

Abstract
Bacillus subtilis oxalate decarboxylase (OxDC) catalyzes the conversion of oxalate into CO2 and formate. The enzyme is composed of two cupin domains, each of which contains a Mn(II) ion. Although there is general agreement that Mn(II) in the N-terminal domain mediates OxDC-catalyzed decarboxylation, legitimate questions have been raised concerning the function (if any) of the Mn(II) bound in the C-terminal cupin domain. We have investigated this problem using a series of OxDC mutants in which Mn(II) binding is perturbed by mutagenesis of Glu-101 and Glu-280, which coordinate the metal in the N-terminal and C-terminal domains, respectively. We now demonstrate that decarboxylase activity and total manganese content are sensitive to modifications in either metal-binding glutamate residue. These findings, in combination with EPR measurements, raise the possibility that the C-terminal Mn(II) center can catalyze the decarboxylation reaction. Further support for this conclusion has been provided from a combination of in vivo and in vitro strategies for preparing wild-type OxDC in which Mn(II) is incorporated to a variety of extents. Kinetic characterization of these variants shows that OxDC activity is linearly correlated with manganese content, as might be expected if both sites can catalyze the breakdown of oxalate into formate and CO2. These studies also represent the first unequivocal demonstration that OxDC activity is uniquely mediated by manganese.
Oxalate decarboxylase (EC 4.1.1.2, OxDC)1 catalyzes the decarboxylation of oxalic acid to yield formic acid and carbon dioxide (Scheme 1) (1). This transformation is chemically interesting because the C–C bond in the substrate is relatively unreactive (2, 3). Efforts to delineate the catalytic mechanism employed by the enzyme (4) have been facilitated by the isolation of a native, bacterial form of OxDC from Bacillus subtilis (5) and the subsequent development of methods to express the catalytically active, wild-type (WT) enzyme in large amounts (6). X-ray crystal structures of B. subtilis OxDC and several site-specific mutants (7-9) confirm that the enzyme is a bicupin (10), as initially proposed from primary sequence alignment studies (11). Evidence from inductively coupled plasma mass spectrometry (ICP-MS) (12) and EPR spectroscopy shows that recombinant, wild-type (WT) B. subtilis OxDC contains Mn(II) when expressed in Escherichia coli (6, 13).
Scheme 1.
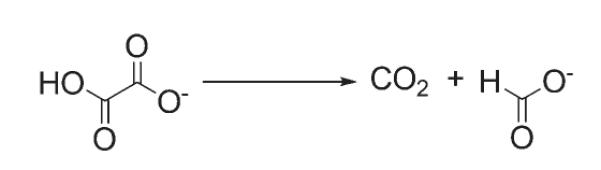
Reaction Catalyzed by B. subtilis Oxalate Decarboxylase
The OxDC monomer is composed of two cupin, β-barrel domains, each of which contains a metal-binding site (Figure 1A). For samples of recombinant, WT B. subtilis OxDC with high Mn(II) occupancy, EPR studies have suggested that only a single metal center interacts with acetate or formate (13). Three observations support the hypothesis that this Mn(II) center, and hence the OxDC active site, is located in the N-terminal domain. First, this domain contains a “substrate channel”, which can be “open” thereby permitting oxalate to diffuse into the Mn(II)-binding site from solution or “closed” to exclude solvent during catalysis. The interconversion of these two “states” is mediated by conformational rearrangement of a tetrapeptide “lid” segment comprising residues 161–165 (8). Second, a molecule of formate is coordinated to the N-terminal metal ion in one of the OxDC crystal structures (7). Third, recent work using OxDC mutants has shown that (i) site-directed mutagenesis of Glu-162 (14) (Figure 1B) and (ii) modification of the N-terminal active site lid (15) abolish decarboxylase activity.
Figure 1.

(A) Cartoon representation of the B. subtilis OxDC monomer. The N- and C-terminal cupin domains are colored green and purple, respectively, and the N-terminal segment that contributes to the secondary structure of the C-terminal domain is colored red. The yellow spheres show the locations of the two Mn centers. (B) Residues defining the Mn-binding (and putative catalytic) sites in the N-terminal (PDB entry 1UW8) (left) and C-terminal (PDB entry 1P58) (right) domains of OxDC. The residue numbering is for the enzyme encoded by the OxdC gene in B. subtilis. For the sake of clarity, hydrogen atoms bound to carbon atoms have been omitted. Atom coloring: C, black; H, white; N, blue; O, red; Mn, silver. These structures were visualized using CAChe Worksystem Pro version 6.5 (Fujitsu America Inc., Beaverton, OR).
Although there is general agreement that Mn(II) in the N-terminal domain mediates OxDC-catalyzed decarboxylation, legitimate questions have been raised concerning the function (if any) of the Mn(II) bound in the C-terminal cupin domain. We have investigated this problem using a series of OxDC mutants in which Mn(II) binding is perturbed by mutagenesis of Glu-101 and Glu-280, which coordinate the metal in the N-terminal and C-terminal domains, respectively (Figure 1B). We now demonstrate that decarboxylase activity and total manganese content are sensitive to modification of either metal-binding glutamate residue. These findings, in combination with EPR measurements, raise the possibility that the C-terminal Mn(II) center can catalyze the decarboxylation reaction. Further support for this conclusion has been provided from a combination of in vivo and in vitro strategies for preparing wild-type OxDC in which Mn(II) is incorporated to a variety of extents. Kinetic characterization of these variants has allowed us to determine that OxDC activity is linearly correlated with manganese content, as might be expected if both sites can catalyze the breakdown of oxalate into formate and CO2. Moreover, these results constitute the first unequivocal demonstration that OxDC activity is uniquely mediated by manganese, in contrast to the behavior of homologous, evolutionarily related bicupins, such as quercetin 2,3-dioxygenase, in which several metals appear to be able to substitute for Mn(II) in catalysis (16-18).
MATERIALS AND METHODS
Materials
Unless otherwise stated, all chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) and were of the highest available purity. Protein concentrations were determined using a modified Bradford assay (Pierce, Rockford, IL) for which standard curves were constructed with bovine serum albumin (19). All DNA primers were obtained from Integrated DNA Technologies, Inc. (Coralville, IA), and DNA sequencing was performed by the core facility in the Interdisciplinary Center for Biotechnology Research (ICBR) at the University of Florida.
Expression and Purification of Recombinant, Wild-Type OxDC
Recombinant wild-type B. subtilis OxDC was expressed and purified using a modified literature procedure (4). Thus, Luria-Bertani broth (50 mL) containing 50 μg/mL kanamycin was inoculated with oxdC:pET-9a/BL21(DE3) and incubated overnight at 37 °C. An aliquot (4 mL) of this stationary phase culture was then used to inoculate Luria-Bertani broth (5×400 mL), and the resulting cultures were incubated at 30 °C until an OD600 value of 0.6 was attained. At this time, the bacteria were heat-shocked at 42 °C for 10 min before the addition of isopropyl thiogalactoside and MnCl2 to final concentrations of 1 and 5 mM, respectively. The induced cells were then grown at 30 °C for 4 h, with shaking to ensure maximal aeration. Cells were harvested by centrifugation (5100g for 20 min at 4 °C), and the pellets were resuspended in 50 mM imidazole-HCl (pH 7.0) (100 mL) before sonication. The lysate was clarified by centrifugation (12000g for 20 min at 4 °C) and stored overnight at 4 °C. Lysis pellets were resuspended in 50 mM imidazole-HCl (pH 7.0) containing 1 M sodium chloride, 10 μM MnCl2, 0.1% Triton X-100, and 10 mM 2-mercaptoethanol (total volume of 100 mL), and the resulting mixture was stirred overnight at 4 °C. After centrifugation (12000g for 20 min at 4 °C), the solubilized extract was combined with the lysate and diluted 7-fold before being applied to a DEAE-Sepharose Fast Flow column (2.5 cm × 25 cm) equilibrated with 50 mM imidazole-HCl (pH 7.0) (buffer A). Elution was performed using a 500 mL linear gradient from buffer A to buffer A containing 1 M NaCl. Fractions containing OxDC were pooled, and solid (NH4)2SO4 was added to a final concentration of 1.7 M. The resulting solution was applied to a phenyl-Sepharose Hi-Performance column (2.5 cm × 18 cm) (GE Healthcare, Piscataway, NJ) equilibrated with 50 mM imidazole-HCl (pH 7.0) containing 1.7 M (NH4)2SO4 (buffer B). Bound proteins were eluted using a 500 mL linear gradient from buffer B to buffer A, and fractions containing purified OxDC were pooled and concentrated by ultrafiltration in an Amicon stirred cell (Millipore, Billerica, MA) to a final volume of 10 mL before being exhaustively dialyzed against 20 mM hexamethylenetetraamine hydrochloride (HMTA) (pH 6.0) containing 0.5 M NaCl. The dialyzed enzyme was then concentrated to approximately 9 mg/mL and stored in aliquots at −80 °C.
