis tool 2.0 (BAT 2.0). The default threshold score was set at 0.05 and was used as a cutoff for SNPs to be excluded. Cell files were processed further to generate genotypes with the BAT 2.0 Affymetrix genotyping console and locally written Perl scripts.
Linkage Analyses
SNP genotypes were incorporated into the family pedigree files to generate PED files with Plink software.14 Pedigrees were analyzed assuming an autosomal-dominant mode of inheritance, 0.02 disease allele frequency, and 90% penetrance by MERLIN algorithms.15 SNP allele frequencies from an ethnically matched control population were obtained from HapMap. The critical interval on 9p was delineated by genotyping STR markers; primers and conditions available upon request. Two point linkage analyses were performed with the FASTLINK version of MLINK from the LINKAGE Program Package.16 Maximum LOD scores were calculated with ILINK under an autosomal-dominant model with a 0.02 disease allele frequency and 90% penetrance. The marker order and distances between the markers were obtained from the Marshfield database and the NCBI chromosome 9 sequence maps. Equal allele frequencies were assumed for the initial genome scan, while for fine mapping allele frequencies were derived from 96 unrelated and unaffected individuals.
Mutation Screening
Primer pairs for individual exons of TCF8 were designed with Primer3. The sequences and amplification conditions are available upon request. Each amplicon was purified and sequenced bidirectionally with Big Dye Terminator Ready Reaction Mix according to manufacturer's instructions (Applied Biosystems).
Construction of the TCF8 Vector
We cloned the TCF8 ORF into a pCS2+ plasmid as follows. The 3375 bp TCF8 ORF was amplified from HEK293T cDNA in two separate amplification reactions. In the first step, the 1875 bp fragment was amplified, digested with BamHI and BglII restriction endonucleases, and cloned into BamHI/BglII-digested pCS2+ plasmid. The orientation of the clone was detected by restriction digestion with BamHI, BglII, and XhoI. In the second step, a 1775 bp fragment was amplified, digested with BglII and XhoI restriction endonucleases, and cloned into BglII/XhoI digested pCS2+ plasmid harboring the N-terminal TCF8 fragment in the correct orientation. The sequence of TCF8 was confirmed by Big Dye Terminator Ready reaction mix according to manufacturer's instructions (Applied Biosystems).
Generation of Site-Directed Mutants
All five missense mutations identified in this study, as well as a previously identified mutation N696S,17 were cloned in the TCF8-pCS2+ vector with QuikChange Site-Directed Mutagenesis Kit according to manufacturer's instructions (Stratagene, La Jolla, CA). The presence of each mutation was confirmed with bidirectional sequencing. Additionally, the entire TCF8 ORF for each clone was sequenced to rule out the presence of any mutations that may have been incorporated during the mutagenesis procedure.
In Vitro Transcription
The pCS2+ plasmids harboring the wild-type and mutant alleles of TCF8 were digested with NotI to linearize the plasmids. These linearized plasmids were used for in vitro transcription with mMessage mMachine transcription kit utilizing the SP6 promoter according to the manufacturer's instruction (Ambion Austin TX).
In Vivo Analyses
We used an antisense translation blocking morpholino (MO) to suppress translation of zebrafish tcf8. The MO was injected into one- to two-cell stage embryos. The exact concentration of MO necessary was determined by a dose-response curve. Embryos were raised to somite stages (approximately 14 hr) and scored for early developmental defects. Embryo phenotypes were rescued by coinjecting mRNA encoding full-length wild-type human TCF8 mRNA and the efficiency of rescue of the zebrafish tcf8MO was measured. Approximately 100 embryos were injected with either MO alone, MO and wild-type TCF8 mRNA, or MO and mRNA bearing one of each of the missense mutations and were scored blind to the injection cocktail.
