

1. Preface
Over the last decade it has become clear that the gene mutations that initiate carcinogenesis are an inevitable aspect of aging. By age 70, the incidence of cancerous lesions in the thyroid has been estimated to approach 100%, and a high percentage of histologically positive cancers can also be detected in colon, prostate, and breast1. Yet, only a small percentage of these carcinomas in situ ever progress to cause frank disease, creating a dilemma for determining which will eventually become life-threatening malignancies that require treatment. Lifestyle changes may decrease the incidence of some of these cancers, and chemoprevention research will hopefully further decrease their initiation. However, for the near future, more benefit may come from understanding what distinguishes malignant cancer from a benign carcinoma in situ.
As an alternative to conventional cytotoxic chemotherapy, biological therapies that target physiological processes required for malignant tumor growth have attracted much recent attention. Pioneering studies by the late Dr. Judah Folkman introduced the concept that recruitment of a blood supply is critical to solid tumor growth2. Benign lesions show the enhanced cell proliferation characteristic of cancer but fail to grow beyond a size limit set by the ability of nutrients and oxygen to diffuse into the lesion. In this state, cancerous cells can exist for many years in self-limiting cycle of proliferation and death3.
One way that such premalignant lesions progress toward malignant cancers is to initiate recruitment of blood vessels, a process known as the angiogenic switch. Much progress has been made toward understanding the molecular basis for this switch4. In addition to increasing production of pro-angiogenic factors, the angiogenic switch requires shutting off expression of endogenous anti-angiogenic factors. A number of the identified pro- and anti-angiogenic factors are proteins. Among the former, vascular endothelial growth factor (VEGF) and its receptor VEGFR2 have been successfully targeted by pharmaceutical companies using VEGF neutralizing antibodies and small molecule kinase inhibitors of the receptor. Several of these agents are now FDA approved anti-angiogenic drugs and show efficacy to extend the survival of cancer patients 5–7. The hope was that these drugs would convert malignant cancer to a survivable benign disease. This ideal has been realized in some animal tumor models8,9, but in clinical practice anti-angiogenic drugs only extend the lifespan of advanced cancer patients on average by less than 1 year. This may be due in part to the plasticity of tumors to induce alternate pro-angiogenic factors that bypass the targets of the existing drugs. Therefore, we need either to develop drugs to target all possible angiogenic factors produced by the tumor or to identify conserved aspects of the signal transduction pathways used by these factors that can be the targets for universal angiogenesis inhibitors. Such targets are viewed by systems biologists as signaling nodes10.
This review focuses on a group of angiogenic signaling nodes that are of increasing interest as targets for anti-angiogenic drug development. Subsequent to its discovery as a paracellular signaling molecule that is responsible for endothelium-dependent vasodilation, nitric oxide (NO) was found to also be a mediator of proangiogenic signaling by VEGF and other angiogenic growth factors11–13. We will discuss the sources and downstream targets of NO that play critical roles in angiogenesis and its regulation by the endogenous angiogenesis inhibitor thrombospondin-1 (TSP1). In addition to NO, two other bioactive gases are becoming recognized as important regulators of angiogenesis: carbon monoxide (CO) and hydrogen sulfide (H2S). Two additional redox-active molecules, superoxide (O2•−) and hydrogen peroxide (H2O2), play important roles in angiogenic signaling, both directly and through their chemical reactions with NO. We will discuss the mechanisms be which redox signaling regulates angiogenesis and prospects for targeting these signaling pathways for therapeutic prevention and control of tumor angiogenesis and growth.
Finally, studies in animals have shown that angiogenesis inhibitors can synergize with other standard modes of cancer treatment. A number of clinical trials are ongoing using angiogenesis inhibitors in combination with chemotherapeutics and radiotherapy14. We will discuss how aspects of redox signaling may contribute to these synergistic activities and may guide the optimization of such therapeutic combinations.
2. Introduction to angiogenesis
Angiogenesis is one of several processes that form new blood vessels in higher animals, but it has received the most research attention and popular interest due to its important roles in cancer and wound repair. During early embryogenesis, the first capillary networks form by a process known as vasculogenesis. Cells in the mesoderm differentiate into vascular endothelial cells and spontaneously connect to form a network of tubes known as a vascular plexus15. In contrast to angiogenesis, embryonic vasculogenesis occurs in the absence of blood flow. This primitive vascular network connects to primitive arteries and veins in the embryo, which establishes blood flow in the developing tissue. The directional flow is one signal that can promote differentiation of the vascular plexus into a hierarchical network of arteries, arterioles, capillaries, venules, and veins16. This differentiation process is known as arteriogenesis. Arteriogenesis is also directed by growth factors released from growing nerves in the embryo, which results in the parallel organization of blood vessels and nerved noted by early anatomical studies17.
During later development and in adult tissues, angiogenesis plays a major role in new blood vessel formation. Angiogenesis is defined as the formation of new blood vessels from an existing perfused vessel bed. This occurs by sprouting of endothelial cells in the vessel wall, either arterial or venous vessels depending on the soluble factors present18,19, which degrade and invade through the underlying basement membrane barrier and then further invade through the underlying extracellular matrix. As the leading cell moves forward, following endothelial cells proliferate and differentiate to form a luminal space. The leading cell eventually finds another vessel, with which it fuses to establish a patent perfused vessel. Further cycles of this process accompanied by arteriogenesis produces a mature vascular network.
In addition to endothelial cells, mature blood vessels require supporting smooth muscle cells. During development, these can be recruited from mesenchymal stem cells or from bone-marrow-derived cells. Arterial vessels develop a thick layer of well organized vascular smooth muscle cells (VSMC) to accommodate the greater hydrostatic pressure in the arterial vasculature. These arterial smooth muscle cells, as will be discussed in greater detail below, also play an important role in adjusting blood flow to specific tissues in response to changing metabolic needs. Veins also have well organized smooth muscle layers, but thinner than those in arteries. The VSMC in capillaries are known as pericytes. In contrast to large vessels, capillary endothelial tubes are not completely covered by pericytes. Rather, the pericytes play important roles in capillary stability and function by secreting factors that regulate endothelial cell function and through direct contact with the adjacent capillary endothelium20.
Due to the positive hydrostatic pressure in perfused vessels, a net flow of water, ions, and small solutes constantly occurs across the vessel wall. This is opposed by an osmotic gradient resulting from the lower macromolecular solute concentration in the interstitial space, but nonetheless, net fluid movement occurs from perfused vessels into the underlying tissue. To maintain a constant blood volume, higher animals have a second vascular network, the lymphatics, that return this fluid to the cardiovascular system21. Lymphatics are a blind ended tree of specialized vascular cells, which form by a process known as lymphangiogenesis.
It has recently become clear that angiogenesis is not the only mechanism responsible for neovascularization of tumors and wounds in the adult22. In adult tissues, vasculogenesis is mediated by recruitment of circulating endothelial precursor cells that differentiate from hematopoietic stem cells in the bone marrow. These along with specialized monocytic stem cells cooperate to form new vessels at sites of injury and in some cancers. The relative contribution of angiogenesis versus vasculogenesis to tumor neovascularization is currently being actively debated, but it is clear that some tumors depend significantly on bone marrow precursor recruitment, whereas this plays a minimal role in others23,24. Likewise, the role of lymphangiogenesis in tumor growth appears to be quite variable, with a subset of tumors being highly dependent on this process25.
This review focuses on the role of redox signaling in angiogenesis and angiogenesis inhibition, but the reader should remain aware that some proangiogenic factors can stimulate vasculogenesis, lymphangiogenesis, and arteriogenesis as well as angiogenesis. Correspondingly, angiogenesis inhibitors can often inhibit more than one of these processes. Therefore, the redox signaling pathways discussed here have been initially defined and are best understood in the context of angiogenesis, but their true function may be more general.
3. Molecular regulation of Angiogenesis
3.1 Vascular endothelial growth factor family
Angiogenesis is stimulated by several protein growth factors and steroids (Table 1). Among these, the vascular endothelial growth factor (VEGF) family plays a major role. VEGF-A is essential for vasculogenesis and angiogenesis during embryonic development and similarly serves as a major angiogenic factor in tumors26. A VEGF-A heterozygous null mutant in mice, retaining one functional copy of the gene, is also embryonic lethal. Therefore, a precisely regulated level of VEGF is critical to this process. Studies using conditional VEGF-A knockouts in mice have further refined the function of VEGF-A in adult mammals. Conditional deletion of VEGF-A in muscle revealed an important role in exercise-induced angiogenesis27. Deletion in kidney podocytes resulted in proteinuria and thrombotic microangiopathy of the kidney28. Deletion in endothelial cells showed that, in addition to its paracrine stimulation of angiogenesis, VEGF-A is a critical autocrine factor for maintaining endothelial function in the adult29.
Table 1.
Major Angiogenic Factors and their Receptors
| Factor | Receptors | Function | References |
|---|---|---|---|
| VEGF-A | VEGFR2, VEGFR1, Neuropilin-1 | Embryonic and adult vasculogenesis, angiogenesis, vascular permeability | 26 |
| VEGF-B | VEGFR1, Neuropilin-1 | Revascularization after myocardial infarction | 34 |
| VEGF-C | VEGFR3, Neuropilin-2, (VEGFR2) | Embryonic lymphangiogenesis | 390 |
| VEGF-D | VEGFR3, Neuropilin-2 | Lymphangiogenesis? | 390 |
| PlGF | VEGFR1 | Tumor- or ischemia-induced vascularization | 35, 391 |
| FGF1 (acidic FGF) | FGFR1, FGFR2, FGFR3, FGFR4 | Tumor angiogenesis? | 42 |
| FGF2 (basic FGF) | FGFR1, αvβ3 integrin, heparan sulfate proteoglycans | Inflammation- and tumor- induced angiogenesis | 42, 43 |
| Angiopoietin-1 (Angiopoietin-2) | Tie2 | Remodeling of embryonic and adult vasculature, arteriogenesis | 392 |
| Adrenomedullin | calcitonin-receptor-like receptor/receptor activity modifying protein | Embryonic and tumor angiogenesis | 54,393, 53 |
| Interleukin-8/CXCL8 | CXCR1 and CXCR2 | Tumor- and ischemia-induced angiogenesis | 49 |
| Platelet-derived growth factor-B | PDGFR | Pericyte recruitment required for embryonic and tumor angiogenesis | 394 |
| Steroids | Estrogen and glucocorticoid receptors, ER46 | Angiogenesis | 102 |
Based on its key role in angiogenesis, VEGF-A has attracted the most attention to date as a molecular target for inhibiting tumor angiogenesis30. The therapeutic antibodies Avastin (bevacizumab) and Lucentis bind to this growth factor and inhibit its activity. VEGF-A stimulates proliferation and motility of endothelial cells by binding to VEGF receptor-2 (VEGFR2) and to a lesser extent VEGFR1. VEGF-A also regulates vascular permeability by regulating endothelial cell-cell junctions and transcytosis through VEGFR231,32. VEGFR2 is a tyrosine kinase receptor, and several therapeutic angiogenesis inhibitors, including the FDA-approved drugs sorafenib and sunitinib, act at least in part by inhibiting this kinase activity33.
In contrast to VEGF-A, VEGF-B is not required for embryonic vascular development, but it plays an essential role in the adult heart for revascularization of ischemic tissue following a myocardial infarct34. VEGF-B signals via VEGFR1, which is also the signaling receptor for placental growth factor (PlGF). Like VEGF-B, PlGF is not required for embryonic angiogenesis, but its absence impedes ischemia-induced angiogenesis and neovascularization of tumors in adult animals35,36.
VEGF-C and VEGF-D play roles in lymphangiogenesis and bind to VEGFR3 expressed on lymphatic endothelium. Deletion of VEGFR3 in mice results in death at embryonic day 10.5, before the emergence of the lymphatic vessels37. VEGF-C is essential for the formation of lymph sacs from embryonic veins, and its absence results in embryonic death of null mice38. In adult tumor-bearing mice, VEGF-C is required for lymphatic metastasis39. In contrast, VEGF-D is not required for embryonic development, possibly because VEGF-C can substitute for its function. However, transgenic expression of VEGF-D can complement some defects in a VEGF-C null mouse40. Therefore, the functions of VEGF-C and VEGF-D may be somewhat redundant.
3.2 Fibroblast growth factor family
Fibroblast growth factor-2 (FGF2, also known as basic FGF) is another major mitogen and motility factor for endothelial cells. Like VEGF-A, FGF2 is sufficient to stimulate a full angiogenic response in a fertilized chicken egg or in the cornea of mice and rats, but it is not necessary for embryonic vascular development41. FGF2 stimulates angiogenesis via its Tyr kinase receptor FGFR1. However, engaging this receptor is not sufficient for signaling, which also requires heparan sulfate proteoglycans and αvβ3 integrin as co-receptors42,43. Angiogenesis of some tumors is dependent on FGF2, prompting interest in antagonists of this factor and its receptor as therapeutic angiogenesis inhibitors44. Several agents that inhibit FGF2 binding to heparan sulfate proteoglycans have progressed to human clinical trials.