Expression and Purification of Site-Specific OxDC Mutants
All site-specific OxDC mutants were constructed using the overlap extension method (20) and the OxdC:pET-9a plasmid containing the gene encoding B. subtilis OxDC (4). Primers for mutagenesis were designed such that the desired mutation was located at the 3′-end and included restriction sites to facilitate cloning into pET9a (Table S1 of the Supporting Information). The regions upstream and downstream of the mutagenesis site were amplified independently, and a third PCR combined these two fragments to yield the full-length gene. The resulting products were digested with BamHI and NdeI and cloned into pET9a. Constructs were transformed into JM109 competent cells, and transformants were screened by restriction enzyme digestions using BamHI and NdeI. Plasmids containing the desired clones were transformed into BL21(DE3) competent cells, and expression of the OxDC mutants was conducted as for the wild-type enzyme. After an initial purification using DEAE-Sepharose Fast Flow column chromatography, as described above, the OxDC mutants could be precipitated from 50 mM imidazole-HCl (pH 7.0) containing 1.7 M (NH4)2SO4. The precipitate was then centrifuged (12000g for 20 min at 4 °C) and resuspended in 20 mM HMTA-HCl (pH 6.0) containing 0.5 M NaCl to yield solutions of the site-specific OxDC mutants at concentrations ranging from 3.5 to 19.6 mg/mL. This truncated purification procedure gave mutant enzymes that were >90% pure, as evaluated by SDS–PAGE.
Metal Content Determination
Divalent cations were removed from 20 mM HMTA-HCl (pH 6.0) containing 0.5 M NaCl by passage through a Chelex 100 column (1.5 cm × 16 cm) (Bio-Rad, Hercules, CA) in its Na+ form (21). Purified samples of WT OxDC and all site-directed OxDC mutants (2.5 mg) were exchanged into this buffer by being washed with 10-fold volumes in Centricon or Centriprep 30 concentrators (Millipore) (22) before the metal content was quantified on the basis of ICP-MS measurements (12) conducted at the University of Wisconsin Soil and Plant Analysis Laboratory.
Steady-State Kinetic Assays
Assay mixtures consisted of 50 mM NaOAc (pH 4.2), 0.2% Triton X-100, 0.5 mM o-phenylenediamine, 1–50 mM potassium oxalate, and either wild-type OxDC (1–4 μM) or the appropriate OxDC mutant (80–120 μM) (100 μL total volume). Reactions were initiated by the addition of substrate, incubated at ambient temperature (21–23 °C), and quenched by the addition of 1 N NaOH (10 μL). The amount of formate product was determined by an end point assay (23) consisting of 50 mM potassium phosphate (pH 7.8), 0.09 mM NAD+, and 0.4–1.0 unit/mg of formate dehydrogenase (1 mL final volume). The absorbance at 340 nm was measured after overnight incubation at 37 °C, and formate was quantified by comparison to a standard curve generated by spiking prequenched OxDC assay mixtures with known amounts of sodium formate. Measurements were taken at specific substrate and enzyme concentrations in duplicate, and data were analyzed to obtain the values of V and V/K by standard computer-based methods (24).
In addition to measuring the rate at which formate was produced by the enzyme-catalyzed decarboxylation of oxalate, we determined the level of oxalate oxidase activity for wild-type OxDC and the series of OxDC mutants at ambient temperatures (21–23 °C). These experiments used a continuous assay in which H2O2 production was coupled to the horseradish peroxidase (HRP)-catalyzed oxidation of 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) (25). Reaction mixtures contained 25 units of HRP, 5 mM ABTS, 50 mM potassium oxalate, and wild-type OxDC or the metal-binding OxDC mutants (at concentrations of up to 35 μg/mL) dissolved in 50 mM sodium acetate (pH 4.0) (1 mL total volume). An extinction coefficient of 10000 M−1 cm−1 for the ABTS radical product was assumed in these experiments. Control samples lacked HRP, so that we could differentiate between H2O2 production and any oxalate-dependent dye oxidation activity by wild-type OxDC or the OxDC mutant.
Size Exclusion Chromatography Measurements
The oligomeric state of the wild-type enzyme was compared with that of the metal-binding mutants by size exclusion chromatography using a BIOSEP-S2000 column (300mm × 7.8 mm with a 75 mm × 7.8 mm guard column) (Phenomenex, Torrance, CA) equilibrated with 20 mM hexamethylenetetraamine hydrochloride (pH 6.0) containing 0.5 M NaCl (buffer C). Calibration was performed using carbonic anhydrase (29.0 kDa), bovine serum albumin (66.0 kDa), alcohol dehydrogenase (150 kDa), β-amylase (200 kDa), apoferritin (443 kDa), and thyroglobulin (669 kDa). The void volume was measured by injecting blue dextran. Samples of recombinant, wild-type OxDC or the site-specific OxDC mutants were then injected onto the column and eluted with buffer C, at a flow rate of 1 mL/min with UV detection at 280 nm, to identify the oligomeric form of the enzyme.
Circular Dichroism (CD) Studies
Recombinant, wild-type OxDC was dialyzed into 25 mM potassium phosphate (pH 7.0) containing 100 mM NaCl, and the protein concentration was adjusted to a final value of 185 μg/mL. A similar procedure was performed for all nine site-specific OxDC mutants. In cases where the protein precipitated (8 of 10 samples), the precipitate was removed by microcentrifugation, The CD spectrum of the protein was then obtained using an Aviv 215 spectrometer (Aviv Associates, Lakewood, NJ) at wavelengths over the range of 190–250 nm (1 mm path length). All spectra were corrected by subtracting the CD spectrum of the buffer over this range of wavelengths.
EPR Spectroscopy
EPR spectra were recorded using samples of wild-type OxDC (12.3 mg/mL) or the E101A (4.5 mg/mL), E101Q (3.9 mg/mL), or E280Q (16.8 mg/mL) mutant enzyme dissolved in 20 mM HMTA-HCl (pH 6.0) containing 0.5 M NaCl (100 μL total volume). The metal contents of wild-type OxDC and the E101A, E101Q, and E280Q OxDC mutants used in these EPR experiments were 1.63, 0.18, 0.11, and 0.73 Mn/monomer, respectively. All high-field EPR experiments were performed using a custom-built spectrometer operating in transmission mode (26). Far-IR radiation was generated by a Gunn source at W-band (94–97 or 100–110 GHz), which was frequency-tripled and/or -quadrupled to achieve frequencies of 320 or 380 GHz, and 400–440 GHz, with a radiation power of 2–10 mW, and transmitted through an oversized waveguide so as to pass through the sample once before being detected by an InSb hot-electron bolometer (QMC Instruments Ltd., Cardiff, U.K.). The analogue signal from the bolometer was fed into a Stanford Instrument SR830 lock-in detector, which was referenced to the field modulation at the sample (typically 50 kHz and up to 25 G). The magnetic field sweep was conducted by sweeping either the main coil or a custom-built auxiliary 1000 G sweep coil. Field calibration was performed using a piece of P-doped silicon, which has a g value of 1.99854 and a hyperfine coupling constant of 117.507 MHz (27, 28). X-Band and W-band spectra were recorded on Elexsys E580 and E680 pulse/cw spectrometers (Bruker Biospin Corp., Billerica, MA), respectively, which were equipped with their standard resonators and cryostats for temperature control. Spectral simulations were performed using the toolbox EasySpin (29) written in the MATLAB computing environment (The MathWorks, Natick, MA). For relaxation rate studies, time-dependent EPR spectra were recorded at X-band with a 5 mm Bruker Flexline dielectric resonator, and samples were cooled using cold helium gas in a Bruker ER4118 (Oxford CF935) cryostat. The temperature during acquisition was controlled with an ITC4 temperature controller and a VC40 gas flow controller (Oxford Instruments, Eynsham, U.K.). Standard pulse sequences were employed in these experiments (30).
