

Abstract
Charcot-Marie-Tooth disease (CMT) is the most common cause of inherited peripheral neuropathy, with an estimated frequency of 1/2500. We studied a large family with 17 patients affected by the axonal form of CMT (CMT2). Analysis of the 15 genes or loci known to date was negative. Genome-wide genotyping identified a CMT2 locus in 16q21-q23 between D16S3050 and D16S3106. The maximum two-point LOD score was 4.77 at θ = 0 for marker D16S3050. Sequencing of candidate genes identified a unique mutation, c.986G>A (p.Arg329His), affecting a totally conserved amino acid in the helical domain of cytoplasmic alanyl-tRNA synthetase (AlaRS). A second family with the same mutation and a different founder was then identified in a cohort of 91 CMT2 families. Although mislocation of mutant Arg329His-AlaRS in axons remains to be evaluated, experimental data point mostly to a quantitative reduction in tRNAAla aminoacylation. Aminoacylation and editing functions closely cooperate in AlaRS, and Arg329His mutation could also lead to qualitative errors participating in neurodegeneration. Our report documents in 18 patients the deleterious impact of a mutation in human cytoplasmic AlaRS and broadens the spectrum of defects found in tRNA synthetases. Patients present with sensory-motor distal degeneration secondary to predominant axonal neuropathy, slight demyelination, and no atypical or additional CNS features.
Main Text
Charcot-Marie-Tooth disease (CMT [MIM 118300]) is the most common cause of inherited peripheral neuropathy, with an estimated frequency estimated of 1/2500. CMT is generally transmitted as a dominant trait and presents a high genetic heterogeneity. Both motor and sensory nerves are affected by a progressive distal degeneration, but motor disability in patients is usually more prominent than sensory disorders and is typically represented by distal amyotrophy of legs and gait disturbances.1 Patients can be classified by electromyographic examination as having a demyelinating form (CMT1) with reduced nerve conduction velocities (NCVs) or an axonal form (CMT2) with conserved NCVs. A value of 38 m/s for the motor NCV in the median nerve is most commonly used to separate CMT1 and CMT2.2 Some families present values between 25 and 45 m/s and are considered to have dominant-intermediate forms of CMT (DI-CMT).3 Fifteen genes or loci are known to date for dominant CMT2 or DI-CMT (Table S1, available online) but mutations have not been identified in most CMT2 families.4
We studied a large French family (family 1) that included 17 individuals affected with autosomal-dominant CMT2 and two unaffected but at-risk individuals (IV.1 and IV.8), aged 50 and 30 yrs, respectively, ascertained by neurological examination (Table 1). Mean age at onset was 28 yrs (range: 6–54 yrs). All patients were ambulatory and presented no pyramidal or cerebellar signs. One patient (IV.7) used a wheelchair for occasional assistance only. One patient (IV.5) had severe asymmetric muscular atrophy in the lower left limb (Figure 1). Electromyographic examination demonstrated a motor and sensory denervation in all patients investigated. Mean motor NCV in the median nerve was 41 m/s (range: 32.4–50 m/s). All patients gave their written consent to genetic investigations in accordance with French legislation.
Table 1.