Results
Mutations in TCF8 Are Sufficient to Cause FCD
The association of SLC4A11 with late-onset FCD11 raised the possibility that genes that cause other corneal endothelial dystrophies might also contribute to FCD. To investigate this hypothesis, we focused on TCF8, a transcription factor that has been causally linked to PPCD, another endothelial dystrophy with some phenotypic similarities to FCD.12,13 We therefore sequenced the entire coding region of TCF8 in 192 unrelated adult-onset FCD patients of northern European descent. In addition to a number of known SNPs, whose frequency in our cohort was indistinguishable from reported HapMap data, we also identified four unique alleles encoding the missense changes p.N78T, p.Q810P, p.Q840P, and p.A905G. Each of these variants affect evolutionary conserved residues (Figure 1) and was not found in 560 unrelated, ethnically matched control chromosomes or in HapMap samples of any ethnicity. Additionally, computational predictions with both SIFT18 and PolyPhen19 suggested that they were pathogenic, raising the strong possibility that missense alleles in TCF8 can contribute to late-onset FCD. To confirm these data, we sequenced a second, independent cohort of 192 sporadic samples of Caucasian descent, ascertained under the same clinical criteria. We found three missense changes: a novel p.P649A that is also evolutionarily conserved and computationally predicted to be pathogenic (Figure 1) and p.N78T and p.Q840P, each of which was also found in the first cohort with no evidence for a shared ancestral haplotype. Six of the seven FCD patients were sporadic and as such, segregation analysis was not possible. The seventh patient bearing the recurring p.Q840P mutation, however, belongs to a large, multigenerational FCD family, acronymed TH. Targeted recruitment within TH (Figure 2) enabled us to further identify 12 individuals (seven females, five males) who fulfill the FCD phenotypic criteria (Table 1). A 13th individual (Figure 2; individual IV:10) exhibited early signs of FCD; however, the patient did not have sufficient guttae for a positive diagnosis. In view of her younger age (38 years) and the number of guttae present bilaterally (<12), she was designated as a subclinical case.
Figure 1.
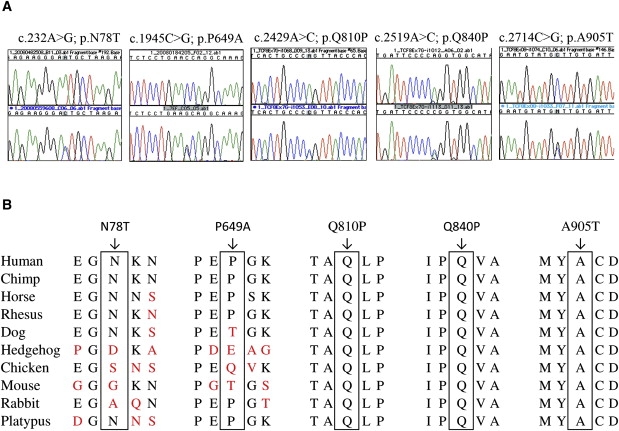
Sequence Analyses and Evolutionary Assessment of Late-Onset FCD Mutants
(A) Sequence chromatograms of the five TCF8 mutations (c.232A>G, c.1945C>G, c.2429A>C, c.2519A>C, c.2714C>G) identified in late-onset FCD patients.
(B) Illustration of the evolutionary conservation of Asn78, Pro649, Gln810, Gln840, and Ala905 in other TCF8 orthologs.
Figure 2.

Pedigree of the Family TH Linked to the FCD4 Locus
Symbols: filled symbol, affected; diagonal line through a symbol, deceased; question mark, affectation status known. Haplotypes of 12 adjacent chromosome 9p microsatellite markers are shown, with alleles forming the disease-bearing haplotype shaded black. The TCF8 mutation; c.2519A>C identified in family TH is shown at the bottom.
Table 1.