Expression and Purification of Co-Substituted, Wild-Type OxDC
Recombinant Co-containing, wild-type enzyme was obtained following the standard protocol for expressing the Mn-substituted enzyme, except that CoCl2 (Fisher Scientific, Pittsburgh, PA) or various CoCl2/MnCl2 mixtures were added to the cell culture in place of MnCl2, after the heat shock step but prior to induction of OxDC expression. After cell lysis, and extraction of the recombinant protein from the crude lysate as described above for recombinant wild-type OxDC, the Co-containing enzyme was purified by DEAE column chromatography. Fractions containing OxDC were pooled and dialyzed for 4 h against 50 mM imidazole-HCl buffer (pH 7.0) (2 L). The resulting sample was then applied to a Q-Sepharose Hi-Performance column (2.5 cm × 18 cm) equilibrated with buffer A and eluted using a 500 mL linear gradient from buffer A to buffer A containing 1 M NaCl. Fractions containing OxDC were pooled and exhaustively dialyzed against 20 mM HMTA-HCl (pH 6.0) containing 0.5 M NaCl. The purified, Co-substituted enzyme was concentrated and stored as described for recombinant, wild-type OxDC.
Preparation of Reconstituted WT OxDC
Recombinant, WT OxDC, prepared as outlined above, was dialyzed against 3.5 M guanidinium hydrochloride (GuHCl), 20 mM Tris-HCl, and 10 mM EDTA (pH 3.1) for 8 h at 4 °C. A second round of dialysis against 2.5 M GuHCl, 20 mM Tris-HCl, and 10 mM EDTA (pH 7.0), was performed, and excess EDTA was removed in a third dialysis against 2.5 M GuHCl containing 20 mM Tris-HCl (pH 7.0). Both of the latter steps were conducted for 8 h at 4 °C. At this stage, a round of dialysis over 8 h, at 4 °C, against 20 mM Tris-HCl (pH 7.0) containing 10 mM MnCl2 was used to reintroduce Mn(II) into the enzyme before exhaustive exchange into 20 mM HMTA-HCl (pH 6.0) containing 0.5 M NaCl at 4 °C.
RESULTS
Characterization of OxDC Active Site Mutants
To determine whether catalytic activity is solely associated with the N-terminal Mn-binding site of OxDC (8, 9), we expressed and purified a series of site-specific OxDC mutants in which residues that coordinate Mn(II) in either the N- or C-terminal domains (Glu-101 or Glu-280, respectively) were modified (Figure 1B). Prior work had shown that modifying any of the histidine residues coordinating the Mn(II) in Flammulina velutipes OxDC gave only inactive enzyme (31). The existence of stable, metal-free cupins lacking metal-binding residues suggested, however, that the absence of Mn(II) in one domain of OxDC or the other was unlikely to perturb the three-dimensional fold of the β-barrel structure (32, 33). By analogy to similar experiments on the Mn-dependent 3,4-dihydroxyphenylacetate 2,3-dioxygenase from Arthrobacter globiformis, which revealed the metal-binding ligands (34), we anticipated that reducing the metal affinity of either binding site in OxDC by mutating Glu-101 or Glu-280 would yield a variant in which Mn(II) was incorporated into a single domain. The resulting enzymes were then expected to exhibit approximately 50% activity relative to WT OxDC if metal incorporation proceeded to give 1.0 Mn/monomer and catalysis were to be mediated independently at both Mn(II) sites. We elected to perform all mutagenesis and metal binding (see below) studies using untagged wild-type OxDC and site-specific OxDC mutants, rather than the His-tagged OxDC variants used in several previous studies (7-9, 15), which generally contained only 0.86–1.14 Mn/monomer. On this point, we note that untagged enzyme samples exhibiting a metal content of 1.8 Mn/monomer are routinely obtained with the inclusion of a chromatographic step employing phenyl-Sepharose (4).2 At this level of Mn incorporation, the samples of recombinant, untagged WT OxDC exhibited a specific activity of 61 units/mg, as measured in an end point assay employing formate dehydrogenase (23).3 When Glu-101 was replaced with alanine, aspartate, or glutamine, the resulting OxDC mutants contained only approximately 0.1–0.2 Mn/monomer even though the metal-binding residues in the C-terminal domain were unchanged (Table 1). Hence, somewhat unexpectedly, efforts to weaken the ability of Mn(II) to bind within the N-terminal site resulted in low levels of total Mn(II) incorporation rather than the anticipated amount of approximately 1 Mn/monomer. Even so, the E101D and E101Q OxDC mutants retained respectable levels of activity after correction of the steady-state kinetic parameters for Mn(II) content (Table 1). This was not the case, however, for the E101A OxDC mutant, for which substantially lower decarboxylase activity was observed (Table 1), raising the possibility that the observed activity of the E101D and E101Q OxDC mutants might not be solely due to Mn(II) bound within the C-terminal sites of these enzymes. Thus, small amounts of Mn(II) might still be bound within their N-terminal sites giving rise to the decarboxylase activity. On this point, we note that replacing a metal-binding glutamate did not completely abolish incorporation of Mn in the active site of Mn-dependent 3,4-dihydroxyphenylacetate 2,3-dioxygenase (34). We then characterized a similar series of OxDC mutants in which Glu-280 was replaced with alanine, aspartate, and glutamine, which exhibited essentially no decarboxylase activity despite having reasonable levels of Mn incorporation (0.64–0.73 Mn/monomer) relative to those observed for the E101A, E101D, and E101Q OxDC mutants (Table 1). None of the six mutant enzymes was found to exhibit oxalate oxidase activity (data not shown), at least as assayed with a dye oxidation method for monitoring oxalate-dependent hydrogen peroxide formation (25). In addition, metals other than manganese were not incorporated into these OxDC mutants to any significant extent (Table S2 of the Supporting Information).
Table 1.
Mn Incorporation and Steady-State Kinetic Parameters for Metal-Binding OxDC Mutantsa
| enzyme | Mnb | Km (mM) | kcat (s−1) | kcat/[Mn] (s−1) | kcat/Km (M−1 s−1) | kcat/Km/[Mn] (M−1 s−1) |
|---|---|---|---|---|---|---|
| WT OxDC | 1.87 | 8.4±0.7 | 53± 1.5 | 28.3±0.8 | 6309 | 3374 |
| E101A | 0.18 | 2.9±0.3 | 0.046±0.002 | 0.26 ±0.01 | 16 | 89 |
| E101D | 0.09 | 3.4±0.1 | 0.49±0.01 | 5.4±0.1 | 144 | 1600 |
| E101Q | 0.11 | 4.0±0.2 | 0.62±0.01 | 5.6±0.1 | 155 | 1409 |
| E280A | 0.67 | 3.0±0.2 | 0.019±0.001 | 0.028±0.002 | 6 | 9 |
| E280D | 0.64 | 5.4±0.4 | 0.14±0.01 | 0.22±0.01 | 26 | 41 |
| E280Q | 0.73 | 10.1±0.6 | 0.62±0.01 | 0.85±0.01 | 61 | 84 |
| E101Q/E280Qc | 0.07 | 2.9±0.3 | 0.012±0.0003 | 0.17±0.004 | 4 | 57 |
Complete details of the metal content of these enzyme preparations are provided as Supporting Information (Table S2).