Clinical Features and Electroneuromyographic Data
| Fam. | ID | Age at Onset of First Signs (Yrs) | First Signs in LL before UL | First Presenting Features | Age at Clinical Exam (Yrs) | Weakness Dist. | Sensory Loss Dist. | Reflex Loss Dist. | Use of Walking Stick (Yrs) | Use of Wheelchair (Yrs) | ENMG: Motor or Sensory Alteration | Motor NCV Median Nerve (m/s) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | III.1 | >50 | + | Repeated ankle sprains | 77 | LL | LL | LL | - | - | MS | 32.4 |
| 1 | III.3 | 50 | + | LL weakness | 71 | LL | LL | LL+UL | - | - | MS | 35 |
| 1 | III.5 | 15 | = | Repeated ankle sprains | 67 | LL+UL | LL+UL | LL+UL | - | - | MS | 35.7 |
| 1 | III.6 | 17 | + | Walking difficulties | 61 | LL+UL | LL | LL | 56 | - | MS | NA |
| 1 | IV.1 | AS | AS | AS | 50 | AS | AS | AS | AS | AS | N | |
| 1 | IV.2 | 54 | + | Mild distal LL amyotrophy | 54 | NA | NA | NA | - | - | MS | 45.7 |
| 1 | IV.3 | 45 | + | Foot deformation | 47 | NA | NA | NA | - | - | MS | 40.8 |
| 1 | IV.4 | 45 | = | Repeated ankle sprains | 49 | LL+UL | LL | LL+UL | - | - | MS | 50 |
| 1 | IV.5 | 6 | + | Repeated ankle sprains | 54 | LL | LL | LL | - | - | MS | 45 |
| 1 | IV.7 | 26 | + | Pain and LL weakness | 38 | LL | - | LL+UL | 40 | 42 | MS | 44.5 |
| 1 | IV.8 | AS | AS | AS | 30 | AS | AS | AS | AS | AS | N | |
| 1 | IV.9 | 18 | + | LL and hand weakness | 32 | LL+UL | LL+UL | LL+UL | - | - | MS | 36.5 |
| 1 | IV.10 | 10 | + | Repeated ankle sprains | 30 | LL | LL | LL | - | - | MS | NA |
| 1 | IV.11 | 25 | + | Repeated ankle sprains | 41 | LL | LL | LL+UL | - | - | MS | 40.5 |
| 1 | IV.12 | NA | + | LL weakness | 41 | LL | LL | LL | - | - | MS | NA |
| 1 | IV.13 | 25 | = | LL weakness | 32 | LL+UL | - | LL | - | - | MS | 42.4 |
| 1 | IV.16 | 22 | + | Cramps in LL | 34 | LL | LL | LL | - | - | MS | 43.5 |
| 1 | V.1 | AS | AS | AS | 9 | AS | AS | AS | AS | AS | MS | 46 |
| 1 | V.2 | 15 | + | Repeated ankle sprains | 28 | LL | LL | LL+UL | - | - | NA | |
| 2 | II.1 | 14 | + | Walking difficulties | 32 | LL | LL | LL | - | - | MS | 35–39 |
Abbreviations are as follows: AS, asymptomatic; Dist., distribution; ENMG, electroneuromyography; Fam., family; LL, lower limbs, M, pure motor, MS, motor and sensory; N, normal; NA, not available; UL, upper limbs.
Figure 1.
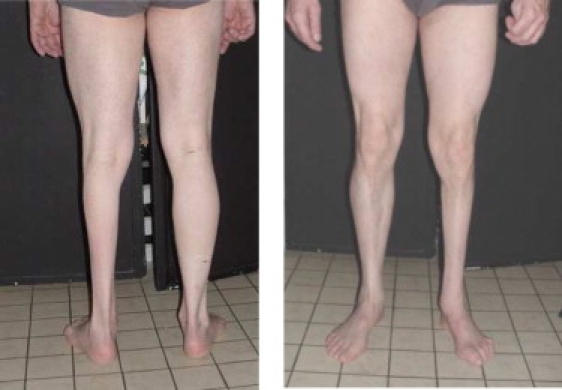
Patient IV.5 of Family 1
As a typical case history, we describe the clinical findings of patient IV.13 (family 1). The first signs of the disease were noticed by this female patient at the age of 25, during her first pregnancy. Symptoms consisted of mild to moderate motor disability of the lower right limb and occasional motor disability of the right hand. Neurological examination was first performed when the patient was 30 yrs of age. Her gait was normal, with no ataxia, but standing on the heels was difficult for her. A bilateral distal motor weakness in the lower limbs was noticed, with a muscular force test score of 4/5 (normal score = 5) for the extensor digitorum brevis muscles. There was no amyotrophy or foot deformation. Vibration sense was mildly diminished in the right lower limb. Deep tendon reflexes were weak at the knees and absent at the ankles. Examination of the upper limbs was normal. There were no tremor or pyramidal-tract signs, and no Argyll-Robertson pupil sign was observed. Motor NCVs were 43.4 m/s on the right median nerve and 41.4 m/s on the left median nerve (elbow-wrist), with reduced amplitudes, respectively, of 15% and 11%. Right ulnar nerve motor NCV was 47.5 m/s, with normal amplitudes. Sensory NCV was 50 m/s on the right radial nerve. Motor and sensory responses were absent in the lower limbs (in popliteal nerves). The patient is now 32 yrs old. Clinical findings are unchanged.