Clinical Characteristics of Affected Individuals of Family TH
| Individual ID | Sex | Age at Time of the Examination (Years) | Age at Time of the Penetrating Keratoplasty (Years) | FCD Krachmer Grading | TCF8 Mutation | FCD4 Disease Haplotype |
|---|---|---|---|---|---|---|
| III:5 | F | 78 | 71 | PK bilaterally | p.Q840P | yes |
| IV:1 | M | 55 | N.A. | OD; +1.0/ OS; +1.5 | p.Q840P | no |
| IV:2 | F | 57 | N.A. | OD; +2.5/ OS; +2.5 | wild-type | yes |
| III:7 | F | 76 | 70 | PK bilaterally | p.Q840P | yes |
| IV:4 | F | 49 | N.A. | OD; +1.0/ OS; +1.0 | p.Q840P | yes |
| IV:5 | M | 53 | N.A. | OD; +1.0/ OS; +1.5 | wild-type | yes |
| IV:6 | M | 46 | N.A. | OD; +1.0/ OS; +1.0 | p.Q840P | yes |
| IV:7 | F | 42 | N.A. | OD; +1.0/ OS; +1.0 | wild-type | yes |
| III:9 | M | 72 | 68 | PK bilaterally | p.Q840P | yes |
| IV:8 | F | 47 | N.A. | OD; +1.0/ OS; +1.0 | wild-type | yes |
| IV:9 | F | 45 | N.A. | OD; +1.0/ OS; +1.0 | wild-type | yes |
| IV:10 | F | 37 | N.A. | trace bilaterally | wild-type | yes |
| III:11 | F | 70 | N.A. | OD; +2.5/ OS; +2.5 | p.Q840P | no |
Individuals III:5, III:7, and III:9 (Figure 1) had bilateral corneal transplants. Penetrating keratoplasty was performed on each eye separately and only once the severity of disease had reached 6 on the modified scale proposed by Krachmer and colleagues.5 Abbreviations: PK, penetrating keratoplasty; F, female; M, male; N.A., not applicable.
Identification of a Fourth FCD Locus
Segregation analysis showed that the p.Q840P allele is sufficient but not necessary for disease pathogenesis. None of the seven unaffected individuals carry the 840P allele, whereas the mutation is present in 7/12 affecteds (Figure 2), suggesting that a second, independent genetic event might account for the phenotype in the other five patients. To investigate this possibility, we performed a genome-wide screen by genotyping all family members with an Affymetrix 5.0 array. Approximately 481K SNPs passed quality control and were considered for linkage. However, because of the unattainable computational needs required to perform linkage with the entire marker set, we selected every 5th accepted SNP to simplify the data while maintaining uniform coverage across the genome and performed two-point linkage on ∼96K SNPs. Initial analyses under various disease models failed to detect LOD scores even above 2.0 when considering the entire family. We therefore took into consideration the TCF8 p.Q840P allele and recomputed linkage conditional to this mutation. This model predicated that (1) all affected individuals negative for the p.Q840P mutation must harbor a pathogenic allele elsewhere in the genome; and (2) affected individuals heterozygous for the p.Q840P mutation may not necessarily harbor the pathogenic allele, with the exception of individuals with affected offspring who lack the p.Q840P mutation. Under this model, we found suggestive linkage to a single region in the genome on chromosome 9p (highest two-point LOD score of 2.43 with rs1410041). Importantly, a “reverse” genome scan was also consistent with a two-locus model. When we conditioned for the 9p risk allele (affected individuals or individuals with affected offspring negative for the 9p allele must harbor a second pathogenic allele elsewhere in the genome, whereas the rest of the affected individuals may not necessarily conceal this allele), we obtained the highest genome-wide two-point LOD score of 1.93 at TCF8 (rs1148233), close to the theoretical maximum.
Because SNPs are biallelic, they have reduced informativeness. Therefore, we genotyped all members of family TH with a series of 10 evenly spaced STR microsatellite markers (Figure 2). We observed LOD scores of 3.09 and 3.20 at θ = 0 with markers D9S168 and D9S256, respectively (Table 2), close to the theoretical maximum attainable with this family, thereby defining a fourth FCD locus, FCD4. Recombinants place the mutation in a 14 Mb interval between D9S1681 proximally and D9S1684 distally. Reconstruction of the disease haplotype was consistent with a two-locus model, in that all affected individuals negative for the p.Q840P TCF8 mutation harbor the disease-transmitting 9p22.1-p24.1 haplotype (Figure 2). Notably, individual IV:10, with a borderline diagnosis, carries the disease haplotype as predicted by the presence of preclinical FCD findings. After considering her as affected, the LOD score is elevated to 3.48 (Table 2).
Table 2.