Number of manganese ions per OxDC monomer.
OxDC double mutant in which Glu-101 and Glu-280 are both replaced with glutamine residues.
Quaternary Structure and CD Spectroscopy of the OxDC Mn-Binding Mutants
Two approaches were undertaken to confirm that modification of either metal-binding glutamate residue did not cause large perturbations in enzyme structure. First, size exclusion chromatography was used to investigate the quaternary structures adopted by the series of OxDC mutants. Although X-ray crystal structures show that B. subtilis OxDC is a hexamer, in which two trimers are packed face to face so that the complex possesses 32 (D3) point symmetry (7) (Figure 2), under our experimental conditions, the recombinant, wild-type OxDC eluted as both a hexamer and oligomers of higher apparent mass, corresponding to complexes formed from approximately 12–18 monomers. Similar studies on the site-specific OxDC mutants showed that they also possessed quaternary structures similar to that of WT OxDC, at least on the basis of their elution behavior (Table S3 of the Supporting Information).
Figure 2.

Cartoon representations of the B. subtilis OxDC trimer and hexamer showing representative Mn–Mn distances. (A) Representation of the OxDC trimer showing distances between Mn ions in the same and adjacent OxDC monomers. Monomers are colored red, green, and purple, and the Mn ions are shown as purple (N-terminal domain) and yellow (C-terminal domain) spheres. (B) Representation of the OxDC hexamer showing the distance between the Mn ions (yellow spheres) in the layered OxDC trimers. One monomer is colored red to emphasize the role of α-helical regions in mediating monomer–monomer interactions.
In a complementary set of experiments, the extent of secondary structural changes resulting from site-specific replacement of the metal-binding glutamate residues was studied by circular dichroism (CD) measurements (Figure 3). As expected from the X-ray crystal structures, the CD spectrum of WT OxDC showed features consistent with the largely β-strand character of the cupin domains together with minima at 222 and 205 nm, which are presumably associated with the α-helices that mediate monomer–monomer contacts. The OxDC mutants in which Glu-101 was replaced with other residues (E101A, E101D, and E101Q) gave CD spectra that resembled that of the WT enzyme, albeit with decreased molar ellipticities observed at wavelengths between 215 and 230 nm, consistent with similar levels of β-strand, secondary structure. Interestingly, the spectrum seen for the E101Q OxDC mutant resembles that reported for the Thermococcus litoralis phosphoglucose isomerase, a cupin for which activity is thought to be Fe-dependent (38). Thus, it is likely that these N-terminal OxDC mutants retain much of their secondary structure, permitting Mn(II) bound in the C-terminal site to exhibit catalytic activity. The CD properties of the E280A, E280D, and E280Q OxDC mutants, however, differed more from those of the WT enzyme, suggesting that the lack of Mn(II) in the C-terminal domain had a greater impact on the overall bicupin structure of these proteins.
Figure 3.
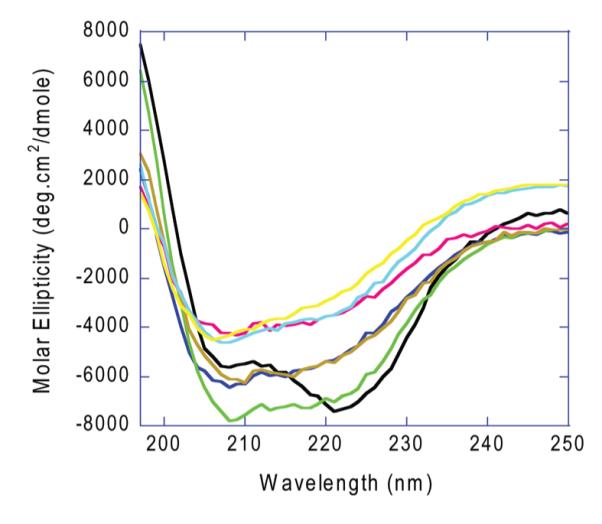
CD spectra of recombinant, wild-type OxDC and the metal-binding OxDC mutants. All samples were of equal concentration (0.185 mg/mL) and were dissolved in phosphate buffer (pH 7): wild -ype OxDC, black; E101A, dark blue; E101D, orange; E101Q, light green; E280A, red; E280D, yellow; E280Q, cyan.
EPR Properties of the Mn(II) Centers in the OxDC Mutants
In a previous multifrequency EPR study of WT OxDC (13), we tentatively assigned two Mn(II) species (assumed to be present in equal amounts) to the two metal-binding sites (N-terminal and C-terminal) of the enzyme. EPR characterization of the E101A, E101Q, and E280Q OxDC mutants, at both high fields and X-band, was undertaken to ascertain the (i) homogeneity of their Mn(II) binding sites and (ii) the extent to which the ligand geometries about the bound metal might resemble those in WT OxDC. If Mn(II) binding were to be precluded in one of the two domains by replacement of the appropriate glutamate ligand, we anticipated that the signal from the corresponding Mn(II) species would be absent in the OxDC mutant. When the EPR spectra of WT OxDC and the three OxDC mutants in the g ≈ 2 region, taken at 406.4 GHz, were overlaid (Figure 4A), however, the six-line Mn(II) signals for all samples exhibited a strong resemblance: the isotropic g factor (giso = 2.00087) was the same for all preparations within experimental accuracy (±0.0001), and the hyperfine coupling constants extracted from the experimental data were also similar (approximately 250 MHz). The spectra also revealed very little g and A strain for the bound Mn(II) ions in all three OxDC mutants, which was observed in our earlier studies of WT OxDC (13). These spectra for the single-site OxDC mutants were simulated assuming at least two slightly different sets of fine structure parameters (see the Supporting Information), with the observed fine structure parameter ∣D∣ being less than 3000 MHz in all cases, as expected for an octahedral coordination environment of the bound Mn(II) ions. Pentacoordinate Mn(II) centers, such as that seen in Mn-SOD (39), exhibit D values that are typically 1 order of magnitude larger than those observed for the main component of the spectra for the OxDC mutants. A lack of Mn(II) species with large fine structure values was also evident in the X-band spectra of the mutant enzymes (Figure S8 of the Supporting Information). The E280Q OxDC mutant exhibited smaller fine structure parameters than WT OxDC (D = 825 MHz; E = 247 MHz), perhaps reflecting a subtle change in either the electronic or structural properties of the Mn(II) ion bound within the N-terminal site. Structural changes in the C-site mutants were already indicated by changes of their CD spectra compared to that of WT OxDC. Because of the low level of Mn(II) incorporation, only tentative values for the D and E parameters of the N-terminal domain mutants could be assigned (Figures S6 and S7 of the Supporting Information). In both mutants, ∣D∣ was less than 3000 MHz and the simulated values were comparable to those observed for WT OxDC (13). No evidence for a pentacoordinate Mn(II) species exhibiting a large zero-field splitting was obtained from these spectra, consistent with the observation of octahedral coordination as modeled in the original X-ray crystal structure of OxDC (7).
Figure 4.