Sequencing of coding exons and linkage analysis were negative for all CMT2 and DI-CMT genes known to date. Twelve patients (III.1, IV.1, IV.2, IV.4, IV.7, IV.8, IV.9, IV.10, IV.11, IV.12, IV.16, and V.2) were selected for SNP genotyping. A genome-wide scan was performed with the Affymetrix Genome-Wide Human SNP 6.0 Array. A total of 889,406 SNPs were genotyped for each of the patients studied, and a unique contig of 18 SNPs was found to be coherent with transmission of the disease in 16q21-q23 between SNPs rs16962499 and rs446695 (59.3 Mb and 76.6 Mb in NCBI build 36.3). Microsatellite haplotypes for the whole family (Figure 2) showed closer recombinations (patients III.5, IV.3, and IV.8) and refined the new CMT2 locus as being a region of 7.1 cM and 10.2 Mb between markers D16S503 and D16S3018 (62.5 Mb–72.7 Mb in NCBI build 36.3). A maximum two-point parametric LOD score of 4.77 was observed at θ = 0 for marker D16S3050 (Table S2).
Figure 2.
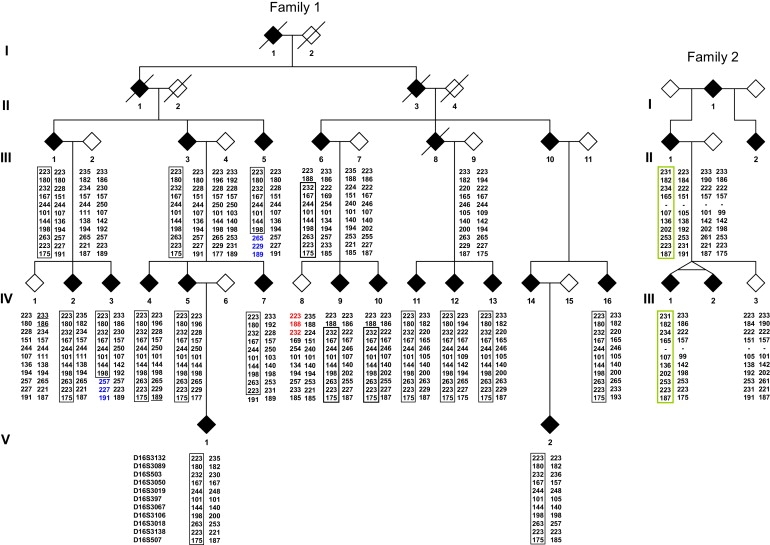
Pedigrees and Haplotypes
Filled symbols indicate affected individuals. Empty symbols indicate unaffected individuals. Identical alleles on the disease-causing haplotypes are boxed for each family. Closest recombinants in family 1 are shown in red (centromeric side) or blue (telomeric side).
In a cluster of ∼130 expressed sequences or genes, the following were considered candidate genes for CMT2 and were sequenced: DYNC1LI2 (dynein cytoplasmic 1 light intermediate chain 2 [MIM 611406], 65.3 Mb in NCBI build 36.3), HSF4 (heat shock transcription factor 4 [MIM 602438], 65.8 Mb), TPPP3 (tubulin polymerization-promoting protein family member 3, no MIM entry, 66.0 Mb), and AARS (cytoplasmic alanyl-tRNA synthetase [MIM 601065], 68.8 Mb). The lysyl-tRNA synthetase gene (KARS [MIM 601421]) located at 74.2 Mb in 16q23 was also sequenced, although it was excluded by recombination events. We identified only one missense mutation, c.986G>A (p.Arg329His), in exon 8 of AARS (Figure S1). All affected patients were found to be heterozygous for the mutation, and all unaffected individuals showed a normal sequence. An additional series of 91 dominant and unrelated CMT2 cases were then analyzed. For these cases, mutation screening had been unsuccessful in at least MFN2 (MIM 608507), GJB1 (MIM 304040), and MPZ (MIM 159440). A second French family (family 2) was identified as having the same c.986G>A mutation, which also co-segregated with the disease. Microsatellite analysis demonstrated a different founder for the two families (Figure 2). The mutation was not found in 500 unrelated anonymous DNA samples used as controls (1000 chromosomes).