Two-Point LOD Scores Generated with 9p STR Markers
| Marker | cM | Mb | 0.00 | 0.01 | 0.05 | 0.10 | 0.20 | 0.30 | Zmax | θmax |
|---|---|---|---|---|---|---|---|---|---|---|
| D9S1686 | 12.50 | 4.63 | 0.672 | 0.658 | 0.598 | 0.567 | 0.520 | 0.357 | 0.672 | 0.00 |
| 0.670 | 0.656 | 0.596 | 0.565 | 0.519 | 0.356 | 0.670 | 0.00 | |||
| D9S1681 | 18.60 | 5.27 | −0.484 | −0.321 | 0.091 | 0.216 | 0.346 | 0.500 | 0.500 | 0.30 |
| −1.417 | −1.225 | −0.646 | −0.448 | −0.223 | 0.155 | 0.155 | 0.30 | |||
| D9S281 | 15.33 | 6.71 | −0.092 | 0.047 | 0.357 | 0.436 | 0.501 | 0.475 | 0.501 | 0.20 |
| −0.094 | 0.046 | 0.358 | 0.436 | 0.502 | 0.476 | 0.502 | 0.20 | |||
| D9S168 | 20.40 | 10.57 | 3.094 | 3.038 | 2.807 | 2.689 | 2.507 | 1.862 | 3.094 | 0.00 |
| 3.375 | 3.314 | 3.067 | 2.939 | 2.744 | 2.049 | 3.375 | 0.00 | |||
| D9S256 | 22.80 | 11.01 | 3.201 | 3.145 | 2.916 | 2.798 | 2.617 | 1.972 | 3.201 | 0.00 |
| 3.482 | 3.422 | 3.175 | 3.048 | 2.854 | 2.160 | 3.482 | 0.00 | |||
| D9S1869 | 26.50 | 14.26 | 1.988 | 1.969 | 1.874 | 1.816 | 1.719 | 1.321 | 1.988 | 0.00 |
| 2.268 | 2.246 | 2.133 | 2.066 | 1.955 | 1.508 | 2.268 | 0.00 | |||
| D9S1684 | 34.42 | 19.60 | −5.918 | −2.870 | −1.394 | −0.839 | −0.745 | −0.232 | −0.232 | 0.30 |
| −0.204 | −0.185 | −0.125 | −0.102 | −0.075 | −0.022 | −0.022 | 0.30 | |||
| D9S1846 | 37.58 | 21.62 | −3.221 | −2.397 | −1.403 | −1.148 | −0.873 | −0.375 | −0.375 | 0.30 |
| −4.299 | −2.220 | −1.169 | −0.919 | −0.658 | −0.221 | −0.221 | 0.30 |
LOD scores were calculated at different θ values for each marker with individual IV:10 as unaffected (top) and affected (bottom row).
Genetic Interaction between TCF8 and FCD4
Within family TH, five affected individuals are heterozygous for both the p.Q840P mutation and the disease haplotype on 9p22.1-p24.1 (Figure 2). We therefore wondered whether the presence of two pathogenic alleles influences the clinical phenotype and severity of the disease. To compare across the same age, we examined the clinical records of individuals III:5, III:7, III:9, and III:11 (all ages > 70). Individuals III:5, III:7, and III:9 harbor both p.Q840P mutation and the FCD4 disease allele, whereas individual III:11 harbors only the p.Q840P mutation. The clinical records confirmed that all three individuals with both mutations have a severe FCD phenotype (modified Krachmer grading of 6 bilaterally) and have undergone corneal transplantations, bilaterally. In sharp contrast to his siblings, individual III:11 has a milder FCD phenotype (Krachmer grading of +2.5 bilaterally). Progression of disease for individuals in family TH with a single pathogenic allele (TCF8 p.Q840P or disease haplotype on 9p) shows as a linear relationship between age and severity with an r2 value of 0.84 (Figure 3). Notably, the severity of individuals III:5, III:7, and III:9, harboring pathogenic alleles in both TCF8 and FCD4, is not consistent with the progression pattern of family TH, and based on these observations, we predict that individuals IV:4 and IV:6 (both in their 40s at time of the examination) will develop a severe FCD phenotype compared with other affected siblings.
Figure 3.
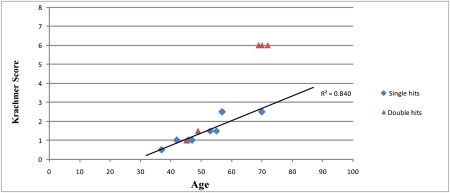
Progression of Late-Onset FCD with Age in Family TH
The diamonds represent the individuals with single pathogenic allele (TCF8 p.Q840P or disease haplotype on 9p), whereas the triangles symbolize individuals harboring both pathogenic alleles. The age of each individual was recorded at the time of the examination or at the time of bilateral keratoplasty.