3.3 Angiopoietins
Angiopoietins are another family of growth factors that play essential roles in embryonic vascular development. Mice lacking either the angiopoietin-1 (Ang1) or its receptor Tie2 die between embryonic day 9.5 and 12.5 due to lack of remodeling of the primary vascular capillary plexus44,45. Ang1-signaling via Tie2 mediates remodeling and stabilization of cell–cell and cell–matrix interactions and plays a role in the recruitment of pericytes to the nascent vessels. Ang2(−/−) mice show defects in developmental remodeling of lymphatic vessels46,47. In contrast, the absence of Ang2 has more subtle effects on vascular development. Ang2(−/−) mice show defects in developmental remodeling of lymphatic vessels46,47. Lack of Ang2 also causes defective regression of the fetal vasculature in the vitreous body of the eye and disorganization and hypoplasia of the intestinal and dermal lymphatic capillaries. In adult mice, Ang2 was up-regulated in response to femoral artery ligation, and subsequent vascular remodeling in the ischemic limb was impaired by a specific Ang2 inhibitor, L1-1048. The authors proposed that Ang2 promotes arteriogenesis in this wound repair model. In vitro evidence indicates that Ang2 has both stimulatory and inhibitory effects on angiogenic responses.
Interleukin-8 is another angiogenic cytokine49. Recent evidence suggests that it also mediates the stimulation of endothelial cell proliferation and migration by Ang150. Thus, IL-8 may mediate some pro-angiogenic activities of Ang1. The angiopoietin family has been further expanded by the discovery of a family of related factors. These include mouse Ang3, human Ang4, and seven angiopoietin-like proteins51. Different members of this family have pro- or anti-angiogenic activities in vitro, but their pathophysiological functions in angiogenesis in vivo are still under investigation.
3.4 Adrenomedullin
Adrenomedullin is a 52 amino acid peptide that is produced by proteolytic cleavage of its precursor preproadrenomedullin52. Adrenomedullin signals by binding to a cell surface seven transmembrane G-protein-coupled receptor. Adrenomedullin is highly expressed in certain cancers and is an important angiogenic factor for these tumors53. Deletion of adrenomedullin in mice is embryonic lethal at day 13.5–14 due in part to disorganization of endothelial cells and their underlying basement membrane54. Therefore, adrenomedullin is necessary for embryonic vascular development.
3.5 Steroids
Steroid hormones also play important roles in angiogenesis55. Glucocorticoids are an essential growth factor for endothelial cells in vitro. Estrogens also stimulate endothelial cell growth in vivo, and induce NO signaling56. Effects of estradiol on endometrial angiogenesis in vivo include induction of VEGF, FGF2, Ang1, PlGF, eNOS, and soluble guanylate cyclase (sGC)57. Therefore, estrogens coordinately induce upstream and downstream elements of NO/cGMP signaling under conditions where they stimulate angiogenesis. The estrogen metabolite 2-methoxyestradiol has been tested in clinical trials as an angiogenesis inhibitor58. However, its anti-tumor activity may be partially independent of inhibiting angiogenesis or antagonism of estrogens57.
3.6 Pro-angiogenic factors in cancer
The diversity of angiogenic factors combined with the ability of cancers to change their expression of specific factors creates a challenge for therapeutic control of tumor angiogenesis. It is clear from experimental animal tumor studies and from clinical experience with existing angiogenesis inhibitors in cancer patients that tumors become resistant to specific angiogenesis inhibitors59. Thus, the current FDA-approved drugs typically extend cancer patent survival by 3–6 months, but long term control of cancer growth by angiogenesis inhibitors as single agents is rare. Current clinical trials are exploring whether combinations of angiogenesis inhibitors are more effective, but at present such efforts are limited by the small number of inhibitors available. We do not know how many angiogenic factors a specific tumor can make, nor can we predict which will become dominant when one pro-angiogenic pathway is inhibited. In developing new anti-angiogenic therapies, it is important to consider whether drug combinations can be found that will effectively inhibit all major tumor angiogenic factors, or whether multiple angiogenic factors share some common downstream signaling pathway that would be a more effective drug target than the individual growth factors or their receptors.
4. Endogenous angiogenesis inhibitors
One way to approach this problem for cancer therapy is to study how angiogenesis is normally controlled during development and in adults and how this process becomes dysregulated in nonmalignant disease. Angiogenesis is highly controlled during embryonic development, with vascular density closely matched to the metabolic needs of a given tissue. In healthy adult individuals, angiogenesis is stimulated in a controlled manner during wound healing, cyclically in the uterine decidual lining during the menstrual cycle, and in specific muscle beds in response to exercise training. Yet, with increasing age and chronic conditions such as diabetes, the ability to stimulate angiogenesis becomes impaired, and tissues can become ischemic due to lack of adequate blood flow60. Conversely, excessive angiogenic responses are factors in growth of keloids and uterine fibroids61–63. It is becoming clear that these diseases can not be explained merely by an excess or deficit in specific angiogenic factors but must be understood in terms of a net balance between angiogenic factors and endogenous angiogenesis inhibitors.
4.1 Thrombospondins
The first identified endogenous angiogenesis inhibitor was TSP1. A 140 kDa protein secreted by an immortalized hamster cell line was found to block angiogenesis in vivo, and its expression was controlled by a tumor suppressor gene64. Loss of the tumor suppressor was accompanied by loss of the secreted protein and acquisition of angiogenic activity. The 140 kDa protein was purified and identified to be a proteolytic fragment of TSP1. Independently, two other groups reported in the same year that native TSP1 purified from platelets potently inhibits endothelial cell proliferation and chemotaxis stimulated by FGF265,66. Subsequent studies confirmed the ability of TSP1 to inhibit angiogenesis in the rat cornea and the chick rat chorioallantoic membrane developmental angiogenesis assays64,67. This activity of TSP1 was extended to tumor angiogenesis by re-expressing TSP1 in a tumorigenic human melanoma cell line MDA-MDB-43568. Transfected clones over-expressing TSP1 formed slower growing tumors in athymic mice that exhibited decreased densities of tumor blood vessels. Similar results in vivo were found when TSP1 was re-expressed in hemangioma, v-src-transformed NIH 3T3, cutaneous squamous cell carcinoma, glioblastoma, and hematopoietic tumor cell lines69. Furthermore, over-expression of TSP1 in the skin or mammary glands of tumor- mice suppressed formation and angiogenesis of carcinogen-induced premalignant epithelial hyperplasias and spontaneous mammary tumors, respectively70. Conversely, mice lacking TSP1 showed increased tumor growth when crossed with mice carrying cancer promoting mutations in APC(Min/+) or mice lacking the tumor suppressor p5371. In a xenograft model of tumor dormancy, high expression of TSP1 was characteristic of nonangiogenic tumor cells that maintained prolonged dormancy when implanted in mice72.
A second member of the thrombospondin family, TSP2, was subsequently shown to be an angiogenesis inhibitor 73. As found for TSP1, over-expression of TSP2 in tumor cells suppressed tumor growth and angiogenesis in mice74, and mice lacking TSP2 showed increased susceptibility to skin carcinogenesis and earlier switching to an angiogenic phenotype75.
4.2 Angiostatin
Many additional angiogenesis inhibitors have been and continue to be discovered (Table 2). Unlike TSP1, which is active in its native form, several of these inhibitors are derived by proteolysis of proteins that serve other physiological functions. Angiostatin is a proteolytic fragment of plasminogen, a precursor of an important protease for resolution of blood clots 76. Plasminogen lacks anti-angiogenic activity, but several of its Kringle repeats are potent inhibitors when released by proteolysis or expressed as recombinant proteins77.
Table 2.
Endogenous Angiogenesis Inhibitors and their Receptors
| Calibri | Calibri | Calibri | Calibri |
|---|---|---|---|
| Thrombospondin-1 | CD36, CD47, HSPG | 150, 395, 153, 146 | |
| Thrombospondin-2 | CD36 | 396 | |
| Platelet factor-4 | CXCR3-B, HSPG | 397 | |
| Interferon-α, β | IFN-α/β receptor (IFNAR1/IFNAR2) | 398, 399 | |
| chondromodulin-I, tenomodulin | ? | 400 | |
| Pigment epithelium- derived factor | ? | 401 | |
| TIMP2 | α3β1 integrin | 402 | |
| angiostatin | plasminogen | Fo-F1 ATPase, angiomotin, αvβ3 | 76,403 |
| endostatin | collagen XVIII | Nucleolin, other | 78,404 |
| Soluble VEGFR1 (sFlt1) | VEGFR1 | Decoy receptor for VEGF | 26 |
| Arresten, Canstatin and Tumstatin | Type IV collagen | Integrins α1β1, α3β1, α6β1, αvβ3 | 81 |
| Vasoinhibins | Prolactin, growth hormone, placental lactogen | ? | 405 |
| Vasostatin | calreticulin | ? | 406 |
| NK4 | Hepatocyte growth factor | c-Met, other? | 407 |
| Endorepellin | Perlecan | α2β1 integrin | 82 |
4.3 Inhibitors derived by proteolysis of extracellular matrix
Several extracellular matrix collagens contain noncollagenous domains that have cryptic anti-angiogenic activities. The first identified inhibitor in this family was endostatin, which is a fragment of type XVIII collagen78. The protein was first identified as a circulating angiogenesis inhibitor in tumor bearing mice. It has been expressed as a recombinant protein and progressed to human clinical trials as a therapeutic angiogenesis inhibitor79. Endostatin levels are controlled by the activities of proteases that release it from its parent collagen as well as by H2O2 and NO/cGMP signaling 80. Similar inhibitors have been identified as proteolytic fragments derived from three subunits of basement membrane type IV collagen81 and as the endorepellin fragment derived from the large basement membrane proteoglycan perlecan82.
5. NO signaling in the cross-talk between pro- and anti-angiogenic factors
5.1 Pro-angiogenic signal transduction
Angiogenic growth factors typically engage plasma membrane receptors that have tyrosine kinase activity26,42,83. The signal transduction pathways activated by growth factors binding to these receptors can be quite complex (Fig. 1). In addition to direct signaling through receptor kinase activation, lateral cross talk involving other membrane components play important roles. Syndecans and other heparan sulfate proteoglycans play important roles in FGFR dimerization, activation and signaling. Cross talk with neuropilins, integrins, and VE-cadherin are important for VEGF receptor signaling84,85.
Figure 1.
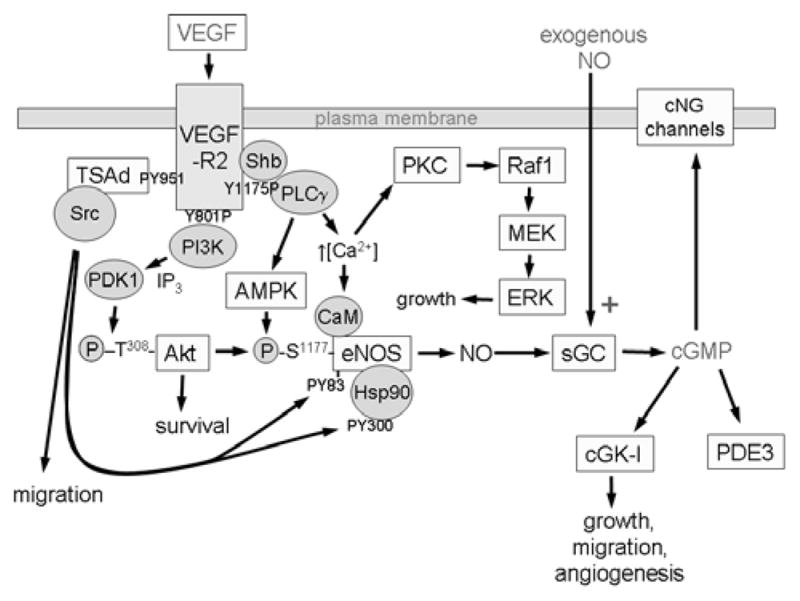
VEGF binding to VEGFR2 on endothelial cells activates its Tyr kinase activity and results in autophosphorylation of the receptor at several cytoplasmic sites. The phosphorylated Tyr serve as docking sites for specific signaling molecules. Phosphatidyinositol 3-kinase (PI3K) is recruited at phosphorylated Tyr801, increasing inositol 1,4,5-triphosphate (IP3) formation, which in turn activates 3-phosphoinositide-dependent protein kinase-1 (PDK1) to phosphorylate and activate Akt, which phosphorylates human eNOS at Ser117712,13. This phosphorylation activates eNOS and decreases its calcium dependence. VEGFR2 phosphorylation at Tyr951 recruits TSAd and Src, which phosphorylates heat shock protein 90 (Hsp90) at Tyr300, which induces Hsp90 binding to eNOS and activation of NO synthesis 89, and phosphorylates eNOS at Tyr83, which is also required for eNOS activation 90. Phosphorylation of VEGFR2 at Tyr1175 recruits phospholipase-Cγ (PLCγ), which mobilizes intracellular Ca2+ and thereby further activates eNOS in a calmodulin (CaM)-dependent manner. PLCγ also increases AMP kinase (AMPK)-mediated eNOS phosphorylation at Ser117788. NO produced by eNOS binds to the heme on soluble guanylate cyclase (sGC) to stimulate cGMP synthesis. cGMP in turn activates cGMP-dependent protein kinase (cGK-I) and cGMP-gated channels to regulate downstream targets that increase endothelial cell proliferation, migration, survival, and permeability127. cGMP also binds to and regulates several phosphodiesterases that terminate that cGMP signal or mediate cross talk with cAMP signaling by hydrolyzing that second messenger. Additional parallel signaling through Src, Akt, and the protein kinase C-mitogen-activated protein kinase pathway (PKC-Raf1-MEK-ERK) synergizes with NO/cGMP signaling to support each of these endothelial cell responses.