Highly conserved and specific aminoacyl-tRNA synthetases attach amino acids to their cognate tRNAs and are key enzymes for the exact translation of the genetic code into proteins. A single G3:U70 base pair in its acceptor arm marks tRNAAla for alanine charging by aminoacylation in the catalytic site of alanyl-tRNA synthetase (AlaRS).5 Unlike almost all amino synthetases, AlaRS recognizes tRNAAla uniquely by its acceptor arm and not by additional determinants in the anticodon stem.5 Particularly challenging is the differentiation among alanine, serine, and glycine in the catalytic domain, and, early in evolution, AlaRSs integrated a modular editing domain to hydrolyse-mischarged tRNAAla.6–8 Aminoacylation and editing domains closely cooperate and link to the acceptor arm of the same tRNA, using both sides of the G3:U70 determinant.9 Conservation of sequence and of modular organization from E. coli to H. sapiens AlaRSs is one of the highest observed among all synthetases.10 From the N teminal to the C terminal, E. coli and H. sapiens AlaRS present an aminoacylation domain (AD), a middle helical domain (HD) and an editing domain (ED)9,11 (Figure 3). In E. coli, a so-called C-Ala domain is responsible for efficient editing by bringing together AD and ED12 (Figure 3). H. sapiens AlaRS is monomeric10 or homodimeric13 and differs mostly from E. coli AlaRS by its C-terminal sequence downstream ED. Human cytoplasmic and mitochondrial AlaRS are encoded by two different nuclear genes, AARS and AARS2 (MIM 612035), respectively.14 AARS is composed of 21 exons and codes for 968 amino acids. Primers and conditions for AARS sequencing are in Table S3.
Figure 3.
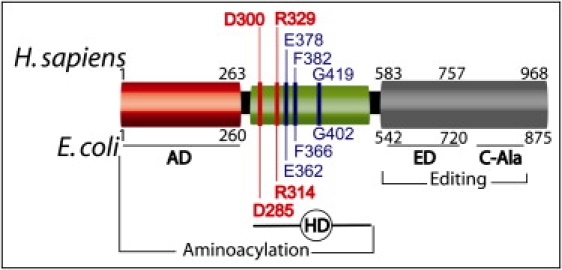
Modular Organization of H. sapiens and E. coli AlaRS
Abbreviations are as follows: AD, aminoacylation domain; HD, helical domain; ED, editing domain. In E. coli, the C-Ala domain brings together AD and ED in docking models, and an N-terminal fragment of 461 amino acids (AD+HD) is sufficient for complete and specific aminoacylation of tRNAAla.
Arginine 329 is located in the middle helical domain of H. sapiens AlaRS and is the ortholog of R314 in E. coli (Figure 4). From crystal structure, it was deduced that E. coli R314 directly interacts with the aromatic ring of F366 (human F382) and has water-mediated interactions with E362 (human E378) and G402 (human G419).11 The network composed of these four amino acids is strictly conserved in Archeobacteria and Eubacteria,11 as well as from E. coli to H. sapiens (Figure 4). Extensive mutagenesis experiments demonstrated that among 37 conserved candidate amino acids with side chains that are able to mediate hydrogen-bound interactions in the E. coli AlaRS helical domain (Figure 4), D285 and R314 are the two major determinants for binding and efficient aminoacylation of tRNAAla.15 The R314A mutant shows a ∼700-fold reduction in aminoacylation as a result of a reduced binding of tRNAAla by its acceptor arm in the catalytic site. Mutants R314A and R314D were unable to complement AlaRS null strains, and the R314K mutant only partially rescued cell growth.15
Figure 4.