The fact that three affected individuals in family TH underwent a corneal transplant was surprising because the majority of the families in our cohort rarely have multiple affecteds that had corneal transplants. Therefore, we wondered whether the presence of pathogenic alleles at two loci might have contributed to the higher incidence of corneal transplants in family TH. To examine this possibility, we interrogated our entire cohort setting the following two stringent criteria: (1) we restricted our analyses to families with three or more affecteds; and (2) we only considered affecteds with bilateral corneal transplants. We were able to examine 33 families and found a remarkably low frequency of bilateral transplants (9/246) which, despite the relatively modest sample size, was significantly different compared to 3/12 in family TH (χ2 = 10.618; p = 0.0006). Even under reduced stringency criteria, where all transplant events were considered in our cohort, the enrichment in family TH remained significant (χ2 = 8.17; p = 0.0042). As such, the most parsimonious explanation for these observations is that in family TH the p.Q840P allele interacts with the FCD4 causal mutation to modulate the severity of the phenotype.
In Vivo Functional Assessment of TCF8 Mutations
Taken together, our genetic data implicate missense mutations in TCF8 in the development of late-onset FCD. All mutations affect conserved residues and have been found exclusively in patients (two of them recurrent). Importantly, phasing pedigree TH for the presence of the p.Q840P allele maps a new FCD locus supportive of a two-locus model. Although TCF8 null mutations have been shown to cause PPCD, an earlier onset endothelial corneal dystrophy, we have determined that missense alleles in FCD are of potential mechanistic consequence. We considered two possibilities: (1) loss-of-function alleles lead to PPCD and dominant-negative or gain-of-function mutations cause FCD, or (2) both disorders can be induced by relative TCF8 loss. To investigate these possibilities and to obtain further evidence for the functional relevance of the mutated residues, we took advantage of an in vivo complementation assay in zebrafish. Upon reciprocal BLAST, we identified the sole ortholog of TCF8 in the zebrafish genome (81% similarity at protein level), also known as kheper. Previous studies have shown that expression of dominant-negative tcf8 RNA results in severe defects during zebrafish gastrulation and neurulation.20 Therefore, under the reasonable premise that morpholino (MO)-induced tcf8 suppression should reproduce such phenotypes, we initiated a series of in vivo complementation studies. Specifically, to interrogate the mechanisms by which mutations in TCF8 cause FCD in humans, we sought to use these developmental defects as a functional readout and ask whether (1) suppression of endogenous tcf8/kheper produces similar phenotypes early in development; (2) whether wild-type (WT) human TCF8 mRNA can rescue such phenotypes; (3) whether expression of human TCF8 mRNA bearing each of the five point mutations can rescue the phenotype; and (4) whether expression of the same mutants might induce early developmental defects (indicative of a dominant-negative or gain-of-function effect).
To suppress expression of tcf8 in embryos, we designed a translational blocking morpholino (tbMO). Injection of 2.5 ng of tbMO at the one- to two-cell stage resulted in defects in early development consistent with published observations in approximately 80% of embryos,20 which could be categorized into two severity classes. Class I embryos survived gastrulation but had shortened body axis and pronounced detachment of cells along the dorsal axis at mid-somitic age, whereas class II embryos exhibited complete disruption of embryo morphology during gastrulation (Figure 4A). These phenotypes could be rescued efficiently by coinjection of human WT TCF8 mRNA. Coinjection of 100 pg of human mRNA with 2.5 ng of tbMO resulted in a complete rescue of the morphant phenotypes (97.5% of embryos, n = 100–150 embryos scored blind) (Figure 4B). To determine whether mutations in TCF8 result in disruption of protein function, we coinjected each mutant mRNA and assessed its ability to rescue morphant phenotypes. This assay further supported our human genetics evidence in that each of the five mutations reduced or abolished TCF8 function. Two mutations, p.N78T and p.Q810P, partially rescued the MO phenotype, indicating that these are probably hypomorphic, whereas rescue with human TCF8 mRNA encoding either 649A, 840P, or 905G were indistinguishable from tbMO-injected embryos, suggesting that these alleles are severe (Figure 4B). The assay is not promiscuous; injection of tbMO with human mRNA encoding an N696S mutation reported previously in a Chinese FCD patient and reported to be benign17 fully rescued the morphant phenotype (Figure 4B). Notably, though injection of wild-type mRNA resulted in defects in early development, injection of each of the mutant mRNAs alone did not produce the same severity of defects, suggesting that these mutations are unlikely to exert a dominant-negative or gain-of-function effect (Figure 4B). As such, although caution must be exercised in translating the function of alleles in a developmental assay to late-onset corneal degeneration phenotypes, it is likely that a loss-of-function mechanism underlies the FCD-associated TCF8 mutations.
