

Abstract
We report a large Chinese family with X-linked postlingual nonsyndromic hearing impairment in which the critical linkage interval spans a genetic distance of 5.41 cM and a physical distance of 15.1 Mb that overlaps the DFN2 locus. Mutation screening of the PRPS1 gene in this family and in the three previously reported DFN2 families identified four different missense mutations in PRPS1. These mutations result in a loss of phosphoribosyl pyrophosphate (PRPP) synthetase 1 activity, as was shown in silico by structural analysis and was shown in vitro by enzymatic activity assays in erythrocytes and fibroblasts from patients. By in situ hybridization, we demonstrate expression of Prps1 in murine vestibular and cochlea hair cells, with continuous expression in hair cells and postnatal expression in the spiral ganglion. Being the second identified gene associated with X-linked nonsyndromic deafness, PRPS1 will be a good candidate gene for genetic testing for X-linked nonsyndromic hearing loss.
Main Text
X-linked deafness is a clinically and genetically heterogeneous disorder that accounts for ∼5% of all congenital deafness1 and < 2% of nonsyndromic hearing loss (NSHL).2 Despite its relative rarity as compared to dominant and recessive forms of NSHL, the first nuclear gene implicated in hereditary NSHL was POU3F4 (MIM 300039), identified in 1995 as the cause of X-linked deafness at the DFN3 locus (MIM 304400).3 The DFN2 locus (MIM 304500), which was mapped before the DFN3 locus, is associated with NSHL that is phenotypically complex. Several families mapping to this locus segregate postlingual progressive NSHL,4–6 although a large, four-generation, British American pedigree segregating congenital profound NSHL was the first DFN2 family to be identified. Obligate female carriers in this family have mild to moderate NSHL that is more pronounced in the higher frequencies.
Manolis and colleagues reported the second DFN2 family, an American family with five affected males and two obligate female carriers. The affected males all showed an upward-sloping audio profile, with severe hearing impairment in the low and middle frequencies and better hearing in the high frequencies. The loss, which was postlingual and progressive, was initially noted between 7 and 20 years of age. The obligate female carriers in this family had less hearing impairment.5 In 2004, Cui and colleagues identified a large Chinese family with X-linked postlingual NSHL that also mapped to the DFN2 locus.6
In this study, we present another large Chinese family (GZ-Z052) segregating X-linked NSHL. This family spans five generations and includes 106 members. Fourteen persons with hearing loss and 29 normal-hearing persons participated in this research, which was approved by the ethics committee of the Chinese PLA General Hospital. Seventeen family members (nine males and eight females) had hearing impairment that ranged from mild to profound in degree. Age at onset of hearing impairment was between 5 and 15 years for males and in the fifth decade for females.
Affected males exhibited symmetric, progressive, severe to profound hearing loss with flat-shaped audio profiles at 24–50 years of age (Figure 1A and Table 1). One affected male (IV-21) also had a ten-day history of gentamicin exposure (dosage unknown) for bronchitis at age 5 and first noted bilateral hearing impairment the following year. However, from his audio profile, which is identical to that of the other affected males in the pedigree, it appears that his hearing loss is unlikely to be secondary to aminoglycoside exposure.
Figure 1.
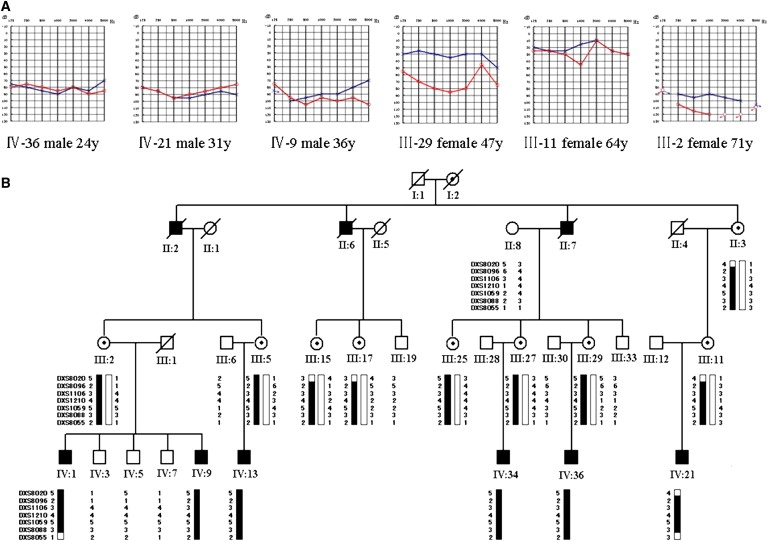
Chinese Family GZ-Z052 with X-linked Heriditary NSHL
(A) Audiograms of some affected male and female subjects (red, right ear; black, left ear). IV-21, who has a history of the use of aminoglycosides, had exactly same audiogram pattern as that of IV-9 and IV-36, suggesting that his hearing impairment was not due to aminoglycoside exposure. The hearing impairment of female carriers is progressive but not completely linked with increasing age.