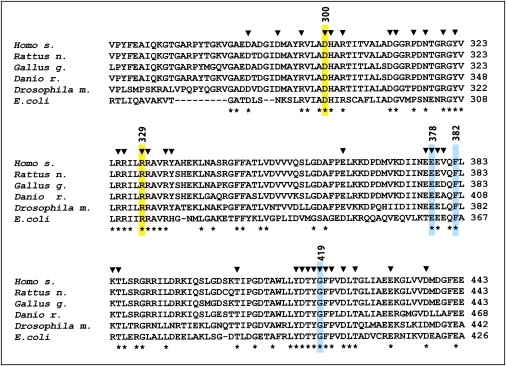
Alignment of AlaRS Helical Domains from E. coli to H. sapiens
GenBank access for proteins were as follows: NP_001596 (Homo sapiens), ACT28094 (Escherichia coli), NP_001093987 (Rattus norvegicus), NP_001005836 (Gallus gallus), NP_001037775 (Danio rerio), and AAF05593 (Drosophila melanogaster). Fully conserved amino acids are indicated with an asterisk (∗) below the alignment. Arrowheads (▾) on top of the alignment indicate amino acids that have been studied by mutagenesis.15E. coli D285 and R314 (H. sapiens D300, and R329) are indicated in yellow. E. coli R314 interacts with E362, F366, and G402, shown in blue (H. sapiens E378, F382, and G419).
To our knowledge, no amino acid other than arginine has been described in the three kingdoms of life as playing the key role of R329 in the middle helical domain of AlaRS. In two independent CMT2 families, we found a substitution of R329 by histidine. Arginine 329 is located in the AlaRS α10 helix.8 In α helices, arginine and lysine side chains have comparable properties and are significantly more flexible than histidine side chain.16,17 Moreover, arginine and lysine are strong proton acceptors, whereas histidine is a variably protonated imidazole ring. Given that even replacement of R314 by lysine is deleterious15 and none of the adjacent arginines can correct a defect of the fully conserved R314,15 we postulate that the R329H substitution is deleterious in H. sapiens AlaRS. Crystallographic studies support the hypothesis that the deleterious effect is likely to be related to modifications within the strictly conserved network R329-E378-F382-G419.11
tRNA synthetase deficiencies have already been implicated in neurological disorders such as leukoencephalopathy, spinal cord disturbances, and distal spinal muscular atrophy.13 Rare CMT cases have been related to defects in glycyl-tRNA synthetase18 (GlyRS [MIM 601472]) and in tyrosyl-tRNA synthetase19 (TyrRS [MIM 608323]). Exact roles of GlyRS and TyrRS mutants in CMT are still being investigated.13 Among other possibilities, defective aminoacylation and abnormal distribution in axons are postulated for both GlyRS13,20 and TyrRS19 mutants. In our observation, functional assays strongly suggest a modified affinity for tRNAAla and a reduced aminoacylation efficiency.15 However, findings in GlyRS and TyrRS indicate that aminoacylation function could be differentially affected by CMT-causing mutations and therefore might not be a major determinant in the disease.13 Thus, additional functional studies are necessary to analyze loss of function or neuron-specific effects resulting from a R329H mutation. The hypothesis of a deleterious mislocalization of mutant R329H AlaRS in axons remains to be evaluated. Charging errors could also be considered, because defective binding of the tRNAAla acceptor arm in the catalytic site for R314 and R329 mutants15 could modify the cooperation between aminoacylation and editing. A mouse model showed that a homozygous defect in AlaRS editing is responsible for mischarging of tRNAAla with serine or glycine and leads to protein misfolding and neurodegeneration, indicating that AlaRS fidelity errors are sufficient for neurodegeneration.21 R314A-AlaRS also has a residual binding of tRNAAla that is not distinguishable from a nonspecific background of tRNALys binding,15 suggesting that binding of noncognate tRNAs could be another source of qualitative errors for R314 and R329 mutants.