Figure 4.
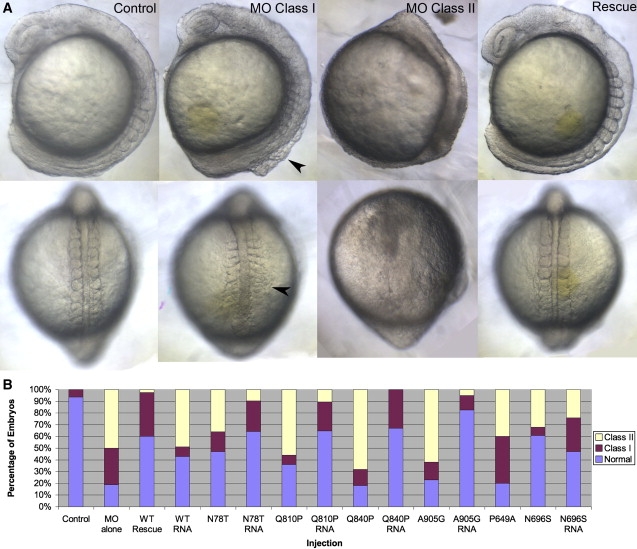
Suppression of tcf8 Produces Specific Defects in Zebrafish Embryos
(A) Embryos injected with MO against tcf8 have early developmental defects including short body axes and detachment of cells along the dorsal embryonic axis (arrowhead). These defects can be rescued by coinjection with full-length human mRNA.
(B) Proportions of unaffected embryos are rescued almost completely in coinjected embryos as compared to those injected with MO alone.
Discussion
Here, we report five novel missense mutations in TCF8 identified in two cohorts of patients diagnosed with late-onset FCD. These mutations affect evolutionary conserved alleles and were absent from ethnically matched control samples. Additionally, we identified a novel locus for late-onset FCD, FCD4, in a large family, with the critical interval mapped to chromosome 9p. Haplotype analyses refined the interval to a 14.3 Mb (15.8 cM) on 9p24.1-22.1 with a maximum two-point parametric LOD score of 3.20. Although the maximum LOD score of 3.20 is only slightly higher than the traditional limiting value of 3.0, it represents the maximum theoretical obtainable value for this family. Our data reinforce the notion that there is extensive heterogeneity for late-onset FCD, contrary to early-onset that is causally associated with COL8A2, with TCF8 contributing as much as 2% to the genetic load (7/384 unrelated patients), with no evidence for shared haplotypes among unrelated patients with the same mutation. Based on genetic and mutational data from all known FCD loci, FCD2 remains the most common locus, with a predicted contribution of ∼25%, followed by SLC4A11, in which we and others have found mutations in 3%–4% of sporadic late-onset FCD patients.11 Each other mapped FCD locus represents, at present, a private mutation.
Mutations in TCF8 have been associated previously with PPCD,12,13 a dystrophy of the corneal endothelium. Interestingly, all mutations reported in PPCD either are nonsense mutations or result in premature termination of TCF8; in contrast, we found only missense mutations in late-onset FCD patients. We have encountered this phenomenon before: MKS1 (MIM 609883), one of the genes that causes Meckel-Gruber syndrome (MKS [604896]), a neonatal lethal condition, is always found to be null in Meckel patients. Under the hypothesis that hypomorphic mutations in that locus might cause a less severe phenotype, we were able to show that residual activity at this locus is causally related to Bardet-Biedl syndrome.21 Given the fact the late-onset FCD represent a milder pathogenic phenotype compared with PPCD, the discovery of only missense mutations suggests that functionally deficient TCF8 alleles are responsible for late-onset FCD phenotype, whereas nonsense mutations lead to PPCD, most probably because of haploinsufficiency.