(B) Haplotype analysis of the Chinese family GZ-Z052. The segregating haplotype associated with the NSHL is indicated by a black bar, delimited by markers DXS8020 and DXS8055 on chromosome Xq22.
Table 1.
Summary of Clinical Data for Some of the Affected Individuals of Chinese Family GZ-Z052
| Patient | Sex |
Age (Yrs) |
Use of Amino-Glycosides |
Hearing Test PTA (dB) |
Tinnitus |
Uric Acid Level (μMOL/L) |
Visual Impairment Test | Peripheral Neuropathy Test | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| At Testing | At Onset | Left Ear | Right Ear | Plasmaa | Urineb | ||||||
| IV-1 | Male | 50 | 10 | No | 106 | 106 | No | 231 | 3846 | Normal | Normal |
| IV-9 | Male | 36 | 14 | No | 88 | 96 | No | NT | NT | NT | NT |
| IV-13 | Male | 30 | 8 | No | 69 | 71 | Yes | 289 | 4148 | Normal | Normal |
| IV-21 | Male | 31 | 6 | Yes | 89 | 84 | Yes | 399 | 2785 | Normal | Normal |
| IV-34 | Male | 28 | 5 | No | 82 | 87 | No | 322 | 3138 | Normal | Normal |
| IV-36 | Male | 24 | 8 | No | 85 | 78 | Yes | 380 | 4624 | Normal | Normal |
| III-2 | Female | 71 | 50 | No | 94 | 113 | No | 288 | 3187 | Normal | Normal |
| III-5 | Female | 56 | 53 | No | 57 | 33 | Yes | 156 | 3541 | Normal | Normal |
| III-11 | Female | 64 | 55 | No | 21 | 27 | No | 310 | 3166 | Normal | Normal |
| III-15 | Female | 57 | 50 | No | 41 | 60 | Yes | NT | NT | NT | NT |
| III-17 | Female | 55 | 51 | No | 63 | 22 | No | 304 | 3535 | Normal | Normal |
| III-25 | Female | 53 | 40 | No | 31 | 61 | No | NT | NT | NT | NT |
| III-29 | Female | 47 | 45 | No | 33 | 70 | No | 298 | 5078 | Normal | Normal |
NT, not tested; PTA, pure tone average.
The normal range is 90-420 μmol/L.
The normal range is 2400-5900 μmol/L.
Obligate female carriers had either symmetric or asymmetric hearing loss that varied from mild to moderate in degree. Asymmetrical and unilateral mild or moderate hearing impairment was observed in individuals III-5, III-11, III-17, III-25, and III-29 at ages ranging from 47 to 64 years, whereas bilateral severe to profound hearing loss was documented in individual III-2 at age 71 (Figure 1A). One female carrier, III-27, had completely normal hearing at age 52.
Distortion products otoacoustic emissions (DPOAEs) were measured in four affected males (IV-1, IV-9, IV-34, and IV-36) and two female carriers (III-27 and III-29) and were found to be significantly below the noise floor bilaterally in the males and in the right ear of female carrier III-29 (data not shown). Female carrier III-27 showed a completely normal result of the DPOAEs test. Three out of six affected males reported low-frequency tinnitus at the onset of hearing loss (Table 1). Computed tomography of the temporal bones in two affected males was normal, as was comprehensive clinical testing including blood and urine analysis, biochemical examination of blood (which specifically excluded gout), B-ultrasonography of the urinary system, and electrocardiography. No vestibular symptoms were reported (Table 1).
Linkage analysis, completed with the use of DNA from 23 persons, generated a two-point LOD score of 2.45 at [θ] = 0 for marker DXS1106 on Xq22. Genotypic data for 11 additional markers flanking DXS1106 resulted in a maximum two-point LOD score of 4.25 at [θ] = 0 with marker DXS8096 (Table S1, available online) and defined the boundaries of the disease interval as DXS8020 (proximal, defined by recombination events occurring in III-11,III-15, III-17, and IV-21) and DXS8055 (distal, defined by a recombination event in IV-1 and IV-21), a 5.4 -cM genetic interval that overlaps DFN2 (Figure 1B).