(A) EPR spectra of the Mn(II) ions in wild-type (black), E280Q (red), E101Q (green), and E101A (blue) OxDC at 20 K. The frequency for all spectra was 406.4 GHz with a modulation amplitude and modulation frequency of 1 G and 50 kHz, respectively. The main coil was slowly swept over a range of 14.4–14.6 T at 0.4 mT/s. (B) Hahn-echo decay experiments with wild-type (black), E280Q (red), E101Q (green), and E101A (blue) OxDC at 5 K. Samples were dissolved in 20 mM HMTA-HCl (pH 6.0) containing 0.5 M NaCl (100 μL total volume), and all spectra were recorded at 3700 G. The microwave frequencies were 9.711620, 9.700938, 9.716131, and 9.708152 GHz for wild-type, E280Q, E101Q, and E101A OxDC, respectively. The Hahn-echo pulse sequence employed π/2 and π pulses of 16 and 32 ns, respectively, and a two-phase CYCLOPS sequence was used. The data shown are the sums of 50 individual pulse trains per CYCLOPS phase (100 for the E101A spectrum), separated by a repetition time of 500 μs. The transients are sums of 5, 15, 50, and 64 averages for the order in which they are listed. Microwave attenuation was set to 12, 10, 7, and 8 dB, respectively, on the basis of visual inspection of the turning point of the Hahn echo as a function of microwave power. All traces were simulated with biexponential decay kinetics. Simulated decay curves are plotted for WT OxDC (orange) and the E101A (green), E101Q (magenta), and E280Q (violet) OxDC mutants, giving the following fit results for the two exponential components: 250 ns (90.6%) and 553 ns (9.4%) for WT OxDC, 423 ns (95.6%) and 1364 ns (4.4%) for E280Q OxDC, 553 ns (79.1%) and 2240 ns (20.1%) for E101Q OxDC, and 581 ns (92.0%) and 2250 ns (8.0%) for E101A OxDC.
Direct effects arising from the absence of Mn(II) in the N- or C-terminal sites were also probed by measuring the relaxation times T1 and T2, which report on direct electron–electron interaction while being less sensitive to subtle changes in environment. The Hahn-echo decay (Figure 4B) and the inversion recovery (Figure S9 of the Supporting Information) of the Mn(II) signals for wild-type OxDC and the mutants were measured at 3700 G where most of the echo-detected spectra had their maximum. In all samples, the observed relaxation times were biexponential, with Tm (T2) values of 250 and 550 ns being obtained for the Mn(II) signal in wild-type OxDC. This presumably reflects the existence of a weak dipolar interaction between metal ions in the adjacent domains of neighboring OxDC in the trimer, which are separated by a Mn–Mn distance of approximately 21 Å (Figure 2A). Similar relaxation enhancements over distances of 15–30Å have been seen in other proteins, including the bacterial photosynthetic reaction center (40) and metmyoglobin (41). The corresponding Tm values for the E280Q OxDC mutant were 420 and 1360 ns, respectively, signaling a considerable change in the relaxation enhancement of the Mn(II) ions when compared to those in WT OxDC, and these differences were more dramatic in the case of the E101Q and E101A mutants, which exhibited Tm times of approximately 550 and 2200 ns for the two components (Figure 4B). Similar findings were obtained in T1 studies of these protein samples (Figure S9 of the Supporting Information). These results are important because they demonstrate that the majority of the EPR-active Mn(II) in all three mutants is present in isolation, as would be case if OxDC monomers contained only one bound Mn(II) ion. This is evident from the crystal structure, which shows the minimum Mn(II)–Mn(II) distances are 41 and 37 Å for singly occupied monomers in the trimer and hexamer, respectively (Figure 2).
Experimental Dependence of Mn(II) Incorporation and OxDC Activity
To date, only indirect evidence suggests that OxDC activity is solely manganese-dependent. For example, the expression of correctly folded OxDC with reasonable catalytic activity requires the presence of Mn(II) in the growth medium (6, 42), and X-ray scattering factors used in refining the structure of recombinant B. subtilis OxDC are fully consistent with this metal being present in both cupin domains (7-9). On the other hand, the native form of OxDC from B. subtilis has not been purified in sufficient quantities for accurate metal analysis (5), and homologous enzymes in the bicupin family seem able to bind and employ a variety of alternate metals in catalysis (16-18). Heterologous expression of metalloenzymes may also yield proteins in which “non-native” metals are incorporated, perhaps because metallochaperones in the native organism are not present in the host cell used for expression of recombinant protein (43, 44). We therefore examined the correlation of Mn(II) and decarboxylase activity, together with the ability of other metals to support catalysis. Thus, the expression of recombinant WT OxDC was performed using CoCl2 (2 mM) as a supplement to the growth medium in place of MnCl2. ICP-MS analysis of the enzyme produced under these conditions showed it to contain 0.80 Co/monomer and 0.05 Mn/monomer (Table 2). The specific activity of this OxDC sample was almost exactly that expected for the low level of Mn(II) incorporation, suggesting that enzyme-bound Co(II) does not catalyze the decarboxylation reaction. The effects of expressing WT OxDC with mixtures of both Co(II) and Mn(II) salts in the growth medium gave samples of the enzyme containing different amounts of Mn per monomer. Interestingly, no obvious relationship existed between the Mn: Co ratio in the medium and the extent to which Mn or Co was incorporated into the recombinant protein (Table 2). For example, although the two metals were incorporated into OxDC in approximately equal amounts when the Mn:Co ratio was 20:1, increasing the amount of MnCl2 relative to CoCl2 in the growth medium did not give wild-type OxDC that contained more Mn(II) than Co(II). These observations almost certainly reflect the complex interplay of cellular mechanisms regulating metal metabolism, especially that of manganese, in bacteria (45, 46).
Table 2.
Effects of MnCl2 and CoCl2 in the Growth Medium on the Metal Incorporation and Specific Activity of Recombinant, WT OxDC
| no. of ions/monomer |
specific activity |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| preparation | [MnCl2] (mM) | [CoCl2] (mM) | Mna | Coa | Zna | Fea | Cua | units/mg | units mg−1 Mn−1 |
| 1 | 5 | 0 | 1.87 | ndb | 0.51 | 0.07 | 0.01 | 61.2 | 32.7 |
| 2 | 5 | 0 | 1.63 | ndb | 0.08 | <0.01 | <0.01 | 40.9 | 25.1 |
| 3 | 0 | 2 | 0.05 | 0.80 | 0.14 | <0.01 | <0.01 | 2.2 | 44.0 |
| 4 | 0.25 | 2 | 0.27 | 1.03 | 0.22 | 0.18 | <0.01 | 8.0 | 29.6 |
| 5 | 1 | 1 | 0.12 | 1.32 | 0.26 | 0.13 | <0.01 | 4.5 | 37.5 |
| 6 | 5 | 0.25 | 0.56 | 0.68 | 0.22 | 0.13 | <0.01 | 13.5 | 24.1 |
| 7 | 5 | 0.05 | 0.47 | 0.09 | 0.19 | 0.06 | <0.01 | 19.0 | 40.4 |
| 8 | -c | -c | 0.90 | ndb | 0.79 | 0.14 | <0.01 | 25.1 | 27.9 |
| 9 | -c | -c | 0.64 | 0.01 | 0.60 | <0.01 | 0.02 | 14.4 | 22.5 |
Expressed as the number of ions per OxDC monomer.
The cobalt content of this sample was not determined.
This sample was obtained by reconstituting WT OxDC using the procedure described in Materials and Methods.
Although these experiments showed OxDC activity to be Mn-dependent (Table 2), the functional form of any correlation between Mn(II) incorporation and catalytic activity could not be determined in the absence of enzyme samples containing between 0.7 and 1.5 Mn/monomer. We therefore set out to prepare such samples by reconstituting the OxDC apoenzyme with Mn(II). In contrast to other bicupins, such as quercetin 2,3-dioxygenase (16-18), it proved to be remarkably difficult to remove manganese from wild-type OxDC. Standard literature protocols (22, 47-51) either failed to remove bound Mn or caused almost complete precipitation of protein. The preparation of Mn-free OxDC was eventually accomplished, however, using a protocol modified from that used to produce the Mn-dependent superoxide dismutase (Mn-SOD) apoenzyme (52-54). Hence, after extensive dialysis of recombinant, wild-type OxDC against EDTA in the presence of GuHCl, manganese could be reintroduced into the apoenzyme in a refolding step with dialysis buffer containing MnCl2. Samples of recombinant OxDC containing 0.64 and 0.90 Mn/monomer were obtained in this manner, with the primary contaminating metal being Zn (Table 2).4 With respect to this point, we note that apo-OxDC appears to have a high affinity for Zn(II) (E. W. Moomaw, unpublished observations), as also reported for Pyrococcus furiosus phosphoglucose isomerase (another member of the cupin family of enzymes) (55). The specific activities of these two reconstituted OxDC samples allowed us to conclude that there is a linear correlation between decarboxylation activity and the level of Mn incorporation (Figure 5).