5.2 NO in VEGF signaling
Autophosphorylation of VEGFR2 at Tyr951 mediates recruitment of T cell-specific adapter (SH2 domain-containing protein 2A), which mediates recruitment of Src kinase26. Autophosphorylation of VEGFR2 at Tyr1175 recruits phospholipase Cγ and Shb. Phosphorylation of Tyr801 is required for recruitment of the p85 subunit of PI 3-kinase and consequent activation of Akt86. These proximal targets in turn activate a number of downstream targets, resulting in increased endothelial proliferation, motility, and permeability. Relevant to the present discussion, all three pathways have been implicated in VEGF-mediated activation of eNOS (Fig. 1). Akt phosphorylates eNOS at Ser1177 and induces NO synthesis12,13. PLCγ signaling increases intracellular Ca2+, which binds to calmodulin to further activate eNOS87. PLCγ signaling also activates AMP kinase, which further activates eNOS by phosphorylation at Ser1177, 88. Src acts on at least two targets to activate eNOS. It phosphorylates heat shock protein-90 (Hsp90) at Tyr300, which induces Hsp90 binding to eNOS and activation of NO synthesis89. Simultaneously, VEGF binding induces Src-dependent phosphorylation of eNOS at Tyr8390. This phosphorylation is also required for eNOS activation.
In mice lacking eNOS, VEGF produces a decreased angiogenesis response relative to wild type mice as assessed using a type I collagen gel implanted under a cranial window91. iNOS null mice show less impairment in VEGF-induced angiogenesis, indicating that eNOS is the major mediator of the proangiogenic activity of VEGF in this assay. NO levels measured in the implanted gels were increased in the respective mice in proportion to the observed angiogenic responses. Therefore, eNOS mediates an NO-dependent angiogenic response to VEGF in vivo.
There are some discrepancies in the literature regarding NO mediating VEGF driven angiogenesis92. These studies employ NO donating molecules at supraphysiological concentrations as well as molecules that have other relevant reactivities in addition to directly releasing NO moieties. The reader is referred to work detailing the characteristics and use of NO donors for additional information on this93. NO is known to have biphasic dependence on concentration for a number of different aspects of cell proliferation and migration92,94,95. However, the responses triggered by NO donors at concentrations appropriate to activate sGC are consistently pro-angiogenic.
5.3 A broader role of eNOS in angiogenic factor signaling
VEGF is not the only angiogenic growth factor that signals via activation of eNOS96. Angiogenic responses to angiopoietin-1 are deficient in eNOS-null mice97. Angiopoietin-related growth factor (AGF) enhances blood flow in a mouse hind-limb ischemia model through induction of angiogenesis and arteriogenesis. In vitro, AGF increases NO production by human umbilical venous endothelial cells98. Furthermore, AGF did not restore blood flow to ischemic hind-limbs of either mice receiving the NOS inhibitor L-NAME or in eNOS knockout mice. Therefore, NO may generally mediate pro-angiogenic activities of angiopoietin family members.
Although adrenomedullin null mice are not viable, heterozygous nulls survive to adulthood. Measurement of NO synthesis by perfused kidneys from adrenomedullin +/− mice showed an approximately 50% decrease in NO levels99. These mice displayed a higher resting mean arterial blood pressure that wild type controls, indicating a functional deficiency in NO activity. This was verified to depend on adrenomedullin regulation of eNOS by treatment with a NOS inhibitor, which resulted in a smaller increase in blood pressure in heterozygous null mice than in wild type. Therefore, endogenous adrenomedullin is an important physiological inducer of vascular NO synthesis.
Lysophospholipids play broad roles in regulating cell behavior, and one of their targets is NO signaling. Sphingosine-1-phosphate (S1P) is an important pro-survival and chemotactic factor for endothelial cells. S1P activates eNOS in endothelial cells via the phosphatidylinositol-3-kinase (PI3K)/Akt pathway96. S1P-stimulated eNOS phosphorylation and NO production is blocked by inhibition of PI3K or Akt100. Similarly, S1P-stimulated capillary growth into subcutaneously implanted Matrigel plugs in mice was significantly reduced in mice that received the NOS inhibitor L-NAME. Lysophosphatidic acid also signals in endothelial cells through G protein-coupled Edg family receptors, and this signaling activates eNOS96.
FGF2 increases the expression of eNOS mRNA and the production of NO in human umbilical vein and calf pulmonary artery endothelial cells when cultured on three-dimensional fibrin gels101. However, other studies suggest that FGF2 stimulates angiogenesis by inducing expression of VEGF-A and its receptors42, so FGF2 may only indirectly regulate endothelial cell NO synthesis.
Proangiogenic signaling by estrogens involves both the conventional nuclear estrogen receptor-α and an N-terminal truncated isoform known as ER46, which is expressed in endothelial cells102. ER46 interacts with Src and mediates rapid activation of eNOS at the plasma membrane. In the presence of 17β-estradiol, phosphorylation of Src at Tyr419 is stimulated, and eNOS becomes phosphorylated at Ser1177. ER46 colocalizes with caveolin-1 in endothelial cells, implying that ER46 associates with the eNOS-caveolin complex in endothelial cells.
Insulin and insulin-like growth factors have pro-angiogenic activities in vivo either alone or in conjunction with other growth factors103,104. Insulin signaling through the insulin receptor tyrosine kinase signals via PI3-kinase and activates eNOS by promoting Ser1177 phosphorylation as well as eNOS de-nitrosylation 96.
5.4 Is NO necessary for angiogenesis?
Although the above studies establish that eNOS is necessary for stimulation of angiogenic responses by several growth factors, it remains unclear that NO itself is essential for angiogenesis. From the perspective of identifying therapeutic approaches to control pathological angiogenesis, this question needs to be addressed separately for developmental and pathological angiogenesis. The eNOS null mouse is viable and lacks major defects in its vascular anatomy except for a decrease in capillary density in the left ventricular myocardium, which is associated with abnormal aortic valve development105. Apart from this tissue, developmental angiogenesis does not appear to require eNOS. Note that this result does not prove that NO is not required for developmental angiogenesis since NO is produced by two other NOS isoforms. Mice lacking all three NOS isoforms also remain viable, and their hemodynamic parameters are similar to those of an eNOS null mouse106. The triple null mice lack any detectable NO synthesis, indicating that embryonic angiogenesis can occur in the complete absence of endogenous NO synthesis. Even in these triple nulls, however, some NO may be present due to nitrite/nitrate reductase activities that can derive NO from dietary nitrate/nitrite107. Furthermore, as discussed below, some additional gasotransmitters may activate sGC to compensate for the loss of NO signaling following complete NOS gene disruption, and VEGF is known to activate synthesis of some of these.
In adult mice, the absence of NOS3 results in specific defects in angiogenic responses to several angiogenic factors including VEGF, Ang1, AGF, angiotensin-II, PlGF, and RANKL. A differential requirement for eNOS was reported for splitting versus sprouting angiogenesis108. These studies may begin to provide insight into why eNOS would be selectively required for certain types of pathological angiogenesis, but more research is needed to identify signaling pathways that are unique to pathological versus developmental angiogenesis.
A role for eNOS in tumor angiogenesis is supported by several studies109. Consistent with this data, treatment with L-NAME but not D-NAME inhibited neovascularization induced by the C3L5 murine mammary adenocarcinoma cell line110. Furthermore, intratumoral vessel density and morphology were specifically altered in B16 melanomas grown in eNOS null mice compared to either wild type or iNOS null mice111. In light of the latter studies in mice, it is interesting that polymorphisms in NOS3 have been associated with increased risk for several cancers in humans112–117. A -786T>C polymorphism is particularly notable because it was associated with increased eNOS expression and significantly increased progression but not the occurrence of prostate cancer118.
5.5 Nitric oxide signaling in cardiovascular physiology
To understand the implications of widespread eNOS regulation by pro-angiogenic and anti-angiogenic factors, it is important to recognize the broader role of NO in vascular biology (Fig. 2). A molecular understanding of the role of NO in mammalian vascular physiology began with the demonstration that organonitrates could activate sGC119 and that NO caused vascular dilation120. The physiological relevance of these observations became obvious when NO was found to be produced by mammalian cells121, and endothelial cells lining of blood vessels were shown to be a source of endogenous NO production122. Near the same time, a specific calcium-dependent enzyme that synthesizes NO was isolated123. Endogenous NO functions to activate many critical survival based pathways in multiple mammalian cell types. These processes derive from the ability of NO to activate the heme protein sGC. Under basal conditions (no NO), sGC catalyzes the production of the second messenger molecule cyclic guanosine monophosphate (cGMP) from GTP. This process is regulated by an allosteric 5-coordinate ferrous heme prosthetic group that is ligated to sGC by an axial Fe2+ - histidine bond124. NO binds to the Fe2+ of sGC activating it over 200-fold. This is mechanistically dependent on NO induced loss of the axial Fe2+ - histidine bond124. This mode of binding is significant in light of the presence of other diatomic heme ligands (O2 and CO) that could compete with NO but prefer to bind reduced hemes in a 6-coordinate fashion. Therefore, these molecules bind to sGC with a much lower affinity than NO (8 and 4 orders of magnitude respectively125) and either do not activate sGC (O2) or do so to a much lesser extent than NO (CO)126. As an intracellular second messenger, cGMP produced by activated sGC amplifies the NO signal and functions to activate a number of signaling pathways that enhance vascular cell survival127. Physiologic levels of NO promote vascular cell proliferation and migration and vascular smooth muscle relaxation via the cGMP signaling pathway. In wound healing environments, sustained NO signaling stimulates angiogenesis. At the level of organ systems NO plays an important acute role to modulate arterial blood flow. By relaxing the contraction of the VSMC of arteries, NO increases vessel diameter, lowers arterial resistance and enhances blood flow to tissues. Thus, endothelial cells lining blood vessels self-regulate their local arterial tone by continuously producing NO.
Figure 2.
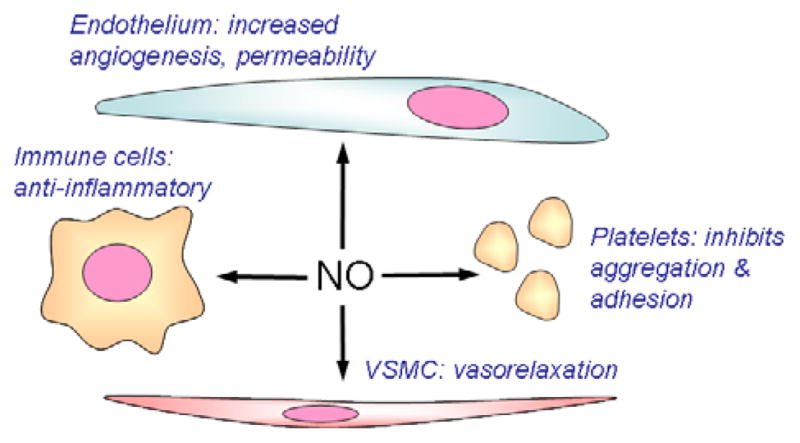
Nitric oxide acts on several target cells in blood vessels that are important for tumor biology. NO acts in endothelial cells to stimulate angiogenesis and increase vascular permeability. The former is important to neovascularization to support tumor growth. The latter contributes to the characteristic leakiness of the tumor vasculature. NO stimulates proliferation and migration of VSMC, which can contribute to angiogenesis. NO also relaxes VSMC, which can either increase or decrease tumor perfusion. NO inhibits platelet adhesion and aggregation. Platelet adhesion to circulating tumor cells plays an important role in metastatic spread of cancers408. Finally, NO has inhibitory activities for immune cells, which can limit host anti-tumor immunity.
Based on its high diffusion coefficient, the signaling activity of NO is not restricted to its site of synthesis, and cell membranes are not a significant barrier. Apart from efficient scavenging by abundant heme proteins128, NO is free to activate down stream signaling throughout the local environment. Thus, until recently, it was unclear whether cells could regulate NO-driven responses other than by hydrolysis of the second messenger cGMP. A family of phosphodiesterases (PDE) hydrolyze cGMP clearly serve this function and have been major targets for pharmacological intervention to enhance NO signaling responses129. Yet it remains a paradox that exogenous NO via inhaled gas or nitroglycerin as a stable form of deliverable NO remains physiologically active, but inhibition of PDE activity does not dramatically alter blood pressure 130,131. These empiric findings suggest that NO signaling is subject to additional regulatory controls.