Of the 119 positional candidate genes in this interval, we selected 14 genes for mutation screening—GLA (MIM 300644), HNRPH2 (MIM 300610), NGFRAP1 (MIM 300361), TCEAL1 (MIM 300237), MORF4L2 (MIM 300409), ARMCX2 (MIM 300363), PLP1 (MIM 300401), RNF128 (MIM 300439), PRPS1 (MIM 311850), MID2 (MIM 300204), COL4A5 (MIM 303630), NXT2 (MIM 300320), PAK3 (MIM 300142), and TIMM8A (MIM 300356)—on the basis of their inclusion in the Morton Cochlear EST database.7 Sequence analysis of seven coding exons of PRPS1 and about 100 base pairs (bp) of their flanking intronic sequences in two affected members (IV-1 and IV-36) and one unaffected member (IV-7) identified a G-to-A transition at position 193 in exon 2 (Figure 2C) that results in an Asp65-to-Asn (D65N) substitution in the α helix at the N terminus. Direct sequence analysis demonstrated that this mutation cosegregated with the deafness phenotype in 43 persons in the extended family.
Figure 2.
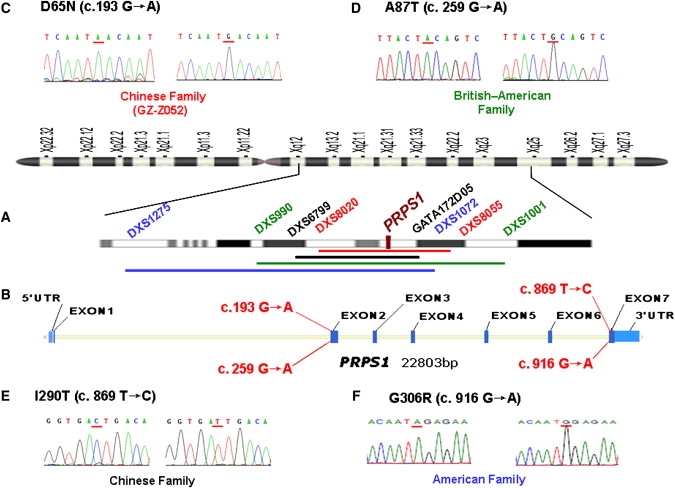
Mapping Intervals and Mutation Analysis of the Four DFN2 Families
(A) Idiogram of the X chromosome with the described DFN2 loci. The linkage intervals and flanking markers of four DFN2 families are indicated in colored fonts and bars (red, Chinese family GZ-Z052; green, British American family; black, Chinese family reported by Cui et al.;6 blue: American family).
(B) The genomic structure of PRPS1 depicting the positions of four DFN2 mutations.
(C–F) DNA sequence chromatograms showing four different missense mutations identified in affected males of four DFN2 families, compared to wild-type controls.
In affected individuals from the family reported by Cui et al.,6 we identified a T-to-C transition affecting the nucleotide position 869 in exon 7 of PRPS1 (Figure 2E) and leading to an Ile290-to-Thr (I290T) substitution at the C terminus. Both the D65 and I290 amino acids are highly conserved from zebrafish to human, and neither mutation was seen in 1025 unrelated control chromosomes of Chinese background (data not shown), supporting the pathogenicity of both mutations.
In the British American DFN2 family reported by Tyson and colleagues, we identified a G-to-A transition at nucleotide position 259 (Figure 2D) that replaces a nonpolar small hydrophobic amino acid—alanine—with an uncharged, polar, larger amino acid— threonine (A87T), and in the American DFN2 family reported by Manolis et al., we identified a G-to-A transition at nucleotide 916 that results in a glycine-for-arginine exchange at codon 306 (G306R) (Figure 2F). Both A87 and G306 are also evolutionarily conserved across species from zebrafish to human and were absent in 1475 control Chinese chromosomes and 450 chromosomes of European descent.