Figure 5.
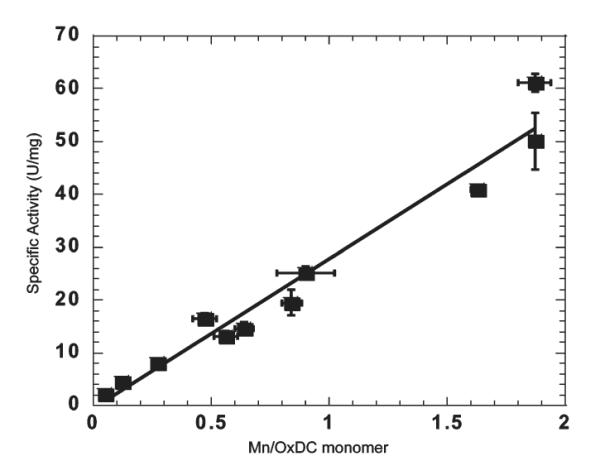
Experimental dependence of OxDC specific activity on Mn incorporation. Note that the points at 0.64 and 0.90 Mn/monomer were obtained using samples of reconstituted WT OxDC, and the expected specific activity assuming a linear correlation with Mn content is indicated by the line.
DISCUSSION
There is strong evidence for decarboxylase activity being associated with the N-terminal domain of the enzyme, especially from mutagenesis studies that have demonstrated (i) the functional role of Glu-162 in catalysis (14) and (ii) the importance of a tetrapeptide loop in controlling the reaction catalyzed by OxDC (15). On the other hand, whether the C-terminal Mn(II) is capable of catalyzing decarboxylation remains an unresolved problem, although early site-directed mutagenesis experiments involving the modification of conserved residues in the C-terminal site gave OxDC mutants with perturbed steady-state kinetic parameters (7). Hence, it has been suggested that full activity of the N-terminal active site requires the presence of bound Mn(II) in the C-terminal domain, i.e., that this site somehow exerts an effect on the overall structure of the enzyme and/or reactivity of the N-terminal metal ion (15).
Assuming that the C-terminal Mn(II) plays no role in catalysis predicts that OxDC variants lacking an N-terminal Mn(II) will not exhibit decarboxylase activity, in contradiction to what we observe for the E101D and E101Q OxDC mutants on the basis of kcat/Km/[Mn] (Table 1). This result seems to imply that Mn(II) ions bound in the C-terminal domain can catalyze the reaction, but this interpretation is complicated by the fact that the E101A OxDC mutant has low activity even though all three enzymes have similar secondary structures on the basis of CD measurements (Figure 3). An alternate explanation is therefore that Mn(II) can still bind in the N-terminal domain of the E101D or E101Q OxDC mutant, albeit to a small extent. If this were the sole explanation for the observed activity, however, then based on published hypotheses (15), one might expect Mn(II) to occupy both sites in the E101D and E101Q OxDC mutants. Both the altered relaxation properties of Mn(II) in either of these OxDC variants (Figure 4B) and the linear dependence of OxDC activity on Mn(II) incorporation (Figure 5), however, do not seem consistent with such a proposal. Indeed, the latter observation is most simply explained by assuming that both sites can catalyze decarboxylation quite independently of each other!
Given the considerable evidence for activity being mediated by the N-terminal Mn(II)-binding site (8, 9, 14, 15), the very low activities exhibited by all of the E280X OxDC mutants were surprising, especially because metal occupancies (Mn per monomer) were significantly higher for these variants than for the corresponding E101X OxDC mutants (Table 1). Once again, EPR relaxation measurements indicated that the majority of the EPR-active Mn(II) in all three mutants is “isolated” (Figure 4B), i.e., that the OxDC monomers contained only one bound Mn(II) ion. That the assumed absence of metal in the C-terminal site does perturb secondary structure in these variants, however, is shown by comparison of the CD properties for the E280X OxDC mutants with those of their corresponding E101X variants (Figure 3). These structural differences may be the reason that these OxDC mutants exhibited very little activity, despite (presumably) binding reasonable levels of Mn(II) in the N-terminal domain.
Taken overall, while our findings support the idea that catalytic activity in the N-terminal site requires the structural integrity of the other domain, they also raise the possibility that the C-terminal Mn(II) center can catalyze the decarboxylation reaction. Such a proposal is not unreasonable given the identical coordination of the Mn(II) ions, and the similarity of the side chains proximal to the metal, in the two active sites (Figure 1B).
Supplementary Material
ACKNOWLEDGMENT
We acknowledge the insightful comments of an anonymous reviewer concerning interpretation of the kinetic data. Vijay Antharam and Omjoy Ganesh provided technical support for the CD studies of WT OxDC and the six metal-binding OxDC mutants, which were performed in the Biophysical Core Facility of the ICBR at the University of Florida.
Footnotes
- OxDC
- oxalate decarboxylase
- ICP-MS
- inductively coupled plasma mass spectroscopy
- EPR
- electron paramagnetic resonance
- WT
- wild type
- DEAE
- diethylaminoethyl
- HMTA
- hexamethylenetetraamine
- HRP
- horseradish peroxidase
- ABTS
- 2,2′-azinobis(3-ethylbenzthiazoline)-6-sulfonic acid
- GuHCl
- guanidinium hydrochloride
- MnSOD
- Mn-dependent, superoxide dismutase
- ZFS
- zero-field splitting
- EDTA
- ethylenediaminetetraacetic acid
- PDB
- Protein Data Bank.
To obtain reproducibly high levels of Mn incorporation, induction must also be undertaken at an optical density lower than that previously reported (4), with cells being grown at a postinduction temperature of 30 °C in an effort to promote the transport of manganese ions into the bacteria (35-37).
Higher values of specific activity have been reported for His-tagged variants of recombinant, wild-type OxDC containing lower levels of Mn(II) per monomer of protein. The activity of these OxDC preparations is reported, however, to vary as a function of time (15). We see no such variation with the untagged wild-type OxDC or OxDC mutants prepared for the studies outlined herein. The basis for differences in the behavior of the tagged and untagged forms of the enzyme remains to be resolved.
These well-defined in vitro conditions for metal substitution and refolding failed, however, to introduce Mn(II) into the E101Q OxDC mutant, raising the possibility that the presence of Mn(II) in the N-terminal cupin domain is required for subsequent loading of the C-terminal site.
SUPPORTING INFORMATION AVAILABLE Primers used in the construction of WT OxDC and metal-binding OxDC mutants (Table S1), full details of the metal content and specific activity for the E101X and E280X OxDC mutants (Table S2), size exclusion chromatographic data for recombinant, WT OxDC and OxDC mutants (Table S3), EPR simulation parameters for the E280Q, E101Q, and E101A OxDC mutants (Tables S4-S6), high-field EPR spectrum of wild-type OxDC (Figure S1), high-field and X-band EPR spectra of the E180Q OxDC mutant (Figures S2-S5), high-field EPR spectra of the E101Q and E101A OxDC mutants (Figures S6 and S7), comparison of the X-band EPR spectra of recombinant, WT OxDC and the E280Q, E101Q, and E101A OxDC mutants (Figure S8), and data for the inversion recovery measurements on WT OxDC and the E280Q, E101Q, and E101A OxDC mutants. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by National Institutes of Health Grant DK61666 to N.G.J.R. and National Science Foundation Grant CHE0809729 to A.A.