5.6 Angiogenic factors as NO-dependent vasodilators
Despite the broad recognition that NO is a pleiotropic regulator of cardiovascular physiology, the idea that angiogenic factors and their inhibitors could have acute cardiovascular effects has attracted little attention. This is slowly changing as the frequency of hypertensive and prothrombotic side effects of therapeutic angiogenesis inhibitors has become clear132–134, 28. The ability of VEGF to induce NO-dependent relaxation of arterial segments was reported in 1993135, but direct demonstration of an acute hypotensive activity of VEGF was only confirmed recently136. This acute vasodilator activity is not unique to VEGF signaling through VEGFR2 because similar vasodilator activity was found for PlGF137. However, PlGF exhibited its NOS-dependent vasodilator activity via VEGFR1 rather than VEGFR2. Furthermore, adrenomedullin acts as a peripheral and coronary vasodilator by stimulating NO signaling138.
5.7 Thrombospondin-1 blocks NO-driven angiogenesis
In studying the anti-angiogenic activity of TSP1 we discovered that the NO/cGMP pathway is an important target of its signaling in endothelial cells139. An in vitro assay model was employed that simulates wound healing. Fresh skeletal muscle biopsies are implanted into a 3D matrix of type I collagen and incubated with growth medium. Within 72 hours under these conditions a predictable degree of vascular cell invasion and migration through the matrix occurs. Tissue samples from TSP1 null mice under the same growth conditions demonstrate enhanced angiogenic response compared to those from wild type mice. More importantly, NO-stimulated angiogenic response is always dramatically greater in explants from null animals compared to wild type. Thus, endogenous levels of TSP1 are sufficient to limit NO-driven angiogenesis. NO-stimulated increases in proliferation, matrix adhesion, and migration of primary human vascular endothelial cells are all potently blocked by TSP1. Concentrations of TSP1 as small as 10 pM are sufficient to block pro-survival responses to NO in endothelial cells. These amounts are well within the demonstrated concentrations of TSP1 in plasma. Similarly, primary endothelial cells from wild type and TSP1 null mice demonstrate that endogenous TSP1 limits NO-stimulated increases in cell proliferation and migration.
Increased numbers of VSMC were found in the vascular cell outgrowth from the muscle explants of TSP1 null mice, suggesting that TSP1 also limits NO-driven responses in VSMC. The activity of TSP1 to inhibit NO-driven responses was confirmed in human aortic VSMC, and murine primary aortic VSMC from TSP1 or CD47 null mice were found to have elevated resting and NO-stimulated cGMP levels140. Whereas TSP1 typically has opposing effects on endothelial and VSMC proliferation in the absence of NO65,141–143, coordinate regulation of VSMC and endothelial cells in the presence of NO may facilitate a balanced of both cell types required for angiogenesis.
5.8 NO signaling is regulated through the TSP1 receptors CD36 and CD47
TSP1 was first identified in 1971 as a major secretory component of activated platelets 144,145, and its anti-angiogenic activity was recognized in 199064–66. Different domains of TSP1 are now known to have pro- or anti-angiogenic activities by engaging at least 9 different receptors on endothelial cells146–150. These include several integrins, heparan sulfate proteoglycans, LDL receptor-related protein, CD36, and CD47.
In chemotaxis assays, CD36 deficient endothelial cells were insensitive to inhibition by TSP1, and re-expression of CD36 restored the inhibitory effect of TSP1150. Lack of TSP1 activity to inhibit corneal angiogenesis in CD36-null mice further indicated that the anti-angiogenic activity of TSP1 requires CD36 binding151. A recombinant portion of the protein that engages CD36 (its type 1 repeats, Fig. 3), a CD36-binding peptide derived from this domain (VTCGGGVQKRSRL), and certain CD36 antibodies (clone SMΦ) can mimic TSP1 by inhibiting NO-stimulated responses in vascular cells in vitro139,140 and angiogenesis in vivo 150,152. Therefore, engaging CD36 is sufficient to inhibit NO signaling. However, TSP1 remains a potent inhibitor of NO signaling in CD36 null vascular cells and muscle explants153. Thus, CD36 interaction with TSP1 is not necessary to block NO signaling in vascular cells.
Figure 3.

Thrombospondin-1 (TSP1) binds to two receptors on endothelial cells that inhibit NO signaling. The second and third central type 1 repeats of TSP1 contain sequences with known CD36-binding activities. The intact protein, recombinant type 1 repeats or synthetic peptides derived from the active repeats bind to CD36 and inhibit uptake of myristate via this plasma membrane fatty acid translocase. Myristate mediates membrane anchoring of a number of signaling proteins including the Src kinases. Membrane anchoring of Src and other undefined targets leads to increased activation of eNOS, which is inhibited by TSP1 160. Simultaneously, myristate activates eNOS in an AMP kinase (AMPK)-dependent manner 158, which may also be inhibited by TSP1. Inhibition of NO signaling via CD36 requires concentrations of TSP1 that exceed those normally circulating in plasma but can be found in plasma of some cancer patients176,177,409. The more potent TSP1 inhibitory pathway involves the signaling receptor CD47. Two CD47-binding sequences have been identified in the C-terminal domain of TSP1155. Engaging CD47 signals through undefined pathways that simultaneously inhibit activation of soluble guanylate cyclase (sGC) and cGMP-dependent protein kinase (cGK-I)157,410. These pathways inhibit NO signaling due either endogenous or exogenous NO.
In contrast, TSP1 failed to inhibit NO-driven angiogenesis in tissue explants from CD47 null mice, and NO-driven responses in vascular cells from CD47 null mice were insensitive to inhibition by TSP1153. Thus TSP1 blockade of physiologic NO-signaling in vascular cells requires CD47. Under basal conditions, vascular cells from CD47 null mice always have elevated cGMP levels compared to wild type cells. Similarly, temporarily suppressing CD47 expression increases basal cGMP levels 154. Thus, TSP1 signaling via CD47, constantly limits the sensitivity of sGC to activation by NO and thereby sets basal intracellular levels of cGMP. TSP1 engages CD47 via the C-terminal domain of the protein (Fig. 3). A recombinant protein containing this domain mimics the potent inhibitory effects of the whole protein on NO-stimulated vascular cell responses. Treating vascular cells with a CD47-binding peptide from the C-terminal module of TSP1 (p7N3, FIRVVMYEGKK155) blocks NO/cGMP signaling. The priority of TSP1-CD47 interaction in blocking NO-driven events in vascular cells is underscored by the fact that 10 pM TSP1 is sufficient to inhibit NO signaling via CD47, whereas >100-fold greater concentrations of TSP1 are required to inhibit via CD36.
5.9 Thrombospondin-1 inhibits NO/cGMP signaling at multiple levels
In murine, porcine and human vascular cells NO-stimulated cGMP accumulation is potently blocked by TSP1/CD47 signaling139,153,154. Because the inhibitory activity of TSP1 was preserved in the presence of cGMP PDE inhibitors, sGC must be a direct target of inhibitory TSP1/CD47 signaling (Fig. 3). The molecular pathways that transmit this signal from CD47 to sGC remain to be determined.
Remarkably, sGC is not the only target of inhibitory TSP1 signaling. Functional responses of endothelial cells to a cell permeable analogue of cGMP are also inhibited by TSP1139. NO decreases the ability of platelets to aggregate and form thrombi 156, and TSP1 blocks this effect of NO, promoting platelet adhesion and aggregation157. The delay of thrombin-induced platelet aggregation by 8-Br-cGMP was also reversed by TSP1. 8Br-cGMP-stimulated phosphorylation of platelet VASP at Ser239 was also inhibited by TSP1. Because this phosphorylation is mediated by cGMP-dependent kinase (cGK), this enzyme appeared to be a second target of TSP1 inhibitory signaling. Regulation of cGK was confirmed using an in vitro kinase assay. 8-Br-cGMP increased Ser phosphorylation of the cGK peptide substrate RKRSRAE, and this stimulated phosphorylation was completely blocked by TSP1 or by a specific cGK antagonist157.
TSP1 control of physiologic NO signaling also extends to events above sGC activation at the level of endogenous NO production (Fig. 3). Zhu et al reported that extracellular myristate activates eNOS in a CD36- and AMP kinase-dependent manner 158. Because CD36 is a major transporter of free fatty acids into cells 159, we proposed that the inhibitory effect of TSP1 mediated by CD36 involves inhibition of its fatty acid translocase activity. Using radiolabeled myristate, we found that TSP1 (and an inhibitory CD36 antibody) block myristate uptake into endothelial cells at concentrations consistent with their activities to inhibit NO/cGMP signaling160. This is associated with a block in myristate-stimulated sGC activation and increases in cellular cGMP. Studies of membrane translocation of the Src kinase Fyn showed that extracellular myristate stimulates Fyn translocation and Src kinase activation in a CD36 dependent manner, and TSP1 inhibits these events. Because Src is known to activate eNOS both by direct phosphorylation and phosphorylation of Hsp90, TSP1 may inhibit eNOS activation via this pathway. Alternatively, Zhu et al showed that the activity of myristate to activate eNOS is AMP kinase-dependent 158. Therefore, a second inhibitory pathway may be through AMP kinase (Fig. 3).
Taken together, these studies show that TSP1 redundantly modulates NO/cGMP signaling in vascular cells at three distinct levels: eNOS activation, sGC activation, and downstream at the level of the cGK (Fig. 3). This should enable TSP1 to be a highly effective physiological antagonist of NO signaling. Because tissue or circulating TSP1 levels are elevated in several disease states, this redundant inhibition must be considered in efforts to improve NO signaling. Therapeutic approaches designed to enhance NO signaling at any one of the levels will not bypass inhibition by TSP1 at downstream sites. A more rational approach to maximize the therapeutic potential of physiologic NO would target the necessary receptor CD47.
5.10 TSP1/CD47 signaling acutely regulates blood flow and tissue survival
Intracellular cGMP, though promoting cell survival in mammalian cells, plays a much more critical acute role in cardiovascular physiology161. Through direct modulation of the contractile apparatus of VSMC cGMP controls blood vessel diameter and thus controls blood flow162. The key protein in the contractile machinery of VSMC is myosin light chain 2 (MLC2) which activates myosin light chain and enhances actin myosin cross-bridge cycling163,164. NO stimulates the de-phosphorylation of MLC2 and thus disrupts actin-myosin cross-bridge cycling and relaxes VSMC. This then leads to vessel dilation and increased blood flow. However, in the presence of TSP1 NO can not dephosphorylate MLC2. In vitro TSP1 blocks NO-stimulated relaxation of contracting VSMC165. However treatment of CD47 null VSMC with TSP1 does not block NO-driven relaxation. These in vitro findings translate directly to regulation of blood flow in the whole animal. Mice treated with NO show a predicted increase in tissue blood flow. However, a similar NO challenge in TSP1 (or CD47) null mice results in over twice the increase in regional blood flow compared to wild type animals. Then endogenous TSP1 is regulating acute blood vessel response to NO165. TSP1-CD47 inhibition of NO stimulated vasodilation and NO-driven increases in blood flow is always present and acts as a rheostat upon NO responses in the vasculature limiting real time the dynamic range of NO effects.
The implications of this discovery are profound. Tissue units exposed to acute ischemic stress via vascular interruption demonstrate enhanced tissue survival and blood flow in the absence of TSP1 or CD47166. Conversely such ischemic stress, in the presence of TSP1-CD47 inhibition of NO signaling, leads to profound loss of tissue blood flow and tissue necrosis. Conversely blocking TSP1-CD47 signaling with monoclonal antibodies or gene silencing techniques dramatically enhances ischemic tissue survival and blood flow in both murine and porcine models of acute tissue ischemia166. Targeting TSP1-CD47 results in immediate effects upon vascular response to vasoactive stress as documented by laser Doppler or EPR tissue oximetry and parallels the immediate enhanced blood flow enjoyed in null mice exposed to ischemic vasoactive stress in soft tissues and hindlimb models.
In a number of acute and chronic pathologic states blood flow becomes interrupted and then restored at a later time. This phenomenon is termed ischemia/reperfusion injury (I/R) and is a major source of tissue and organ damage/loss 167. NO is known to be tissue protective in I/R168. However, administration of NO donors has produced limited therapeutic benefits for I/R injury. We found that TSP1 is rapidly induced following a liver I/R injury, suggesting that TSP1 limits the beneficial activity of NO169. Consistent with this hypothesis, null animals lacking TSP1 or CD47 were remarkably resistant to visceral organ I/R injury. Blocking the TSP1-CD47 pathway in wild type animals also confers dramatic tissue protective effects to I/R injury.