We identified two unrelated families with the same amino acid variation. Because direct neurological examination in family 2 was limited to proband II.1, interfamilial comparison was difficult to establish. Nonetheless, clinical findings for the family 2 proband were similar to those found in several age-matched patients of family 1 (IV.10, IV.12, and IV.16), and identical age at onset together with analogous slight demyelinating signs for the same duration of disease were found in patient IV.9. Monozygous twins III.1 and III.2 are both reported as having only mild motor trouble in the feet and Achilles' tendons areflexia at the age of 13 yrs, which fits well with a disease limited mainly to the lower limbs, as illustrated by family 1. The R329H mutation occurred independently in the two families, indicating that dinucleotide CG in codon 329 could be a mutational hotspot and that data on other families with the same mutation might allow additional interfamilial comparisons. In both families, patients with the Arg329His AlaRS mutation present with sensory-motor distal degeneration secondary to predominant axonal neuropathy and with absent or slight demyelination. Neurodegeneration implicated peripheral nerves with no evident brainstem or spinal cord involvement. Asymmetric amyotrophy is found in one out of the 18 cases reported here but is also observed in 10%–15% of CMT2 patients.4 Distal degeneration of peripheral motor nerves is by now a common phenotypic feature related to mutations in human amino synthetases. Data from additional families would help to confirm and delineate involvement of cytoplasmic AlaRS mutations in CMT or in other neurodegenerative disorders. Other tRNA synthetase genes could be good candidates for other subtypes of CMT disease.
Acknowledgments
We are grateful to the patients in each of the two families for their active cooperation. We thank Muriel Bost, Patrice Bouvagnet, and Antoon Vandenberghe for their help with the project, and we thank Susan Gamon for reviewing the manuscript. This work was supported by the Association pour le Développement de la Neurogénétique (project 09-02).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
easyLINKAGE version 5.02, http://sourceforge.net/projects/easylinkage/
The European Bioinformatics Institute (EMBL-EBI), http://www.ebi.ac.uk/Tools/clustalw2/index.html
Ensembl Homo sapiens assembly, http://www.ensembl.org/Homo_sapiens/Info/Index
Marshfield Center for Medical Genetics Mammalian Genotyping Service, http://research.marshfieldclinic.org/genetics/GeneticResearch/compMaps.asp
NCBI Build 36.3, http://www.ncbi.nlm.nih.gov/projects/mapview/map_search.cgi?taxid=9606&build=previous
NCBI Map Viewer, http://www.ncbi.nlm.nih.gov/mapview/
NCBI Nucleotide Database, http://www.ncbi.nlm.nih.gov/nucleotide
NCBI Protein Database, http://www.ncbi.nlm.nih.gov/protein/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Dyck P.J., Thomas P.K., Lambert E.H., Bunge R. Third Edition. W.B. Saunders; Philadelphia: 1993. Peripheral neuropathy. [Google Scholar]
- 2.Harding A.E., Thomas P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103:259–280. doi: 10.1093/brain/103.2.259. [DOI] [PubMed] [Google Scholar]
- 3.Davis C.J.F., Bradley W.G., Madrid R. The peroneal muscular atrophy syndrome: clinical, genetic, electrophysiological and nerve biopsy studies. I. Clinical, genetic and electrophysiological findings and classification. J. Genet. Hum. 1978;26:311–349. [PubMed] [Google Scholar]
- 4.Bienfait H.M., Baas F., Koelman J.H., de Haan R.J., van Engelen B.G., Gabreëls-Festen A.A., Ongerboer de Visser B.W., Meggouh F., Weterman M.A., De Jonghe P. Phenotype of Charcot-Marie-Tooth disease Type 2. Neurology. 2007;68:1658–1667. doi: 10.1212/01.wnl.0000263479.97552.94. [DOI] [PubMed] [Google Scholar]