PRPS1 encodes phospho-ribosylpyrophosphate synthetase 1 (PRS-I; EC 2.7.6.1), which catalyzes the reaction of ribose-5-phosphate (R5P) with ATP to yield AMP and PRPP (5-phosphoribosyl-1-pyrophosphate), which is necessary for the de novo and salvage pathways of purine-, pyrimidine-, and pyridine-nucleotide biosynthesis. The open reading frame of human PRPS1 consists of 957 bp coding for 318 amino acid residues.8 Eriksen et al. reported the structure of B. subtilis phosphoribosyl pyrophosphate synthetase and analyzed the structural basis of the catalytic mechanism, revealing several regions and residues important for binding of substrates (ATP and R5P) and effectors (ADP, Pi, and Mg2+).9 The enzyme requires phosphate for activation and uses Mg2+ for activity, and it is subject to purine-nucleotide (ADP and GDP) feedback inhibition.10 The crystal structure of human PRS-I reveals that the active site comprises the binding sites for ATP and R5P and consists of three primarily structural elements, namely the flexible loop (residues Phe92–Ser108), the PPi (pyrophosphate)-binding loop (residues Asp171–Gly174), and the flag region (residues Val30–Ile44 of an adjacent subunit), respectively.11 Any structural changes of the above three elements would be likely to influence the catalytic specificity and efficiency of PRS-I and alter its enzymatic activity. On the basis of the structural analysis of the wild-type PRS-I in complex with AMP (PDB ID: 2HCR), we used PyMOL to predict the likely functional impact of D65N, A87T, I290T, and G306R on PRS-I enzymatic activity. Residue Asp65 (on the α2 helix) interacts with the residue Lys34 from loop3 (on the middle of the flag region) by hydrogen bonds and firms the configuration of loop3. The replacement of the negatively charged Asp with a polar and uncharged Asn (D65N) changes the configuration of the loop3 and impairs the ATP-binding ability of the adjacent PRS-I subunit (Figure 3A). Ala87 is located at the middle of the β5 strand (the second β strand in an anti-parallel β sheet, also one of end points of flexible ATP-binding loop). The substitution of the hydrophobic Ala with a polar and uncharged Thr (A87T) may alter the secondary structure of the whole antiparallel beta sheet and would thus depress the ATP-binding efficiency (Figure 3B). The PRS-I protein contains two primary domains that are connected by two short, flexible loops (Leu145-Ala147, loop11; Ile290-Ser293, loop22). The I290T substitution is located at the beginning of loop22. Replacement of a hydrophobic Ile with a polar and uncharged Thr is predicted to change the configuration of loop22, and it may thus alter the enzymatic activity (Figure 3C). The Gly306 residue is located at the middle of loop23, very close to the allosteric site of PRS-I. Replacement of the polar and uncharged residue Gly with a positively charged, much larger residue Arg (G306R) tends to influence the allosteric regulatory mechanism of the enzyme (Figure 3D).
Figure 3.
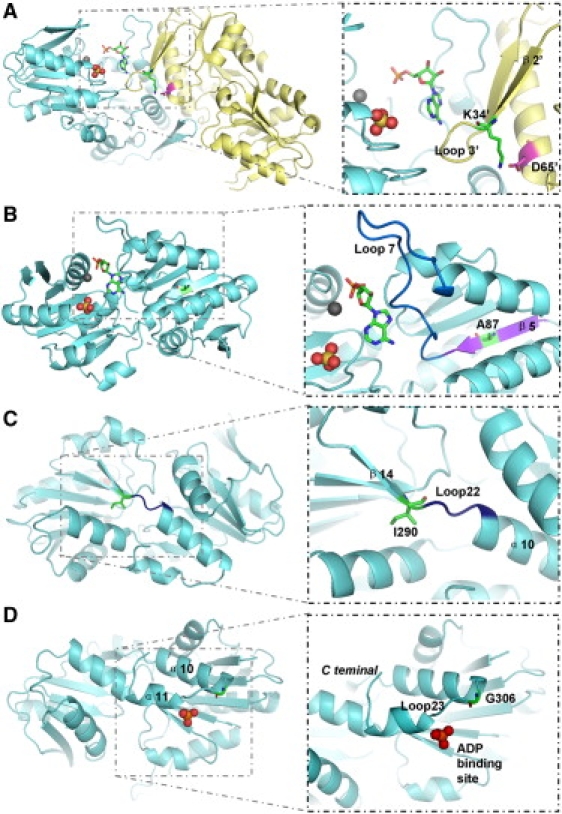
Structure of PRS-I in Complex with AMP
The substrate-binding pocket of PRS-I consists of a metal ion-binding site (gray sphere), an ATP-binding site (represented by AMP moiety of ATP as sticks), and an R5P-binding site (a SO42− ion occupies the position of the 5′ phosphate of R5P). In this figure, two different asymmetric subunits of PRS-I (aquamarine and pale yellow) form a part of the hexamer interface, and all of the structural elements in the pale yellow subunit are tagged with an apostrophe.