TSP1-CD47 signaling also limits tissue survival in conditions of complete absence of blood flow as found in full thickness skin grafting. In wild type mice, full thickness skin grafts experience complete necrosis. In contrast full thickness skin grafts in TSP1 and CD47 null animals enjoy essentially 100% survival170. Agents that interrupt TSP1-CD47 signaling facilitate the effects of endogenous NO and greatly increase full thickness skin graft survival in the wild type animals.
5.11 Can TSP1/CD47 antagonism of NO signaling control tumor perfusion?
Based on the evidence that TSP1/CD47 signaling acutely limits NO-mediated vasodilation in healthy and ischemic tissues, we examined whether this regulation extends to the tumor vasculature. This is an important question for cancer therapy in that many have sought to acutely increase tumor perfusion to enhance responses to intravenous chemotherapy and to enhance radiation killing to tumor cells by increasing the local oxygen tension 171. In contrast to normal tissue, systemic administration of NO results in a net decrease in blood flow in the tumor 172. This is consistent with the known impairment of function in the tumor vasculature 173,174. Thus, the tumor behaves as a passive resistance to blood flow, and vasodilation of other peripheral tissues by NO decreases flow through the tumor. This is generally known as a “steal effect” 175. When TSP1 was over expressed by the tumor cells, we found that the magnitude of this decrease diminished, suggesting that TSP1 secreted by the tumor was generally inhibiting the global vascular response to the administered NO. Conversely, over expression of TSP1 in the tumor also decreased the net increase in tumor blood flow induced by treating the mice with the vasoconstrictor norepinephrine. These observations may provide an explanation that some murine and human cancers are associated with elevated circulating TSP1176,177. The circulating TSP1 may selectively constrict vessel beds outside of the tumor and thereby increase blood flow into the tumor. The growth advantage thus provided would be a selective pressure for maintaining elevated TSP1 expression. Several endogenous angiogenesis inhibitors were first identified based on their elevated circulating levels in tumor-bearing mice, but the reason for their presence was unclear. We propose based on the signaling pathways discussed here that those circulating inhibitors may have similar acute effects to enhance tumor perfusion.
5.12 TSP1/CD47 antagonism of NO signaling controls tissue radiosensitivity
NO donors have known radioprotective activities for whole body irradiation of mice 178 and for gamma or UV irradiation of cells in vitro179,180. This suggested that ablating TSP1/CD47 signaling could protect tissue from radiation injury by enhancing pro-survival NO signaling. This hypothesis was tested by irradiating the hindlimbs of wild type, TSP1 null, and CD47 null mice181. Remarkably, at 25 Gy irradiation both null mice were essentially protected from the effects of irradiation. Hair loss (alopecia) was absent in CD47 null and decreased in TSP1 null mice. Both null mice showed minimal apoptosis in skeletal muscle and bone marrow 24 h following irradiation, and muscle function was preserved 2 months following irradiation. Remarkably, radioprotection in the null mice extended to isolated cultures of vascular cells. These cells survived irradiation at up to 40 Gy, and remained competent to replicate their DNA post-irradiation. This identifies the TSP1/CD47 pathway as a limiting pathway for recovery from radiation injury, and suggests that targeting this pathway could protect adjacent healthy tissue from radiation injury due to radiotherapy of tumors or following accidental or military–related exposure to radiation.
5.13 Do other angiogenesis inhibitors block NO signaling?
TSP1 may be unique in its redundant regulation of the NO/cGMP signaling cascade, but increasing evidence indicates that the same pathway is a target of additional endogenous angiogenesis inhibitors (Fig. 4). Prolactin-derived vasoinhibins were shown to inactivate eNOS via protein phosphatase 2A182,183. A second study indicated that vasoinhibins can further limit NO signaling through down regulation of iNOS184.
Figure 4.
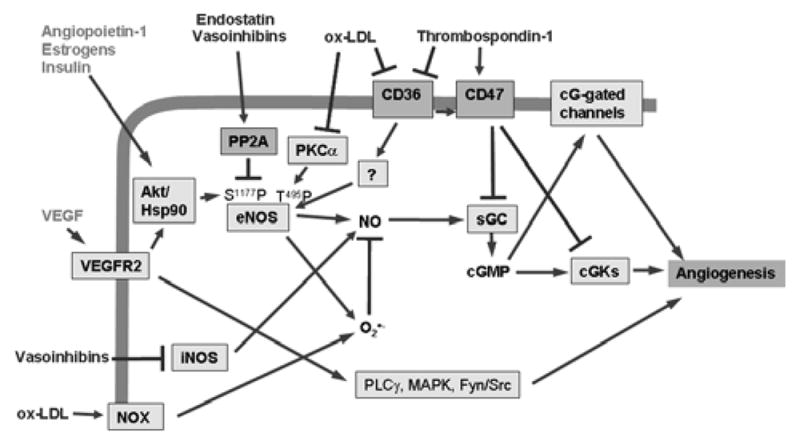
The NO/cGMP pathway is a signaling node for pro- and anti-angiogenic signaling. In addition to VEGF, angiopoietin-1, estrogens, and insulin can activate eNOS to increase NO synthesis in endothelial cells. TSP1 signaling via CD36 and CD47 inhibits downstream elements of this pathway. Oxidized LDL (oxLDL) is another known ligand of CD36 that could inhibit angiogenesis via an overlapping pathway. In addition, oxLDL signaling inhibits protein kinase Cα, which phosphorylates eNOS at Thr495 and activates NADPH oxidase (NOX), and the resulting superoxide depletes NO levels. The angiogenesis inhibitors endostatin and vasoinhibins activate the phosphatase PP2A, which dephosphorylates and thereby inactivates eNOS.
NO signaling has also been identified as a target for the anti-angiogenic activity of endostatin. Endostatin reduces VEGF-induced phosphorylation of eNOS at Ser1177 independent of any change in Akt phosphorylation185. This was attributed to activation of PP2A, which dephosphorylates eNOS at Ser1177. Furthermore, sGC protein levels were suppressed following treatment with endostatin186. The decrease in sGC protein was not associated with a decrease in mRNA levels, indicating that regulation is post-transcriptional. PP2A was also implicated in this response based on abrogation in the presence of okadaic acid.
Oxidized LDL was also shown to inhibit VEGF-induced endothelial cell migration by blocking Akt-mediated phosphorylation of eNOS at Ser1177 and to thereby decrease NO production 187. Subsequent studies showed that oxidized LDL also decreases the phosphorylation of eNOS on Thr495 via PKCα, and this was accompanied by increased O2•− production due to uncoupling of eNOS188. Furthermore, oxidized LDL increases NADPH oxidase activity in endothelial cells, which further increases O2•− production189. This increased O2•− could further lower cGMP signaling by consuming available NO 190. Like TSP1, oxidized LDL is a ligand for CD36191, and TSP1 is known to inhibit eNOS activation by blocking myristate uptake via CD36160. However, whether CD36 is the receptor that mediates the above activities of oxidized LDL has not been established.
6. Hydrogen peroxide and angiogenesis
6.1 H2O2 as a signaling molecule
Reactive oxygen species (ROS) have been negatively associated with many different aspects of cardiovascular disease such as hypertension, atherosclerosis, heart failure, and restenosis192. Recently however, ROS, and particularly hydrogen peroxide, have been recast as important second messenger molecules that respond to a variety of cytokines and growth factors. Examples include tumor necrosis factor α (TNFα)193, platelet derived growth factor (PDGF)194, epidermal growth factor (EGF)195, and insulin196, all of which elicit a transient increase in H2O2. A review of all of the essential roles of H2O2 signaling is outside the scope of this review but is available elsewhere197.
In the context of tumor angiogenesis, the recognized roles of H2O2 in signaling are rapidly expanding and are the subject of active research. Importantly, ROS such as O2•− H2O2 are increased in numerous cancer cells198. Exogenously added as well as cellularly derived H2O2 stimulates angiogenic responses in cultured endothelial cells and smooth muscle cells as well as in tissue and animal models of angiogenesis (reviewed in199). Brauchle et al first documented the ability of H2O2 to directly stimulate the production of VEGF from cultured keratinocytes while investigating the effects of UV irradiation200. The connection between cellular ROS/H2O2 levels and VEGF production was made at about the same time based on both signals being elevated following I/R injury201. H2O2 stimulates the production of VEGF-A protein and mRNA in a variety of cell types including rat VSMC202, rat heart endothelial cells203, C2C12 skeletal myotubes204, human and rat macrophages205, NIH 3T3 cells206, and DU-145 prostate carcinoma cells206. The addition of antioxidants to scavenge O2•− and H2O2 such as N-acetylcysteine and green tea catechins have been shown to inhibit angiogenesis in vivo207.
In addition to VEGF signaling, stromal cell-derived factor 1 (SDF-1) receptor CXCR4 mRNA expression is upregulated maximally in the presence of 10 μM H2O2208. Consistent with this, H2O2 plays a critical role in the mobilization, homing, and angiogenic capacity of bone marrow derived endothelial progenitor cells (EPC) 209. To critically assess the putative and as yet undefined targets of H2O2 in angiogenic signaling, one must first understand the basic chemical biology of H2O2.
H2O2 is the product of biological reduction of dioxygen. Most H2O2 in cells derives from the dismutation of superoxide anion (O2•−). O2•− can be generated by several enzymes including xanthine oxidase, cytochrome p450, uncoupled NOS, and myeloperoxidase, although the main sources of cellular O2•− for the production of H2O2 are NADPH oxidases (NOXs)210 and the mitochondria through electron transport chain-associated enzymes211,212. From here, O2•− can be converted to OONO- by reaction with NO, to hydroxyl radical (HO•) by Fenton or Haber-Weiss processes, or to H2O2 by superoxide dismutase. Alternatively, H2O2 can be formed directly from dioxygen by DuOXs (dual-function oxidases)210 or oxidoreductases such as glucose oxidase213.
6.2 NADPH oxidase in endothelial cells
The main source of O2•− in endothelial cells is the NADPH oxidase system214. NADPH oxidase was first characterized in phagocytic cells (neutrophils) and is a complex enzyme composed of 5 different regulatory subunits. A membrane spanning cytochrome b558 composed of gp91phox (Nox2) and p22phox as well as the cytosolic components p47phox, p67phox, and the small GTPase Rac215,216. NOX2 contains an NADPH binding site on the cytosolic portion of the protein as well as a flavin adenine dinucleotide (FAD) binding site facilitating electron transfer216. Gp91phox uses pairs of histidine residues to bind two hemes in a hexacoordinate low-spin fashion whereby the outer heme can reduce O2 to O2•− through rapid outer-sphere electron transfer when reducing equivalents are available from NADPH217.}218:
| (1) |
This also implies that the hemes of NADPH oxidase cannot be “poisoned” by CO or CN−, though there is one report that CO can interact with the cytochrome b558 heme of Nox2 under nonphysiological conditions219. The cytosolic components are necessary to activate the electron transfer. They facilitate catalysis by translocating to the membrane, in a Rac1 or Rac2 dependent manner, assembling with the plasma membrane subunits216,220. Vascular cell and phagocytic cell NOX differ in the manner in which they produce O2•−. Neutrophil NOX2 produces large concentrations of O2•− in short bursts, while vascular NOXs produce a sustained low level of O2•− that can be enhanced acutely by growth factors and other cell ligands221. Several paralogs of gp91phox (Nox2) that share 30 to 60% sequence homology are expressed in vascular cells as part of the NOX complex including Nox1, Nox4, and Nox5222,223. Each shares the common NADPH, FAD, and heme binding sites. Endothelial cells also express all of the other canonical NOX subunits with similar regulation.
O2•− from NADPH oxidase is produced on the opposite side of a plasma membrane from the electron source (NADPH). This may be into the extracellular space or into a subcellular membrane compartment ranging in size from a group of signaling proteins or containing a major organelle. Unlike other small molecule signaling agents (NO, CO, O2), O2•− is anionic and cannot freely diffuse across membranes. O2•− is then restricted as a signal by being able to move across membranes only with the aid of ion channels or after reduction to neutral diffusible H2O2. O2•− is converted into H2O2 by a group of proteins known as superoxide dismutases:
| (2) |
Mammals express 3 isoforms: a cytosolic CuZnSOD (SOD1), a mitochondrial MnSOD (SOD2), and an extracellular CuZnSOD (SOD3 or ecSOD)224. Although the dismutation of O2•− is spontaneous, these enzymes catalyze the process at rates approaching 109 M−1s−1. While the other two isoforms are ubiquitous, ecSOD is the major isoform found in the vascular extracellular space secreted by endothelial cells, smooth muscle cells, and fibroblasts224. It binds to the extracellular matrix near endothelial cells through interactions with collagens, heparan sulfate proteoglycans, and fibulin-5. The subcellular localization of NADPH oxidase determines the relevance of each SOD isoform. SOD1 is the most relevant to signaling in intracellular vesicles while ecSOD handles O2•− produced into the extracellular space.