REFERENCES
- (1).Shimazono H, Haiyashi O. Enzymatic decarboxylation of oxalic acid. J. Biol. Chem. 1957;227:151–159. [PubMed] [Google Scholar]
- (2).Svedružić D, Jónsson S, Toyota CG, Reinhardt LA, Ricagno S, Lindqvist Y, Richards NGJ. The enzymes of oxalate metabolism: Unexpected structures and mechanisms. Arch. Biochem. Biophys. 2005;433:176–192. doi: 10.1016/j.abb.2004.08.032. [DOI] [PubMed] [Google Scholar]
- (3).Begley TP, Ealick SE. Enzymatic reactions involving novel mechanisms of carbanion stabilization. Curr. Opin. Chem. Biol. 2004;8:508–515. doi: 10.1016/j.cbpa.2004.08.004. [DOI] [PubMed] [Google Scholar]
- (4).Reinhardt LA, Svedruzic D, Chang CH, Cleland WW, Richards NGJ. Heavy-atom isotope effects on the reaction catalyzed by the oxalate decarboxylase from Bacillus subtilis. J. Am. Chem. Soc. 2003;125:1244–1252. doi: 10.1021/ja0286977. [DOI] [PubMed] [Google Scholar]
- (5).Tanner A, Bornemann S. Bacillus subtilis YvrK is an acid-induced oxalate decarboxylase. J. Bacteriol. 2000;182:5271–5273. doi: 10.1128/jb.182.18.5271-5273.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Tanner A, Bowater L, Fairhurst SA, Bornemann S. Oxalate decarboxylase requires manganese and dioxygen for activity. Overexpression and characterization of Bacillus subtilis YvrK and YoaN. J. Biol. Chem. 2001;276:43627–43634. doi: 10.1074/jbc.M107202200. [DOI] [PubMed] [Google Scholar]
- (7).Anand R, Dorrestein PC, Kinsland C, Begley TP, Ealick SE. Structure of oxalate decarboxylase from Bacillus subtilis at 1.75 Å resolution. Biochemistry. 2002;41:7659–7669. doi: 10.1021/bi0200965. [DOI] [PubMed] [Google Scholar]
- (8).Just VJ, Stevenson CEM, Bowater L, Tanner A, Lawson DM, Bornemann S. A closed conformation of Bacillus subtilis oxalate decarboxylase OxdC provides evidence for the true identity of the active site. J. Biol. Chem. 2004;279:19867–19874. doi: 10.1074/jbc.M313820200. [DOI] [PubMed] [Google Scholar]
- (9).Just VJ, Burrell MR, Bowater L, McRobbie I, Stevenson CEM, Lawson DM, Bornemann S. The identity of the active site of oxalate decarboxylase and the importance of the stability of active-site lid conformations. Biochem. J. 2007;407:397–406. doi: 10.1042/BJ20070708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dunwell JM, Purvis A, Khuri S. Cupins: The most functionally diverse protein superfamily? Phytochemistry. 2004;65:7–17. doi: 10.1016/j.phytochem.2003.08.016. [DOI] [PubMed] [Google Scholar]
- (11).Dunwell JM, Khuri S, Gane PJ. Microbial relatives of the seed storage proteins of higher plants: Conservation of structure and diversification of function during evolution of the cupin superfamily. Microbiol. Mol. Biol. Rev. 2000;64:153–179. doi: 10.1128/mmbr.64.1.153-179.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Olivares JA. Inductively-coupled plasma mass spectrometry. Methods Enzymol. 1988;158:205–232. doi: 10.1016/0076-6879(88)58057-6. [DOI] [PubMed] [Google Scholar]
- (13).Angerhofer A, Moomaw EM, Garcia-Rubio I, Ozarowski A, Krzystek J, Weber RT, Richards NGJ. Multifrequency EPR studies on the Mn(II) centers of oxalate decarboxylase. J. Phys. Chem. B. 2007;111:5043–5046. doi: 10.1021/jp0715326. [DOI] [PubMed] [Google Scholar]
- (14).Svedruzic D, Liu Y, Reinhardt LA, Wroclawska E, Cleland WW, Richards NGJ. Investigating the roles of putative active site residues in the oxalate decarboxylase from Bacillus subtilis. Arch. Biochem. Biophys. 2007;464:36–47. doi: 10.1016/j.abb.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Burrell MR, Just VJ, Bowater L, Fairhurst SA, Requena L, Lawson DM, Bornemann S. Oxalate decarboxylase and oxalate oxidase activities can be interchanged with a specificity switch of up to 282000 by mutating an active site lid. Biochemistry. 2007;46:12327–12336. doi: 10.1021/bi700947s. [DOI] [PubMed] [Google Scholar]
- (16).Schaab MR, Barney BR, Francisco WA. Kinetic and spectroscopic studies on the quercetin 2,3-dioxygenase from Bacillus subtilis. Biochemistry. 2006;45:1009–1016. doi: 10.1021/bi051571c. [DOI] [PubMed] [Google Scholar]
- (17).Gopal B, Madan LL, Betz SF, Kossiakoff AA. The crystal structure of a quercetin 2,3-dioxygenase from Bacillus suggests modulation of enzyme activity by a change in the metal active site(s) Biochemistry. 2005;44:193–201. doi: 10.1021/bi0484421. [DOI] [PubMed] [Google Scholar]
- (18).Bowater L, Fairhurst SA, Just VJ, Bornemann S. Bacillus subtilis YxaG is a novel Fe-containing quercetin 2,3-dioxygenase. FEBS Lett. 2004;557:45–48. doi: 10.1016/s0014-5793(03)01439-x. [DOI] [PubMed] [Google Scholar]
- (19).Bradford MM. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- (20).Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- (21).Holmqvist B. Elimination of adventitious metals. Methods Enzymol. 1988;158:6–14. doi: 10.1016/0076-6879(88)58042-4. [DOI] [PubMed] [Google Scholar]
- (22).Gonzalez JC, Peariso K, Penner-Hahn JE, Matthews RG. Cobalamin-independent methionine synthase from Escherichia coli: A zinc metalloenzyme. Biochemistry. 1996;35:12228–12234. doi: 10.1021/bi9615452. [DOI] [PubMed] [Google Scholar]
- (23).Schutte H, Flossdorf J, Sahm H, Kula MR. Purification and properties of formaldehyde dehydrogenase and formate dehydrogenase from Candida boidinii. Eur. J. Biochem. 1976;62:151–160. doi: 10.1111/j.1432-1033.1976.tb10108.x. [DOI] [PubMed] [Google Scholar]
- (24).Cleland WW. Statistical analysis of enzyme kinetic data. Methods Enzymol. 1979;62:151–160. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- (25).Requena L, Bornemann S. Barley (Hordeum vulgare) oxalate oxidase is a manganese-containing enzyme. Biochem. J. 1999;343:185–190. [PMC free article] [PubMed] [Google Scholar]
- (26).Hassan AK, Pardi LA, Krzystek J, Sienkiewicz A, Goy P, Rohrer M, Brunel L-C. Ultrawide band multifrequency high-field EPR technique: A methodology for increasing spectroscopic information. J. Magn. Reson. 2000;142:300–312. doi: 10.1006/jmre.1999.1952. [DOI] [PubMed] [Google Scholar]
- (27).Petrenko A, Maniero AL, van Tol J, MacMillan F, Li YJ, Brunel L-C, Redding K. A high-field study of P700+. in wild-type and mutant photosystem I from Chlamydomonas reinhardtii. Biochemistry. 2004;43:1781–1786. doi: 10.1021/bi035466j. [DOI] [PubMed] [Google Scholar]
- (28).Feher G. Electron spin resonance experiments on donors in silicon. I. Electronic structure of donors by the electron nuclear double resonance technique. Phys. Rev. 1959;114:1219–1244. [Google Scholar]
- (29).Stoll S, Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- (30).Schweiger A, Jeschke G. Principles of Pulse Electron Paramagnetic Resonance. Oxford University Press; Oxford, U.K.: 2001. [Google Scholar]