(A) Location of Asp65′ (magenta sticks) and Lys34′ (green sticks between the β2′ strand and loop3′).
(B) Location of Ala87 (green sticks in the purple β5 strand) and the ATP-binding loop (marine loop7).
(C) Location of Ile290 (green sticks in the β14 strand) and of loop22 (deep blue).
(D) Location of Gly306 (green sticks in the β14 strand) and the allosteric site of PRS-I (an SO42− ion occupies the position of the β-phosphate of ADP).
Expression analysis performed by quantitative real-time PCR on total RNA isolated from leukocytes via the RNA Blood Mini Kit (QIAGEN, Valencia, CA, USA) showed that the average expression level of PRPS1 in four affected males (IV-1, IV-9, IV-34, and IV-36) of family GZ-Z052 was comparable to the expression level in two female carriers (III-27 and III-29), two unaffected males (IV-5 and IV-7), and two unrelated controls (Table S2). To assess the functional consequence of the D65N mutation, we measured PRS-I enzymatic activity in erythrocytes from four affected males (IV-1, IV-9, IV-34, and IV-36), two carrier females (III-27 and III-29), and two unaffected males (IV-5 and IV-7) of family GZ-Z052, as well as two unrelated healthy controls (C-1 and C-2). (Standard deviations were calculated from three experiments.) The assay is based on high-performance liquid chromatography measurement of AMP, which is produced in equimolar amounts with PRPP. The preparation of hemolysate and cultured fibroblasts from skin biopsies, as well as the details of the PRS-I enzymatic activity assay, have been previously described.12–14
Statistically significant reduction in PRS-I activity in erythrocytes was observed (p < 0.05, calculated by CHISS t test) when PRS-I activity in affected males was compared to that in unaffected males and unrelated healthy controls. In the two female carriers that we studied, no significant differences in PRS-I activity were noted in the same comparisons (Table 2). In cultured skin fibroblasts from affected males IV-9 and IV-36, PRS-I activity was 74.24 and 68.07 nmol/mg/hr, respectively, which was lower than the activity observed in unaffected male,IV-7 (129.16 nmol/mg/hr) (Table 3).
Table 2.
PRS-I Activity Assay in Erythrocytes from Family GZ-Z052
| Individual | Sex | Mutation Carrier? | Activity (± SD) (nmol/mg/hr)a | Percentageb |
|---|---|---|---|---|
| Affected Males | ||||
| IV-1 | M | yes | 4.22 (± 1.37) | 38.5 |
| IV-9 | M | yes | 6.17 (± 0.31) | 56.3 |
| IV-34 | M | yes | 6.80 (± 0.21) | 62.1 |
| IV-36 | M | yes | 7.55 (± 0.10) | 68.9 |
| Female Carriers | ||||
| III-27 | F | yes | 11.29 (± 0.21) | 103.1 |
| III-29 | F | yes | 9.73 (± 0.10) | 88.9 |
| Unaffected Males | ||||
| IV-5 | M | no | 9.89 (± 1.78) | 90.3 |
| IV-7 | M | no | 10.27 (± 0.04) | 93.8 |
| Unrelated Controls | ||||
| C-1 | F | no | 10.87 (± 1.26) | 99.3 |
| C-2 | M | no | 12.77 (± 1.16) | 116.6 |
The standard deviations is calculated from three experiments.
Individual PRS-I activity / mean of the PRS-I activity of unaffected males and controls.
Table 3.
PRS-I Activity Assay in Fibroblasts from Family GZ-Z052
| Individual | Sex | Mutation Carrier? | Activity (± SD) (nmol/mg/hr)a | Percentageb |
|---|---|---|---|---|
| Affected Males | ||||
| IV-9 | M | yes | 74.24 (± 1.81) | 57.5 |
| IV-36 | M | yes | 68.07 (± 1.04) | 52.7 |
| Carriers | ||||
| III-27 | F | yes | 107.85 (± 11.36) | 83.5 |
| III-29 | F | yes | 84.03 (± 4.56) | 65.1 |
| Unaffected Male | ||||
| IV-7 | M | no | 129.16 (± 1.19) | 100 |
The standard deviations is calculated from three experiments.
Individual PRS-I activity / PRS-I activity of the unaffected male.