6.3 H2O2 targets in vascular cells
H2O2 is consumed by a number of different enzyme systems. Catalase is a heme protein that uses one molecule of H2O2 as an oxidant to oxidize a second equivalent of H2O2, generating water and molecular oxygen:
| (3) |
| (4) |
Catalase reacts with H2O2 with a second order rate constant of 107 M−1s−1 in order to maintain the level of H2O2 and minimize any promiscuous oxidations225. In addition to catalase, H2O2 is consumed by cellular thiols and is a major contributor to the thiol redox status of the cell. The thiol redox status is controlled by two major systems, the thioredoxin system and the glutathione system. H2O2 interacts with these two systems by reactions with peroxiredoxins (Prx) and glutathione peroxidases (Fig. 5). Prx are dithiol containing enzymes that are converted to the disulfide by H2O2. Prx are reduced by thioredoxin, which is in turn reduced by the selenocysteine and NADPH dependent thioredoxin reductase. Glutathione peroxidase, on the other hand, uses selenocysteine to reduce H2O2, generating a selenic acid that is recycled using glutathione, which is in turn reduced by glutathione reductase in an NADPH-dependent fashion.
Figure 5.

VEGF signaling leads to H2O2 production and positive feedback through inhibition of PTPs. VEGF binding to its receptor (VEGFR) results in autophosphorylation of tyrosine residues leading to downstream kinase activation and angiogenic activity. VEGFR activation also leads to activation of the small GTPase RAC1 which, in conjunction with other cytosolic components (gp47- and p67phox), activates NADPH oxidase. NADPH oxidase reduces dioxygen to superoxide subsequently converted to H2O2 by SODs. H2O2 either directly or through a thiol peroxidase intermediate inactivates PTPs which oppose the actions of VEGFR thus enhancing and sustaining the downstream signal. Another source of superoxide is from the reduction of oxygen by the uncoupling of the mitochondrial electron transport chain (through complex -I or –III). This may contribute to the potentiation of VEGFR signaling as well.
Early studies using rat livers estimated the intracellular H2O2 concentration to range from 0.001 μM to 0.1 μM226,227. The maximum proliferative level was determined to be 0.7μM in Jurkat T cells, above which apoptosis occurs228. It is important to note that there is a concentration gradient from the outside to the inside of the cell when administering H2O2 experimentally due to the membrane permeability and intracellular consumption of H2O2. Thus, the intracellular concentration is approximately 7- to 10-fold less that the extracellular concentration229. Accordingly, stimulated rat brain extracellular H2O2 levels were measured amperometrically to be 2 to 4 μM, which would correspond to 0.2 to 0.4 μM inside the cell and well under the upper physiological limit of 0.7 μM230.
The signaling properties of H2O2 derive mostly from its electrophilic character and ability to react with protein thiols, oxidizing them to sulfenic and then to sulfinic acids231:
| (5) |
| (6) |
H2O2 reacts with thiols (RSH) relatively slowly, but the reaction rate is enhanced by deprotonation of the thiol to a thiolate anion (RS−) with a modest rate of 10–100 M−1s−1. For H2O2 based signal transduction to occur, some thiolate proteins have evolved with an active site pocket that significantly lowers the transition state energy of H2O2-thiolate reaction such that the reaction rate can be as fast as 105 M−1s−1, 232. The thiolate containing proteins having the fastest rate constant for H2O2 of course are the peroxidases, but the body contains other important thiolate-dependent proteins whose primary role is not the degradation of peroxides such as phosphatases, thiol-disulfide isomerases, glutathione S-transferases, dehydrogenases, and transglutaminases233. These proteins react with H2O2 at rates 3–5 orders of magnitude slower that the peroxidases197 and are not expected to interact with H2O2 below the 0.2–0.4 μM levels described in stimulated cells described above (vide supra). Thus, it is more likely that physiological H2O2 signaling occurs in “bursts” that produce in excess of 0.2 μM but less than 0.7 μM H2O2.
6.4 H2O2 regulation of vascular Tyr kinase signaling
One important established role of peroxide signaling is the potentiation of receptor tyrosine kinase signaling by oxidation of protein tyrosine phosphatases (PTP) that negatively regulate their signaling234. A majority of PTPs rely on a cysteine thiolate in their active site for their phosphatase actions235. Reversible oxidation of this moiety by H2O2 can shift the equilibrium in favor of enhanced tyrosine phosphorylation and downstream signaling (Fig. 6). The result is to extend the duration of cellular responses following ligation of Tyr kinase receptors.
Figure 6.

Superoxide and hydrogen peroxide reactivity. Superoxide generated by the mitochondria and NADPH oxidase has three main cellular fates; reaction with NO to form superoxide, reaction with iron-sulfur cluster (Fe-S) proteins to release ferric iron and hydroxyl radical, and reaction with super oxide dismutases (SOD) to form hydrogen peroxide. Hydrogen peroxide is consumed in scavenging and signaling reactions over a wide range of second order rate constants. References: NO411; [Fe-S]412, SOD413, catalase225,414, GPx415, Prx416, Trx417, PTP418, GSH419.
As discussed above, VEGFR2 is a transmembrane receptor activated by ligand stimulated dimerization and trans(auto)phosphorylation of cytoplasmic facing tyrosine residues(Tyr951,996,1054,1059)236. H2O2 is implicated as an important mediator of VEGF angiogenic signaling in endothelial cells through VEGFR2 phosphorylation and enhanced phosphorylation of downstream targets such as c-Src and VE-cadherin237–239. Ushio-Fukai et al have shown also that VEGF-induced VEGFR2 autophosphorylation is inhibited by the H2O2 scavenger N- acetylcysteine, NADPH oxidase inhibitors, and either gp91phox antisense oligonucleotides or over-expression of a dominant negative form of Rac1237. Potentiation of receptor tyrosine kinase signals is common for other angiogenic factors as well. As mentioned above, angiopoietin-1 signals through a receptor tyrosine kinase known as Tie2, and its effects are inhibited by over-expression of catalase240,241. Angiotensin II induces H2O2 dependent phosphorylation of epidermal growth factor receptor (EGFR) tyrosine residues242. Additionally, insulin and PDGF dependent autophosphorylation of their respective receptors is inhibitable with catalase over-expression194,243.
The targets of this signaling are specific PTPs. In the case of VEGFR2, SHP-1, SHP-2, and HCPTPA are known to associate with activated VEGFR2. Reversible redox regulation has been reported for each of these PTPs244–246. For example, SHP-2 oxidation by H2O2 in vitro resulted in active site cysteine oxidation and decreased PTP activity247. Importantly, the sensitivity to oxidation was greater when the protein was without its SH2 domain, which it uses to dock with VEGFR. Under basal (non-signaling) conditions, the SH2 is folded over to protect the catalytic domain from oxidative inactivation. Thus, VEGF-stimulated production of H2O2 constitutes a positive feedback loop that potentiates the actions of it and other similar receptor tyrosine kinase dependent growth factors (Fig. 6).
6.5 H2O2 regulation of vascular matrix metalloproteinases
Matrix metalloproteinases (MMP) are essential to many different facets of angiogenesis. MMPs actively maintain the balance between pro- and anti-angiogenic factors by generating matrix protein fragments in both categories (Table 2) and clearing cytokines from membrane bound states while also using their proteolytic ability to degrade endothelial basement membrane to facilitate cell migration and providing a proper space for endothelial cell lumen development. MMPs belong to a family of zinc-containing endopeptidases, of which at least 28 different isoforms have been identified in humans248. MMPs can be secreted or membrane bound (MT-MMPs). The membrane bound class includes four transmembrane MMPs (MT1-, MT2-, MT3-, and MT5-MMP) and two that are glycosylphosphatidylinositol anchored (MT4- and MT6-MMP). As a group, these MMPs can degrade all recognized mammalian extracellular matrix proteins249,250, while MMP-2, MMP-9, and MT1-MMP are critical to angiogenesis251,252. In addition to MMPs, several metalloproteinase homologs perform important pericellular angiogenic functions. These are known as ADAMs (A Disintegrin and Metalloproteinase Domain) and ADAMTS (A Disintegrin and Metalloproteinase and Thrombospondin motifs).
The MMPs are regulated at multiple levels including gene expression, compartmentalization (pericellular), zymogen activation, and enzyme inactivation. MMPs are not produced in an active state but are rather in a pro-state (zymogen) waiting for an angiogenic signal in the form of wound repair, inflammation, or tumor perfusion to induce their activation. This is due to the structure of MMPs containing a built-in autoinhibitory pro-domain as well as a catalytic domain. The autoinhibitory domain has a critical cysteine residue that coordinates the catalytic site Zn2+ thereby blocking coordination of other peptides. For the enzyme to be active, the thiol- Zn2+ interaction must disrupted. This is known as a cysteine switch as first proposed by Van Wart et al. The switch can happen in three basic ways: 1) proteolytic cleavage of the autoinhibitory domain by another protease (serine protease or MMP); 2) oxidation of the inhibitory cysteine thiol by ROS, heavy metals, or thiol modifiers; 3) allosteric modification of the zymogen resulting in intramolecular proteolytic cleavage of the autoinhibitory domain. H2O2 has the potential to activate at least two of these pathways. Direct oxidation of the inhibitory Cys residue by H2O2 is suggested in work by Grote et al in which mechanical stress induced MMP activation was highly NADPH oxidase dependent253 as well as work citing the inhibitory effects of antioxidants on the activation of MMPs254. H2O2 can also stimulate the proteolytic activation of MMPs by inducing expression of proteinases such as urokinase plasminogen activator and MMP-1 through Ets-1255. Furthermore, H2O2 stimulation leads to enhanced expression of MMPs through a mechanism that is dependent on NADPH oxidase activity.
6.6 Intracellular vascular targets of H2O2
H2O2 is also a direct effector of angiogenic signaling downstream of cell surface receptors via redox sensitive H2O2-dependent activation of transcription factors. These generally have a regulatory cysteine residue that, when oxidized, modulates the transcriptional activity of the protein. Examples of these relevant to angiogenesis are NF-κB256,257, Ets-1258, and p53259.
The reaction of H2O2 with any of the above mentioned signaling proteins is 3–5 orders of magnitude slower than with the H2O2 scavenging proteins (Fig. 5). For efficient signaling to occur, the H2O2 concentration would have to significantly increase (say, 3 to 5 orders of magnitude) or the concentration of scavenging proteins would have to be decreased. Alternatively, a recent review by Ushio-Fukai explains that location may be critical for NADPH oxidase signaling, thereby addressing some of the issues of H2O2 concentrations and scavengers260. During angiogenesis, endothelial cells become polarized, and migration is directed by formation of lamellipodia and filopodia at the leading edge. Migration is also depends on spatially and temporally restricted the localization of signaling molecules including Rac1 (NADPH oxidase generated O2•−) and PI3K (PIP3). Accordingly, NADPH oxidase is localized to signaling complexes at the leading edges of migrating cells, which could spatially modulate the activities of the lipid phosphatase PTEN, PTPs, and receptors such as EGFR260. Another perspective on the mechanism of H2O2 signaling involves the intermediacy of other oxidized thiol protein such as Prx, thioredoxins, or glutathione (see Figure 5) to mediate the signal. An example of this is the reaction of oxidized yeast GPx3 with the transcription factor Yap1261. Oxidation of GPx3 produces the sulfenic acid, which then reacts with a Yap1 cysteine residue forming an intermolecular disulfide that then resolves into an intramolecular disulfide and activates the signal. In this way, Prx acts as the local sensor of H2O2 and extends the lifetime of the signal to find the specific target protein thiol. This suggests that Prx and the like have two functions: scavenging H2O2 and propagating its signal. Proponents of this dual role often refer to a floodgate hypothesis governing where Prx consume low levels of H2O2 and act as signals in the presence of higher concentrations that could over oxidize Prx to the sulfinic acid262,263.
Although it is clear that H2O2 plays a significant role in angiogenic signaling, more work needs to be done to address the specific regulation of NADPH oxidases by angiogenic regulators and to clarify how specificity in regulating downstream targets is achieved.
7. What is the contribution of carbon monoxide to angiogenesis?
Until NO became widely recognized as a key endogenously generated signaling molecule, CO was regarded as an uninteresting byproduct of heme degradation. However recent work suggests that endogenously generated CO also plays a significant physiological role. Numerous cardiovascular and immunological effects have been reported for both endogenous CO and pharmacological CO donors. Unlike NO, however, no specific CO target has been identified that clearly mediates its effects. In the context of angiogenesis, CO may derive some of its effects as a discreet signaling moiety but also (maybe more importantly) via its effects on NO and O2 signaling.
7.1 Introduction to carbon monoxide chemistry and physiology
CO is a colorless gas at room temperature and pressure. Like NO and O2, it favorably partitions into hydrophobic membranes and has a similar water solubility (2.5 mM). However, unlike NO and O2, the physiological chemistry of CO is relatively simple. It is unreactive towards the other diatomic gasses and most biological molecules. CO can be oxidized to CO2 by the mitochondria264, but this is not a major path for CO elimination, which is primarily via exhalation through the lungs 265. The most significant aspect of biological CO chemistry is its ability to bind to transition metals in metalloproteins, particularly reduced heme-proteins (as reviewed in266). CO competes with NO and O2 for binding to some heme proteins, thus affecting their signaling pathways. However, CO binds to these heme proteins in a different manner than O2 and NO. The binding geometry for NO can be bent or linear depending on the electronics of the overall complex, and O2 prefers a bent geometry. CO, on the other hand, binds only in a linear mode. This becomes important when considering the relative affects of CO, NO, and O2 signaling through proteins capable of interacting with all three (such as sGC).