- (31).Chakraborty S, Chakraborty N, Jain D, Salunke DM, Datta A. Active site geometry of oxalate decarboxylase from Flammulina velutipes: Role of histidine-coordinated manganese in substrate recognition. Protein Sci. 2002;11:2138–2147. doi: 10.1110/ps.0206802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Christendat D, Saridakis V, Dharamsi A, Bochkarev A, Pai EF, Arrowsmith CH, Edwards AM. Crystal structure of dTDP-4-keto-deoxy-D-hexulose 3,5-epimerase from Methanobacterium thermoautotropicum complexed with dTDP. J. Biol. Chem. 2000;275:24608–24612. doi: 10.1074/jbc.C000238200. [DOI] [PubMed] [Google Scholar]
- (33).Lawrence MC, Izard T, Beuchat M, Blagrove RJ, Colman P. Structure of phaseolin at 2.2 Å resolution. Implications for a common vicilin/legumin structure and the genetic engineering of seed storage proteins. J. Mol. Biol. 1994;238:748–776. doi: 10.1006/jmbi.1994.1333. [DOI] [PubMed] [Google Scholar]
- (34).Boldt YR, Whiting AK, Wagner ML, Sadowsky MJ, Que L, Jr., Wackett LP. Manganese(II) active site mutants of 3,4-dihydroxyphenylacetate 2,3-dioxygenase from Arthrobacter globiformis strain CM-2. Biochemistry. 1997;36:2147–2153. doi: 10.1021/bi962362i. [DOI] [PubMed] [Google Scholar]
- (35).Kehres DG, Maguire ME. Emerging themes in manganese transport, biochemistry, and pathogenesis in bacteria. FEMS Microbiol. Rev. 2003;27:263–290. doi: 10.1016/S0168-6445(03)00052-4. [DOI] [PubMed] [Google Scholar]
- (36).Kliegman JI, Griner SL, Helmann JD, Brennan RG, Glasfeld A. Structural basis for the metal-selective activation of the manganese transport regulator of Bacillus subtilis. Biochemistry. 2006;45:3493–3505. doi: 10.1021/bi0524215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Que Q, Helmann JD. Manganese homeostasis in Bacillus subtilis is regulated by MntR, a bifunctional regulator related to the diphtheria toxin repressor family of proteins. Mol. Microbiol. 2000;35:1454–1468. doi: 10.1046/j.1365-2958.2000.01811.x. [DOI] [PubMed] [Google Scholar]
- (38).Jeong JJ, Fushinobu S, Ito S, Jeon BS, Shoun H, Wakagi T. Characterization of the cupin-type phosphoglucose isomerase from the hyperthermophilic archaeon Thermococcus litoralis. FEBS Lett. 2003;535:200–204. doi: 10.1016/s0014-5793(02)03900-5. [DOI] [PubMed] [Google Scholar]
- (39).Un S, Dorlet P, Voyard G, Tabares LC, Cortez N. High-field EPR characterization of manganese reconstituted superoxide dismutase from Rhodobacter capsulatus. J. Am. Chem. Soc. 2001;123:10123–10124. doi: 10.1021/ja016258m. [DOI] [PubMed] [Google Scholar]
- (40).Hirsh DJ, Brudvig GW. Long-range electron spin-spin interactions in the bacterial photosynthetic reaction center. J. Phys. Chem. 1993;97:13216–13222. [Google Scholar]
- (41).Zhou Y, Bowler BE, Lynch K, Eaton SS, Eaton GR. Inter-spin distances in spin-labeled metmyoglobin. Biophys. J. 2000;79:1039–1052. doi: 10.1016/S0006-3495(00)76358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Chang CH, Svedružić D, Ozarowski A, Walker L, Yeagle G, Britt RD, Angerhofer A, Richards NGJ. EPR spectroscopic characterization of the manganese center and a free radical in the oxalate decarboxylase reaction. J. Biol. Chem. 2004;279:52840–52849. doi: 10.1074/jbc.M402345200. [DOI] [PubMed] [Google Scholar]
- (43).Tottey S, Harvie DR, Robinson NJ. Understanding how cells allocate metals using metal sensors and metallochaperones. Acc. Chem. Res. 2005;38:775–783. doi: 10.1021/ar0300118. [DOI] [PubMed] [Google Scholar]
- (44).Culotta VC, Yang M, O’Halloran TV. Activation of superoxide dismutases: Putting the metal to the pedal. Biochim. Biophys. Acta. 2006;1763:747–758. doi: 10.1016/j.bbamcr.2006.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Moore CM, Helmann JD. Metal ion homeostasis in Bacillus subtilis. Curr. Opin. Microbiol. 2005;8:188–195. doi: 10.1016/j.mib.2005.02.007. [DOI] [PubMed] [Google Scholar]
- (46).Zaharik ML, Finlay BB. Mn2+ and bacterial pathogenesis. Front. Biosci. 2004;9:1035–1042. doi: 10.2741/1317. [DOI] [PubMed] [Google Scholar]
- (47).Whitfield CD, Weissbach H. Binding of folate substrate to 5-methyltetrahydropteroyltriglutamate-homocysteine transmethylase. J. Biol. Chem. 1970;245:402–409. [PubMed] [Google Scholar]
- (48).Mizuno K, Whittaker MM, Bachinger HP, Whittaker JW. Calorimetric studies on the tight binding metal interactions of Escherichia coli manganese superoxide dismutase. J. Biol. Chem. 2004;279:27339–27344. doi: 10.1074/jbc.M400813200. [DOI] [PubMed] [Google Scholar]
- (49).Yamakura F, Kobayashi K, Ue H, Konno M. The pH-dependent changes of the enzymatic activity and spectroscopic properties of Fe-substituted manganese superoxide dismutase. A study on the metal-specific activity of Mn-containing superoxide dismutase. Eur. J. Biochem. 1995;227:700–706. doi: 10.1111/j.1432-1033.1995.tb20191.x. [DOI] [PubMed] [Google Scholar]
- (50).Quijano C, Hernandez-Saavedra D, Castro L, McCord JM, Freeman BA, Radi R. Reaction of peroxynitrite with Mn-superoxide dismutase. Role of the metal center in decomposition kinetics and nitration. J. Biol. Chem. 2001;276:11631–11638. doi: 10.1074/jbc.M009429200. [DOI] [PubMed] [Google Scholar]
- (51).Li T, Walker AL, Iwaki H, Hasegawa Y, Liu A. Kinetic and spectroscopic characterization of ACMSD from Pseudomonas fluorescens reveals a pentacoordinate mononuclear metallocofactor. J. Am. Chem. Soc. 2005;127:12282–12290. doi: 10.1021/ja0532234. [DOI] [PubMed] [Google Scholar]
- (52).Vance CK, Miller A-F. Spectroscopic comparisons of the pH dependencies of Fe-substituted (Mn) superoxide dismutase and Fe-superoxide dismutase. Biochemistry. 1998;37:5518–5527. doi: 10.1021/bi972580r. [DOI] [PubMed] [Google Scholar]
- (53).Ose DE, Fridovich I. Manganese-containing superoxide dismutase from Escherichia coli. Reversible resolution and metal replacements. Arch. Biochem. Biophys. 1979;194:360–364. doi: 10.1016/0003-9861(79)90628-3. [DOI] [PubMed] [Google Scholar]
- (54).Ose DE, Fridovich I. Superoxide dismutase. Reversible removal of manganese and its substitution by cobalt, nickel, or zinc. J. Biol. Chem. 1976;251:1217–1218. [PubMed] [Google Scholar]
- (55).Berrisford JM, Hounslow AM, Akerboom J, Hagen WR, Brouns SJJ, van der Ost J, Murray IA, Blackburn GM, Waltho JP, Rice DW, Baker PJ. Evidence supporting a cis-enediol-based mechanism for Pyroccocus furiosus phosphoglucose isomerase. J. Mol. Biol. 2006;358:1353–1366. doi: 10.1016/j.jmb.2006.03.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.