By in situ hybridization in the murine cochlea (as described previously15), at embryonic day 18.5 (E18.5) Prps1 expression was observed in hair cells, Claudius cells, and the greater epithelial ridge (GER). Prps1 was also expressed in utricular and crista hair cells (data not shown). By postnatal day 6 (P6), expression of Prps1 could be detected in the spiral ganglion cells in addition to hair cells and Claudius cells, although expression in the GER was reduced (Figure 4).
Figure 4.
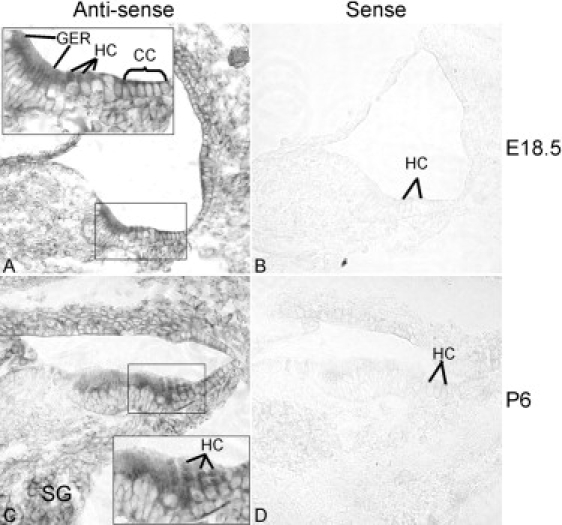
Prps1 Expression in the Mouse Inner Ear
(A) With an antisense probe, Prps1 was observed at E18.5 as robustly expressed in the cochlear hair cells (HC), the Claudius cells (CC), and the greater epithelial ridge (GER), but not in other cochlear supporting cells.
(B) A sense control probe did not produce any signals.
(C) At postnatal day 6, HC and CC expression of Prps1 was maintained, whereas expression in the GER was reduced. Prps1 was also expressed in the spiral ganglions (SG) at this stage.
(D) A sense control probe did not produce any signal. Magnification 20×.
To date, seven missense mutations (D51H, L128I, N113S, D182H, A189V, H192L, and H192Q) have been identified in PRPS1 as causing PRS-I superactivity, which results in PRPS1-related gout (MIM 311850).16,17 The disorder is characterized by purine nucleotide and uric acid overproduction and, occasionally, neurological problems.18 PRS-I superactivity has also been reported to cause sensorineural hearing loss and developmental delay.19 Interestingly, recent studies have provided evidence that missense mutations in PRPS1 can lead to decreased PRS-I enzymatic activity in several syndromes. Two missense mutations (Q133P and L152P) in PRPS1 have been described in Arts syndrome (MIM 301835), characterized by mental retardation, early-onset hypotonia, ataxia, delayed motor development, hearing impairment, and optic atrophy.14 E43D missense mutation was identified in a syndromic form of inherited peripheral neuropathy known as Rosenberg-Chutorian syndrome (RCS), and M115T mutation was identified in an X-linked form of Charcot-Marie-Tooth inherited neuropathy (CMTX5 [MIM 311070]).13 The CMTX5 syndrome is associated with a symptom triad of sensorineural hearing loss, visual impairment, and peripheral neuropathy, identical to that reported in RCS. It is of note that CMTX5 and RCS are allelic disorders sharing a common disease gene.
Our study extends the spectrum of PRPS1-related disorders to include NSHL, seen in DFN2. Four missense mutations (D65N, A87T, I290T, and G306R) have been identified in four DFN2 families as resulting in a loss of PRS-I activity, as was shown in silico by structural analysis and was shown in vitro by enzymatic activity assays in erythrocytes and cultured fibroblasts from patients. None of the mutations that we report are predicted to result in a major structural change in the PRS-I protein, which may explain why the disease phenotype is limited to NSHL. Molecular modeling showed that the D65N (DFN2), M115T (CMTX5), and Q133P (Arts syndrome) mutations probably affect the ATP-binding pocket. The analysis predicted that the D65N mutation has a much less severe effect on the binding of ATP, as compared with the Q133P14 mutation, which has more severe consequence than the M115T mutation.13
Further evidence comes from the comparison of the severity of reduction of the PRS-I activity observed in patients with DFN2, CMTX5, and Arts syndrome, with a diverse spectrum of phenotypes in affected males. In the family with Arts syndrome, there was no detectable PRS-I activity in erythrocytes from the affected males. The PRS-I activity of three patients was reduced 13-fold in comparison to that in fibroblasts from controls. The phenotype of Arts syndrome exhibits mental retardation, early-onset hypotonia, ataxia, delayed motor development, hearing impairment, and optic atrophy. In the CMTX5 family, the PRS-I activity in fibroblasts from affected males decreased by 62% (38% activity remained) in comparison to that of an unaffected male and unrelated healthy controls. The phenotype of CMTX5 syndrome is reduced to hearing loss, visual impairment, and peripheral neuropathy. Similar to that in the DFN2 family, the PRS-I activity in erythrocytes and cultured fibroblasts from affected males has decreased by 44%–45% (55%–56% activity remained) in comparison to that of normal male family members and unrelated control subjects. The phenotype of DFN2 is limited to NSHL.