The molecular mechanism of TCF8 pathogenicity leading to two different corneal dystrophies remains unclear. TCF8 is a transcription factor acting in some instances as an enhancer and in other instances as a repressor.22 The two zinc finger domains present in the TCF8 define the DNA-binding specificity. Ikeda and colleagues investigated the DNA-binding properties of TCF8 protein and identified G/TT/GCACCTGT and C/TACCTG/TT as the optimal binding sites for the N- and C-terminal zinc finger motifs, respectively.23 Interestingly, Krafchak and colleagues identified these consensus sequences present in the promoter regions of COL4A1 (MIM 120130), COL4A2 (MIM 120090), COL4A3 (MIM 120070), COL4A5 (MIM 303630), COL4A6 (MIM 303631), and COL8A2.12 Although TCF8 transcripts have been detected in corneal samples,12 it remains to be seen whether deficiency in the DNA-binding properties of TCF8 leads to late-onset FCD.
Our data provide the first familial evidence for mutations in adult-onset FCD and indicate that loss of function of TCF8 can induce a range of corneal dystrophy phenotypes. Recently, it has been proposed that loss-of-function mutations in SLC4A11, a gene mutated in CHED2, might also contribute to adult-onset FCD.11 Taken together, these data suggest the presence of a continuum across several clinically distinct corneal dystrophies, where the nature of mutations might determine the severity and onset of the phenotype, a phenomenon we have encountered in other disorders.21 As such, genes identified in CHED2, PPCD, and FCD represent bona fide candidates for all three clinical presentations. Moreover, our data also suggest that genetic interactions between FCD loci probably interact to modify the expressivity of the disorder; understanding the molecular basis of such interaction will be important both in terms of disease mechanism and also in the context of patient management and prognosis.
Web Resources
The URLs for data presented herein are as follows:
International HapMap Project, http://www.hapmap.org
Marshfield Clinic Research Foundation, http://www.marshfieldclinic.org/research/pages/index.aspx
National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov
Primer3, http://frodo.wi.mit.edu/primer3
Acknowledgments
We thank all family members for their enthusiastic participation in this study. This study was supported in part by National Eye Institute Grants R01EY016835 (J.D.G.) and R01EY016514 (G.K.K.), the Kwok Research Fund (J.D.G.), and the National Institute of Child Health and Development Grant R01HD04260 (N.K.). The Gottsch and Katsanis laboratories contributed equally to this work.
References
- 1.Wilson S.E., Bourne W.M. Fuchs' dystrophy. Cornea. 1988;7:2–18. [PubMed] [Google Scholar]
- 2.Klintworth G.K. Corneal dystrophies. Orphanet J. Rare Dis. 2009;4:7. doi: 10.1186/1750-1172-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lorenzetti D.W., Uotila M.H., Parikh N., Kaufman H.E. Central cornea guttata. Incidence in the general population. Am. J. Ophthalmol. 1967;64:1155–1158. [PubMed] [Google Scholar]
- 4.Mannis M.J., Holland E.J., Beck R.W., Belin M.W., Goldberg M.A., Gal R.L., Kalajian A.D., Kenyon K.R., Kollman C., Ruedy K.J., Cornea Donor Study Group Clinical profile and early surgical complications in the Cornea Donor Study. Cornea. 2006;25:164–170. doi: 10.1097/01.ico.0000164832.69668.4b. [DOI] [PubMed] [Google Scholar]
- 5.Krachmer J.H., Purcell J.J., Young C.W., Bucher K.D. Corneal endothelial dystrophy. A study of 64 families. Arch. Ophthalmol. 1978;96:2036–2039. doi: 10.1001/archopht.1978.03910060424004. [DOI] [PubMed] [Google Scholar]
- 6.Gottsch J.D., Sundin O.H., Rencs E.V., Emmert D.G., Stark W.J., Cheng C.J., Schmidt G.W. Analysis and documentation of progression of Fuchs corneal dystrophy with retroillumination photography. Cornea. 2006;25:485–489. doi: 10.1097/01.ico.0000178726.11693.14. [DOI] [PubMed] [Google Scholar]