- 5.Giegé R., Sissler M., Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–5035. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beebe K., Ribas De Pouplana L., Schimmel P. Elucidation of tRNA-dependent editing by a class II tRNA synthetase and significance for cell viability. EMBO J. 2003;22:668–675. doi: 10.1093/emboj/cdg065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahel I., Korencic D., Ibba M., Söll D. Trans-editing of mischarged tRNAs. Proc. Natl. Acad. Sci. USA. 2003;100:15422–15427. doi: 10.1073/pnas.2136934100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sokabe M., Ose T., Nakamura A., Tokunaga K., Nureki O., Yao M., Tanaka I. The structure of alanyl-tRNA synthetase with editing domain. Proc. Natl. Acad. Sci. USA. 2009;106:11028–11033. doi: 10.1073/pnas.0904645106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beebe K., Mock M., Merriman E., Schimmel P. Distinct domains of tRNA synthetase recognize the same base pair. Nature. 2008;451:90–93. doi: 10.1038/nature06454. [DOI] [PubMed] [Google Scholar]
- 10.Schimmel P., Ripmaster T. Modular design of components of the operational RNA code for alanine in evolution. Trends Biochem. Sci. 1995;20:333–334. doi: 10.1016/s0968-0004(00)89067-2. [DOI] [PubMed] [Google Scholar]
- 11.Swairjo M.A., Otero F.J., Yang X.L., Lovato M.A., Skene R.J., McRee D.E., Ribas de Pouplana L., Schimmel P. Alanyl-tRNA synthetase crystal structure and design for acceptor-stem recognition. Mol. Cell. 2004;13:829–841. doi: 10.1016/s1097-2765(04)00126-1. [DOI] [PubMed] [Google Scholar]
- 12.Guo M., Chong Y.E., Beebe K., Shapiro R., Yang X.L., Schimmel P. The C-Ala domain brings together editing and aminoacylation functions on one tRNA. Science. 2009;325:744–747. doi: 10.1126/science.1174343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antonellis A., Green E.D. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet. 2008;9:87–107. doi: 10.1146/annurev.genom.9.081307.164204. [DOI] [PubMed] [Google Scholar]
- 14.Chihade J.W., Brown J.R., Schimmel P.R., Ribas De Pouplana L. Origin of mitochondria in relation to evolutionary history of eukaryotic alanyl-tRNA synthetase. Proc. Natl. Acad. Sci. USA. 2000;97:12153–12157. doi: 10.1073/pnas.220388797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribas de Pouplana L., Buechter D., Sardesai N.Y., Schimmel P. Functional analysis of peptide motif for RNA microhelix binding suggests new family of RNA-binding domains. EMBO J. 1998;17:5449–5457. doi: 10.1093/emboj/17.18.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stapley B.J., Doig A.J. Free energies of amino acid side-chain rotamers in alpha-helices, beta-sheets and alpha-helix N-caps. J. Mol. Biol. 1997;272:456–464. doi: 10.1006/jmbi.1997.1250. [DOI] [PubMed] [Google Scholar]
- 17.Koch K., Zöllner F., Neumann S., Kummert F., Sagerer G. Comparing bound and unbound protein structures using energy calculation and rotamer statistics. In Silico Biol. 2002;2:351–368. [PubMed] [Google Scholar]
- 18.Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordanova A., Irobi J., Thomas F.P., Van Dijck P., Meerschaert K., Dewil M., Dierick I., Jacobs A., De Vriendt E., Guergueltcheva V. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- 20.Antonellis A., Lee-Lin S.Q., Wasterlain A., Leo P., Quezado M., Goldfarb L.G., Myung K., Burgess S., Fischbeck K.H., Green E.D. Functional analyses of glycyl-tRNA synthetase mutations suggest a key role for tRNA-charging enzymes in peripheral axons. J. Neurosci. 2006;26:10397–10406. doi: 10.1523/JNEUROSCI.1671-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J.W., Beebe K., Nangle L.A., Jang J., Longo-Guess C.M., Cook S.A., Davisson M.T., Sundberg J.P., Schimmel P., Ackerman S.L. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.