By immunohistochemistry staining, we have shown that Prps1 was expressed in both vestibular and cochlea hair cells in early developing and postnatal mice and could also be detected in the spiral ganglion cells in mice at P6. The Prps1 distribution pattern suggests a role in inner ear development and maintenance. Basic physiological knowledge suggests that DPOAEs can be frequency-specific tests of cochlear status whenever pathology involves outer hair cells. In family GZ-Z052, there was absence of detectable DPOAEs in affected males and female carriers that were tested, which indicate that hair cells are probably the main site of pathology responsible for the hearing loss in DFN2. There were no symptoms of auditory neuropathy involving either the spiral ganglion cells or their axons, nor was there any visual impairment from optic neuropathy. Thus, it is possible that the deafness observed in DFN2 patients may be the consequence of defects of hair cell origin. However, additional experiments are still needed for elucidation of the exact pathological mechanisms that lead to hearing loss in families with DFN2 mutations.
In summary, we have identified PRPS1 as the second gene to be implicated in X-linked NSHL, by (1) identifying missense mutations in PRPS1 in four DFN2 families, (2) showing that these changes are within functional domains of the PRS-I protein and thereby alter protein function (shown for the D65N mutation), and (3) showing that the changes are uniformly conserved from zebrafish to human. The audio profile and clinical presentation of persons with DFN2 is easily distinguished from that of persons with DFN3 (POU3F4), which will facilitate the genetic testing of these two types of X-linked NSHL.
Acknowledgments
We sincerely thank all of the family members for their participation in and support of this study. These investigations were supported by the National High Technology Research and Development Program of China (863 Program) (2007AA02E466 to H.Y.), the National Natural Science Foundation of China (30571018 to H.Y., 30528025 to X. Liu, 30872862 to P.D., and 30728030 to Z.-Y.C.), the U.S. National Institutes of Health (R01 DC005575 to X. Liu and R01 DC006908 to Z.-Y.C.), the National Key Technology R&D program of China (2007 BAI18B12 to H.Y.), the Federick and an Ines Yeatts Inner Ear Hair Cell Regeneration fellowship (to A.K.). We thank Sawsan Khuri for her assistance with molecular modeling.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
Hereditary Hearing Loss homepage, http://webhost.ua.ac.be/hhh
Morton Cochlear EST Database, http://www.brighamandwomens.org/bwh_hearing/human-cochlear-ests.aspx
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PyMOL, http://pymol.org/
UCSCGenomeBrowser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Reardon W. Sex linked deafness: Wilde revisited. J. Med. Genet. 1990;27:376–379. doi: 10.1136/jmg.27.6.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen M.M., Gorlin R.J. Epidemiology, etiology, and genetic patterns. In: Gorlin R.J., Toriello H.V., Cohen M.M., editors. Hereditary hearing loss and its syndromes. Oxford University Press; Oxford: 1995. pp. 9–21. [Google Scholar]
- 3.de Kok Y.J., van der Maarel S.M., Bitner-Glindzicz M., Huber I., Monaco A.P., Malcolm S., Pembrey M.E., Ropers H.H., Cremers F.P. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science. 1995;267:685–688. doi: 10.1126/science.7839145. [DOI] [PubMed] [Google Scholar]
- 4.Tyson J., Bellman S., Newton V., Simpson P., Malcolm S., Pembrey M.E., Bitner-Glindzicz M. Mapping of DFN2 to Xq22. Hum. Mol. Genet. 1996;5:2055–2060. doi: 10.1093/hmg/5.12.2055. [DOI] [PubMed] [Google Scholar]
- 5.Manolis E.N., Eavey R.D., Sangwatanaroj S., Halpin C., Rosenbaum S., Watkins H., Jarcho J., Seidman C.E., Seidman J.G. Hereditary postlingual sensorineural hearing loss mapping to chromosome Xq21. Am. J. Otol. 1999;20:621–626. [PubMed] [Google Scholar]