CO interacts with several heme proteins that regulate important signaling pathways as reviewed in Wu, 2005267. The one known example of CO being an agonist for these targets is the NO binding protein sGC, where CO is an activator, albeit much weaker than NO. The affinity constants are 2.6×10−4 for CO125 and 4.2×10−12 for NO268,269. CO can elicit cardiovascular effects such as vasorelaxation and inhibit platelet aggregation by directly activating sGC 270. Due to this competition, periods of hypoxia or oxidative stress can modulate CO binding to heme protein signaling pathways. However, CO is unlikely to be a significant antagonist of NO for sGC activation because its binding to sGC is too weak.
7.2 Biogenesis of CO
Biological generation of CO is largely due to the enzymatic degradation of heme by heme oxygenase enzymes (Fig. 7). These enzymes catalyze the primary and rate limiting step in heme catabolism using NADPH and dioxygen to produce one equivalent of CO, biliverdin-IXα, and Fe2+ per equivalent of heme271. Mammalian cells express two isoforms of heme oxygenase: a highly inducible (HO-1) 32kDa isoform and a noniducible (HO-2) 36 kDa isoform. A third isoform (HO-3) has recently been cloned from rat brain cDNA, but is much less active than HO-1 or HO-2. For reviews on heme oxygenases, see 267,270,272,273. HO-2 is constitutively found in the brain, intestines, testis, and endothelium. HO-1 is responsible for metabolizing the heme released during red blood cell turnover, and as a result, it is constitutively found in the spleen and liver. However, HO-1 can also be induced in most tissues in response to a diverse set of stress signals. These include ROS, UV irradiation, heat shock (HO-1’s alternate name is HSP32), hypoxia, hyperoxia, heavy metals, NO, inflammatory cytokines, heme, and ethanol273. The common feature shared by all these stimuli seems to be that they induce oxidative cell stress. Therefore, HO-1 induction is thought to be part of the anti-oxidant response. Indeed, HO-1 induction protects against a number of pathophysiological insults including myocardial reperfusion toxicity 274, organ transplant rejection 274, hypoxia-induced pulmonary hypertension 275, and TNFα-mediated endothelial cell apoptosis 276. It is important to note that although the co-products of HO-1 (biliverdin, bilirubin, and Fe2+) are bioactive themselves, most actions of HO-1 in these stress models can be recapitulated by CO.
Figure 7.

CO biosynthesis. Carbon monoxide is synthesized by the heme oxygenases (HOs). HOs bind heme and use it to activate oxygen to catalyze its own oxidation to biliverdin and CO. biliverdin is subsequently reduced by biliverdin reductase to bilirubin. CO signaling is exclusively due to interaction with reduced heme proteins including those that are targets of NO and O2.
7.3 CO as a stimulator of angiogenesis
In addition to its anti-oxidant, anti-inflammatory, anti-apoptotic, and vasorelaxant effects, HO-1 and its product CO have recently gained interest as mediators of angiogenesis, nowhere more importantly than in the area of tumor biology. Notably, HO-1 expression is increased in a number of human solid tumors including renal cell 277 and prostate carcinoma 278 as well as some experimental solid tumors such as rat hepatoma AH136B 279 and mouse Sarcoma 180 280. Over-expression of HO-1 in a mouse pancreatic cancer model correlated with significant increases in tumor size, angiogenesis, lung metastasis, and mortality 281. Correspondingly, the first suggested link between HO-1 and angiogenesis came from correlating HO-1 over-expression with enhanced endothelial cell proliferation282. Likewise, endothelial cells made HO-1 deficient using an antisense oligonucleotide exhibited decreased proliferation, in vitro capillary formation, and cell cycle progression, which could be rescued using a CO donor283. The effect of the CO donor reinforces the view that the HO-1 product relevant to angiogenesis is CO.
HO-1/CO’s influence on angiogenesis is not limited to vascular cells. In vivo studies of human gliomas have linked enhanced HO-1 expression in tumor associated macrophages with greater small vessel density284. This finding is supported by work showing increased expression levels of HO-1 in macrophages associated with melanomas but not in the associated melanocytes, fibroblasts, or keratinocytes285. The cell type-specific expression may be an important caveat when considering HO-1/CO mediated tumor angiogenesis. The in vivo aspects of HO-1 induced angiogenesis were first demonstrated by infusion of an adenoviral vector expressing HO-1 into the femoral artery of a rat, resulting in a significant increase in blood flow and vessel density286. These effects could be inhibited using the HO-1 inhibitor Zn-protoporphyrin286.
7.4 HO-1 up-regulation increases expression of angiogenic factors
As discussed above, VEGF-A is a potent stimulator of angiogenesis through increasing endothelial cell mitogenesis and migration. A growing number of recent studies provide links between VEGF expression and activities of HO-1. In this way, CO may be beneficial to the growth of tumors and play a role in the angiogenic switch for tumor progression.
Inhibition of HO-1 using SnPPIX completely inhibited the release of VEGF from rat VSMC stimulated with IL-1β and hypoxia, whereas NO synthase inhibitors failed to completely inhibit VEGF release 287. VEGF production was increased by hemin (a stimulator of HO-1 activity) as well as by HO-1 over-expression287. Inducers of HO-1 (endotoxin, H2O2, and a metalloproteinase) all led to increased in VEGF production from rat lung microvascular endothelial cells transfected with an HO-1 expression plasmid288. Of the products of HO-1 catalysis, CO (1% atmosphere) was the only moiety able to recapitulate the increase in VEGF seen with HO-1 stimulation or over expression287. Likewise in macrophages, SnPPIX and ZnPPIX significantly decreased the production of VEGF by RAW264.7 cells unstimulated or stimulated with lipopolysaccharide289,290.
15-deoxy-Delta(12,14)prostaglandin-J2 (15d-PGJ2) is the most potent endogenous ligand for PPARγ activation and is known to up-regulate VEGF expression in VSMC290,291, coronary endothelial cells292, and macrophages290,293. When PPARγ binds a stimulator such as 15d-PGJ2 or polyunsaturated fatty acid metabolites, it dimerizes with retinoid X receptor, and this complex binds to DNA at specific elements known as PPAR response elements294. HO-1 is under the control of this element295,296, and induction of HO-1 in turn stimulates VEGF production. In contrast, 15d-PGJ2 can also stimulate HO-1 expression via the antioxidant response element (ARE) in a PPARγ independent manner297–300. Interestingly, 15d-PGJ2 is part of a larger group of cyclopentenone prostanoids that are capable of Michael addition chemistry to nucleophilic thiols such as those reported in Ras301 leaving open the possibility of direct 15d-PGJ2 interaction with KEAP/Nrf2. Either way that the HO-1 stimulation occurs, 15d-PGJ2 stimulation of HO-1 leads to VEGF production, and HO-1 inhibitors (SnPPIX) prevent 15d-PGJ2 induced VEGF production302,303. This is suggested to occur through the phosphorylation of ERK1/2303 downstream of 15d-PGJ2 induction of HO-1.
VEGF expression in human umbilical vein endothelial cells (HUVEC) is also stimulated by HO-1 activity as shown with over expression and chemical stimulation289. The ability of an NO donor (SNAP) to stimulate VEGF production in HUVEC was also dependent on the activity of HO-1.304 High glucose impairs the ability of cells to produce VEGF and is thought to contribute to the decrease in wound healing observed in diabetic patients305. Expression of HO-1 restores VEGF levels in keratinocytes cultured under hyperglycemic conditions306. VEGF may not be the only angiogenic factor regulated by CO because HO-1 induction also stimulates the release of the angiogenic cytokine IL-8 from human microvascular endothelial cells307.
7.5 Stimulators of HO-1 and CO production leading to VEGF
Endothelial cells given a positive angiogenic signal (prolactin) increase HO-1 expression associated with endothelial cell proliferation and angiogenesis308,309.
A possible feedback system exists whereby VEGF can stimulate the expression of HO-1 to reinforce the angiogenic response. This is suggested in work by Fernandez and Bonkovsky where they observed increased HO-1 expression consistent with angiogenesis 48 h post-stimulation of chicken embryo chorioallantoic membranes with VEGF310. The effect was inhibited by ZnPPIX. Interestingly, VEGF driven HO-1 expression was Ca2+ dependent and inhibitable using a PKC inhibitor (staurosporine)310. This is consistent with the previously known Ca2+/PKC dependence of TNFα and IL-1 induced HO-1 expression311, although staurosporine is not a specific PKC inhibitor.
NO has an established role as a mediator of angiogenesis312. NO triggers prosurvival and proangiogenic activities in the endothelium by promoting growth and differentiation by activation of the endothelial isoform of NO synthase (eNOS) and signaling through sGC to elevate cGMP levels (see section 4). However, in other cell types such as smooth muscle, tumor cells, keratinocytes, macrophages, and mesangial cells, expression of the inducible form of NO synthase (iNOS) led to HO-1 dependent production of VEGF313. NO and CO have complicated relationship in vivo as they are known to regulate the production of one another (reviewed in314,315). Low (nM) concentrations of NO consistent with pro-angiogenic signaling do not induce expression of HO-1 and appear to inhibit constitutively expressed HO-2316. However, higher NO fluxes such as those produced from iNOS317, rapidly decomposing NO donors318, or organic nitrites319 are known to induce HO-1 expression and thereby increase the cellular levels of CO. Over expression of iNOS in smooth muscle cells led to an enhancement of IL-1β induced VEGF expression that was dependent on HO-1 activity320. Treatment of endothelial cells with an NO donor (SNAP) increased the synthesis of VEGF in an HO-1 dependent manner304.
7.6 HO-1 and CO mediate the pro-angiogenic actions of SDF-1
The proangiogenic effects of SDF-1 (aka CXCL12) and its receptor CXCR4 have recently been linked to HO-1 and CO321,322. SDF-1 is a major chemokine responsible for the mobilization and targeting of endothelial progenitor cells to sites of injury and new vessel formation. Bone marrow derived vascular stem cells will migrate to areas presenting a high concentration of SDF-1 such as nascent wounds or tumors323. In fact, inactivation of SDF-1 or CXCR4 in mice is embryo lethal due to impaired vascular development324,325. SDF-1/CXCR4 have also been implicated in the targeting of metastatic breast cancer cells to the brain326 and the impairment of EPC recruitment in coronary artery disease and vascular pathology of diabetes327. Human endothelial cells as well as EPC exposed to SDF-1 up-regulate HO-1 mRNA, and inhibition of HO-1 impairs SDF-1 driven in vitro angiogenesis321. A portion of the angiogenic effect of SDF-1 is through stimulation of VEGF, which in contrast to the examples cited above, appears to be independent of HO-1 321. The HO-1 dependent angiogenic actions of SDF-1 can be mimicked with the addition of CO donors321. The authors attribute the effects of CO to its well known activation of sGC and subsequent activation of the cytoskeletal-associated cGK target protein, VASP321.
7.7 Decreased CO contributes to angiogenesis inhibition
Dysregulation of angiogenesis is a hallmark of preeclampsia, and HO-1/CO have been linked to successful pregnancy in human and animal models 328. The anti-angiogenic proteins soluble Flt-1 (aka soluble VEGFR-1, Table 2) and soluble endoglin are elevated in preeclamptic women 2 – 3 months prior to the clinical manifestation of the disease 329. In pregnant rats, over-expression of soluble Flt-1 induced symptoms mimicking preeclampsia 330. 330. The levels of soluble Flt-1 and endoglin were recently shown to be potentiated in HO-1 deficient mice as well as wild type mouse placental explants treated with an HO-1 inhibitor (SnPPIX)331. Correspondingly, administration of a CO donor (CORM-2) decreased soluble Flt-1 released from cultured HUVECs, whereas adenoviral over-expression of HO-1 in placental explants reduced soluble endoglin331.
The introduction of HO-1 siRNA to mice bearing hepatocellular carcinomas produced a decrease in angiogenesis in the tumors as well as an overall decrease in tumor size growth rate332. This suggests that HO-1 gene suppression could be an effective anti-angiogenic therapy for cancer.