- 7.Goar E.L. Dystrophy of the corneal endothelium (Cornea Guttata), with report of a histologic examination. Trans. Am. Ophthalmol. Soc. 1933;31:48–59. [PMC free article] [PubMed] [Google Scholar]
- 8.Sundin O.H., Jun A.S., Broman K.W., Liu S.H., Sheehan S.E., Vito E.C., Stark W.J., Gottsch J.D. Linkage of late-onset Fuchs corneal dystrophy to a novel locus at 13pTel-13q12.13. Invest. Ophthalmol. Vis. Sci. 2006;47:140–145. doi: 10.1167/iovs.05-0578. [DOI] [PubMed] [Google Scholar]
- 9.Sundin O.H., Broman K.W., Chang H.H., Vito E.C., Stark W.J., Gottsch J.D. A common locus for late-onset Fuchs corneal dystrophy maps to 18q21.2-q21.32. Invest. Ophthalmol. Vis. Sci. 2006;47:3919–3926. doi: 10.1167/iovs.05-1619. [DOI] [PubMed] [Google Scholar]
- 10.Riazuddin S.A., Eghrari A.O., Al-Saif A., Davey L., Meadows D.N., Katsanis N., Gottsch J.D. Linkage of a mild late-onset phenotype of Fuchs corneal dystrophy to a novel locus at 5q33.1-q35.2. Invest. Ophthalmol. Vis. Sci. 2009;50:5667–5671. doi: 10.1167/iovs.09-3764. [DOI] [PubMed] [Google Scholar]
- 11.Vithana E.N., Morgan P.E., Ramprasad V., Tan D.T., Yong V.H., Venkataraman D., Venkatraman A., Yam G.H., Nagasamy S., Law R.W. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum. Mol. Genet. 2008;17:656–666. doi: 10.1093/hmg/ddm337. [DOI] [PubMed] [Google Scholar]
- 12.Krafchak C.M., Pawar H., Moroi S.E., Sugar A., Lichter P.R., Mackey D.A., Mian S., Nairus T., Elner V., Schteingart M.T. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am. J. Hum. Genet. 2005;77:694–708. doi: 10.1086/497348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aldave A.J., Yellore V.S., Yu F., Bourla N., Sonmez B., Salem A.K., Rayner S.A., Sampat K.M., Krafchak C.M., Richards J.E. Posterior polymorphous corneal dystrophy is associated with TCF8 gene mutations and abdominal hernia. Am. J. Med. Genet. A. 2007;143A:2549–2556. doi: 10.1002/ajmg.a.31978. [DOI] [PubMed] [Google Scholar]
- 14.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 16.Lathrop G.M., Lalouel J.M. Easy calculations of lod scores and genetic risks on small computers. Am. J. Hum. Genet. 1984;36:460–465. [PMC free article] [PubMed] [Google Scholar]
- 17.Mehta J.S., Vithana E.N., Tan D.T., Yong V.H., Yam G.H., Law R.W., Chong W.G., Pang C.P., Aung T. Analysis of the posterior polymorphous corneal dystrophy 3 gene, TCF8, in late-onset Fuchs endothelial corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 2008;49:184–188. doi: 10.1167/iovs.07-0847. [DOI] [PubMed] [Google Scholar]
- 18.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sunyaev S., Ramensky V., Koch I., Lathe W., Kondrashov A.S., Bork P. Prediction of deleterious human alleles. Hum. Mol. Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 20.Muraoka O., Ichikawa H., Shi H., Okumura S., Taira E., Higuchi H., Hirano T., Hibi M., Miki N. Kheper, a novel ZFH/deltaEF1 family member, regulates the development of the neuroectoderm of zebrafish (Danio rerio) Dev. Biol. 2000;228:29–40. doi: 10.1006/dbio.2000.9909. [DOI] [PubMed] [Google Scholar]
- 21.Leitch C.C., Zaghloul N.A., Davis E.E., Stoetzel C., Diaz-Font A., Rix S., Alfadhel M., Al-Fadhel M., Lewis R.A., Eyaid W. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 22.Vandewalle C., Van Roy F., Berx G. The role of the ZEB family of transcription factors in development and disease. Cell. Mol. Life Sci. 2009;66:773–787. doi: 10.1007/s00018-008-8465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikeda K., Kawakami K. DNA binding through distinct domains of zinc-finger-homeodomain protein AREB6 has different effects on gene transcription. Eur. J. Biochem. 1995;233:73–82. doi: 10.1111/j.1432-1033.1995.073_1.x. [DOI] [PubMed] [Google Scholar]