- 6.Cui B., Zhang H., Lu Y., Zhong W., Pei G., Kong X., Hu L. Refinement of the locus for non-syndromic sensorineural deafness (DFN2) J. Genet. 2004;83:35–38. doi: 10.1007/BF02715827. [DOI] [PubMed] [Google Scholar]
- 7.Skvorak A.B., Weng Z., Yee A.J., Robertson N.G., Morton C.C. Human cochlear expressed sequence tags provide insight into cochlear gene expression and identify candidate genes for deafness. Hum. Mol. Genet. 1999;8:439–452. doi: 10.1093/hmg/8.3.439. [DOI] [PubMed] [Google Scholar]
- 8.Roessler B.J., Bell G., Heidler S., Seino S., Becker M., Palella T.D. Cloning of two distinct copies of human phosphoribosylpyrophosphate synthetase cDNA. Nucleic Acids Res. 1990;18:193. doi: 10.1093/nar/18.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eriksen T.A., Kadziola A., Bentsen A.K., Harlow K.W., Larsen S. Structural basis for the function of Bacillus subtilis phosphoribosyl-pyrophosphate synthetase. Nat. Struct. Biol. 2000;7:303–308. doi: 10.1038/74069. [DOI] [PubMed] [Google Scholar]
- 10.Zoref E., De Vries A., Sperling O. Mutant feedback-resistant phosphoribosylpyrophosphate synthetase associated with purine overproduction and gout. Phosphoribosylpyrophosphate and purine metabolism in cultured fibroblasts. J. Clin. Invest. 1975;56:1093–1099. doi: 10.1172/JCI108183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li S., Lu Y., Peng B., Ding J. Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site. Biochem. J. 2007;401:39–47. doi: 10.1042/BJ20061066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torres R.J., Mateos F.A., Puig J.G., Becker M.A. Determination of phosphoribosylpyrophosphate synthetase activity in human cells by a non-isotopic, one step method. Clin. Chim. Acta. 1996;245:105–112. doi: 10.1016/0009-8981(95)06178-9. [DOI] [PubMed] [Google Scholar]
- 13.Kim H.J., Sohn K.M., Shy M.E., Krajewski K.M., Hwang M., Park J.H., Jang S.Y., Won H.H., Choi B.O., Hong S.H. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (cmtx5) Am. J. Hum. Genet. 2007;81:552–558. doi: 10.1086/519529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Brouwer A.P., Williams K.L., Duley J.A., van Kuilenburg A.B., Nabuurs S.B., Egmont-Petersen M., Lugtenberg D., Zoetekouw L., Banning M.J., Roeffen M. Arts syndrome is caused by loss-of-function mutations in PRPS1. Am. J. Hum. Genet. 2007;81:507–518. doi: 10.1086/520706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sage C., Huang M., Vollrath M.A., Brown M.C., Hinds P.W., Corey D.P., Vetter D.E., Chen Z.Y. Essential role of retinoblastoma protein in mammalian hair cell development and hearing. Proc. Natl. Acad. Sci. USA. 2006;103:7345–7350. doi: 10.1073/pnas.0510631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Becker M.A., Smith P.R., Taylor W., Mustafi R., Switzer R.L. The genetic and functional basis of purine nucleotide feedback-resistant phosphoribosylpyrophosphate synthetase superactivity. J. Clin. Invest. 1995;96:2133–2141. doi: 10.1172/JCI118267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roessler B.J., Nosal J.M., Smith P.R., Heidler S.A., Palella T.D., Switzer R.L., Becker M.A. Human X-linked phosphoribosylpyrophosphate synthetase superactivity is associated with distinct point mutations in the PRPS1 gene. J. Biol. Chem. 1993;268:26476–26481. [PubMed] [Google Scholar]
- 18.Becker M.A., Puig J.G., Mateos F.A., Jimenez M.L., Kim M., Simmonds H.A. Inherited superactivity of phosphoribosylpyrophosphate synthetase: association of uric acid overproduction and sensorineural deafness. Am. J. Med. 1988;85:383–390. doi: 10.1016/0002-9343(88)90591-8. [DOI] [PubMed] [Google Scholar]
- 19.Simmonds H.A., Webster D.R., Lingam S., Wilson J. An inborn error of purine metabolism, deafness and neurodevelopmental abnormality. Neuropediatrics. 1985;16:106–108. doi: 10.1055/s-2008-1052552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.