7.8 What are the targets of CO in angiogenesis?
No unique direct target of CO has been found that mediates angiogenesis. This is not to mean that there is none, and further effort is needed on this topic. Like NO, CO signaling in vascular cells has been attributed to activation of the heme protein sGC333. However, CO is only a weak activator of sGC and requires levels that are 2 orders of magnitude more than NO to elicit a signal. Consistent with this, the inhibition of sGC has no effect on the anti-inflammatory actions of CO in macrophages334. Aside from a direct activation of sGC, CO could increase the concentration of NO in cells by limiting the scavenging of NO by other heme-proteins or O2•−. As stated above, NO reacts with O2•− at nearly diffusion controlled rates to form OONO-, viewed here as a potential sink for NO but is also a controversial physiological oxidant335,336. CO can potentially bind reduced heme-proteins to limit their ability to scavenge NO. More importantly, it has been shown that heme oxygenase (1 and 2) derived CO up-regulates the expression of ecSOD337,338 to limit the concentration of superoxide. This may be more significant in terms of the interaction of CO and H2O2 angiogenic signaling. This would allow CO to act as a feedback stimulator of H2O2 signaling from the NADPH oxidases by providing more of the component necessary to convert their product into an angiogenic signal. In addition, CO could also directly mediate the production of O2•− (and ultimately H2O2) from the mitochondria. Paradoxically, CO was reported to mediate its anti-inflammatory effects by binding cytochrome c oxidase (complex IV of the mitochondrial electron transport chain), increasing the flux of ROS production from the mitochondria339. CO competes with oxygen for binding to cytochrome c oxidase with a Ki of 0.3 μM, and 20 μM CO inhibited cellular respiration 40% in the presence of 20 μM O2340,341. A role for mitochondrial derived H2O2 in angiogenesis is suggested in work by Chandel et al342. Hep3B cells deficient in mitochondria (termed ρ0) failed to produce VEGF and erythropoietin in response to hypoxia342. Interestingly, these cells retained the ability to produce these angiogenic factors upon H2O2 stimulation with CoCl2 indicating that there are multiple ROS generating systems with a common target342. Connor et al343 using an SOD2 over- expressing fibrosarcoma cell line showed that increases in mitochondrial derived H2O2 induced VEGF synthesis through inhibition of the redox active tumor suppressing phosphatase, PTEN (phosphatase and tensin homolog deleted from chromosome 10). PTEN attenuates PI3K signaling and Akt activation leading to HIF1-α stabilization and VEGF production343. In addition to this, cells producing a higher steady state level of mitochondrial H2O2 produced greater cell sprouting in an in vitro angiogenesis assay as well as an in vivo angiogenic assay using the chicken chorioallantoic membrane343.
The fact that CO is involved in angiogenic signaling is now well established. However, the direct targets of CO mediated angiogenic signaling and their relative contributions to the overall process remain elusive, but actively researched. The recently defined role of a hydrogen sulfide (H2S) biosynthetic enzyme as a physiological CO target is discussed in section 7. Further research is also needed to clarify the broader implications of CO crosstalk with NO and ROS.
8. What is the contribution of hydrogen sulfide?
Until recently, H2S was considered only as a toxic environmental pollutant and associated with the smell of rotten eggs. Indeed, H2S is more toxic than CO or hydrogen cyanide314. Like its so called “gasotransmitter” cousins, H2S is now gaining research interest based on newly reported physiological signaling properties (reviewed in: 344–347.344–347. H2S is categorized with CO and NO because it is a water soluble gas at room temperature and pressure, though all of the signal transduction properties of the “gasotransmitters” are derived from their solute form. Unlike CO and NO, however, H2S has an acidic proton with a pKa of 6.8, making the anionic conjugate base the predominant form (approximately 80%) in biological fluids at physiological pH (7.4). However, equilibrium with the highly lipophilic protonated form enables it to freely penetrate cell membranes.
8.1 H2S biosynthesis
H2S is synthesized endogenously by three enzymes: cystathionine β-synthase (CBS), 3-mercaptopyruvate sulfurtransferase, and cystathionine γ-lyase (CSE)348 (Fig. 8). Two of these enzymes use L-cysteine as a substrate to make H2S but were discovered in different locations and named for a different reaction. CBS is the primary H2S producer in the brain, where it normally makes cystathionine by condensing serine and homocysteine349. H2S production from CBS is stimulated 2–3 fold by Ca2+347 and 2 fold by S-adenosylmethionine349 and requires pyridoxyl phosphate. CBS also contains a non-catalytic heme iron to which CO and NO have been shown to bind and inhibit350. However, CO binds with a 200 fold higher affinity than NO and is much more likely to regulate CBS under physiological conditions350. CBS activity can result in physiological H2S concentrations as high as 50–160 μM in mammalian brains351. CSE, on the other hand, is largely found in peripheral tissues such as the kidney, lungs, and vasculature352, while the liver contains large amounts of both enzymes353. In the rat vasculature, the highest amount of H2S is produced by the tail artery followed by the aorta and mesenteric artery354.
Figure 8.

H2S bio synthesis. H2S is synthesized by 3 different enzymes: cystathionine beta-synthase (CBS), cystathionine γ-lyase (CSE), and mercaptopyruvate sulfurtransferase (MPST). CBS condense cysteine and homocysteine to make cystathionine and H2S. CSE takes cystine and converts it to thiocysteine, pyruvate, and ammonia. Thiocysteine can be reduced by another equivalent of thiol to cysteine and H2S. Cysteine, converted in the mitochondria by aspartate aminotransferase (AAT) to 3-mercaptopyruvate, is converted to H2S, pyruvate, and ammonia by MPST.
8.2 H2S signaling targets
Though H2S is ubiquitously produced and apparently in high concentration in some vascular tuissues, there is no established signaling mechanism. Confounding this, many of its biological effects remain controversial. H2S can stimulate or block apoptosis; can be produced too much or too little in myocardial ischemia; can stimulate or inhibit cell proliferation; and can be pro- or anti-inflammatory in a mouse model of edema. It is more firmly established in the cardiovascular system that H2S acts as a vasodilator relaxing aorta, portal vein, corpus cavernosum, mesenteric and hepatic arteries, but not coronary arteries in vitro344. H2S given intravenously to whole animals decreases blood pressure in a dose-dependent manner355. The mechanism of this hypotensive activity received much attention and was determined to be due to opening of ATP sensitive K+ (KATP) channels in smooth muscle cells by H2S351. Although the direct effect of H2S on KATP channels is not yet known, it is speculated that H2S may reduce a key disulfide that regulates the channel356. A role of H2S as a physiologic vasorelaxant was recently established using CSE−/− and CSE−/+ transgenic mice357. The lack of CSE-generated H2S resulted in hypertensive mice and significantly increased the level of plasma homocysteine. This work clearly establishes H2S as an endothelium-dependent vasodilator that is sensitive to changes in intracellular calcium, much akin to NO/NOS regulation.
Two reports show direct angiogenic effects of H2S. One work concluded that H2S is proangiogenic, having a maximum effect at 10–20 μM358. H2S increased cell growth and migration in cultured endothelial cells in a manner that was dependent on Akt and PI3K. The effect of H2S was also independent of increases in VEGF, FGF, angiopoietin-1, or NO metabolites. Mice treated with 10 and 50 mM kg−1 day−1 showed increased angiogenesis in vivo in Matrigel implants358. In the second study, H2S also stimulated endothelial cell proliferation in vitro, but similar concentrations inhibited vascular outgrowth in a muscle explant angiogenesis assay 359. The latter result suggests that H2S has effects on angiogenesis that extend beyond its direct effects on endothelium.
Angiotensin II (Ang II) is a potent stimulator of NADPH oxidase in VSMC221,242, endothelial cells360,361, adventitia362, and cardiac myocytes363,364. By binding to different receptors, Ang-II can either stimulate or inhibit angiogenesis 365. Ang II is the product of angiotensin converting enzyme, a zinc metallopeptidase that cleaves angiotensin I to angiotensin II (a vasoconstrictor) while also degrading bradykinin (a vasodilator). H2S is an inhibitor (KI ~100 μM) of angiotensin-converting enzyme in HUVEC with and without phorbol 12-myristate 13-acetate (PMA)366. The effect of H2S was abrogated in the presence of Zn2+, indicating that H2S may be interacting with the Zn2+ containing active site of angiotensin-converting enzyme366. Consistent with this is the fact that H2S biosynthesis is increased in tissues of streptozotocin-induced diabetic rats367, correlating the impaired vascular responses and angiogenesis/wound healing attributed to diabetes.
8.3 H2S and Down Syndrome
Down syndrome, which is a result of having an additional copy of chromosome 21, has been linked to overproduction of H2S due to CBS being encoded on this chromosome368,369. Overproduction of H2S has been hypothesized to be a cause of the gradual mental retardation associated with afflicted individuals370. Interestingly, the incidence of solid tumors and hemangiomas is significantly lower in Down syndrome individuals indicating that they may have impaired angiogenesis371. Excess H2S may not be the only cause because others have attributed impaired angiogenesis to an extra copy of the type XVIII collagen gene, which increases the circulating concentration of endostatin 372.
8.4 H2S and homocysteinemia
Hyperhomocysteinemia, a condition of elevated homocysteine in the blood, may be a marker of impaired CSE activity or expression and, therefore, of reduced H2S biosynthesis (Fig. 8). This is evident in the CSE−/− transgenic mouse where H2S levels are down 80%, while plasma homocysteine is up over 18-fold357. Indeed H2S and homocysteine are associated with opposing effects in cardiovascular studies 373. Homocysteine is elevated following balloon injury (a model of angioplastic restenosis) in rats as H2S and CSE levels are lowered, leading to significant neointimal thickening374. Introduction of H2S relieved the effects of balloon injury induced hyperplasia, supporting its role as an anti-angiogenic mediator374. Also, lowering plasma homocysteine levels in patients with peripheral artery disease and diabetes mellitus resulted in a significant lowering of circulating VEGF without significantly altering endostatin levels375. Many additional reports associate changes in homocysteine levels with angiogenic and vascular outcomes, highlighting the potentially integral yet poorly defined role played by the transsulfuration pathway.
Despite some evidence supporting a role for H2S in angiogenesis, this remains an infant field. In a broader context, H2S is gaining traction due to exciting cardiovascular and neurological effects of the “third gasotransmitter”. It will be important to define how H2S integrates into the signaling actions of the other agents (NO, CO, ROS/H2O2, etc.) discussed here in terms of angiogenesis but also in more broad physiological terms. For instance, studies have reported the regulation of CO production by H2S in activated macrophages376 or the mutual modulation of NO and H2S in the vasculature351,377.
9. HNO and angiogenesis
There has been recent excitement about the possible physiological generation of the one electron reduced and protonated form of NO, nitroxyl (HNO) (most recently reviewed in378. HNO has important cardiovascular effects that are both shared379,380 and distinct from NO380–382. It is produced from NOS in vitro as well as by direct reduction of NO. Another often overlooked pathway is the reaction of an S-nitosothiol with another thiol:
| (7) |
However, because of the lack of direct detection methods its physiological generation has not been conclusively demonstrated. Despite this, a number of HNO donor molecules have been used to explore its pharmacological functions as well as establish biological reactivity that departs from NO379. HNO prefers to bind to ferric heme proteins but will form complexes with ferrous hemes383. HNO is extremely thiophilic, leading to reversible (N-hydroxy sulfonamide) and irreversible (sulfinamide) modifications384. Unlike NO, it does not react with superoxide385. Consistently, the main biological targets of HNO are low pKa thiols386 and ferric heme proteins386, although it can directly bind and activate the ferrous heme protein sGC to elicit NO-like activation (TWM unpublished results).
Although HNO differs empirically from NO in only a proton and an electron, it may elicit opposite angiogenic effects from NO. Mice treated with an HNO donor showed decreases in xenograft tumor size as well as decreased tumor vessel density387. Accordingly, circulating VEGF and HIF1α levels were decreased. The authors speculate that the effect of HNO on angiogenesis is dependent on potent inhibition of glycolysis (GAPDH) by HNO, leading to decreased HIF1α and decreases in VEGF.
10. Conclusions
Studies of developmental and tumor angiogenesis have uncovered important signaling roles for gasotransmitters and other redox-active small molecules. They play key roles as cytoplasmic and intercellular mediators of both pro- and anti-angiogenic signaling. To date, NO is the best understood of these mediators, and its role in role in developmental versus pathological angiogenesis is fairly well understood. Efforts to directly limit NO synthesis using NOS inhibitors have shown some ability to limit tumor angiogenesis in animal models, but the pleiotropic role of NO in cardiovascular physiology to control blood pressure and hemostasis would likely prevent any long term therapeutic use of such inhibitors in cancer patients. However, recognizing that NO signaling plays several roles in the cardiovascular system may be crucial to developing more effective therapeutic angiogenesis inhibitors. All of the currently FDA approved anti-angiogenic drugs have hypertensive and prothrombotic side effects134,388, which are shared by some endogenous angiogenesis inhibitors165,389, and the role of NO signaling in the etiology of these side effects is increasingly being recognized (Fig. 9). Defining the angiogenesis-specific versus general targets of cardiovascular NO signaling may allow tumor angiogenesis to be controlled without these adverse side effects. One recent example that applies this strategy employed NO-independent vasodilators to overcome the hypertensive activity of an experimental VEGFR2 kinase inhibitor without impairing its ability to inhibit tumor angiogenesis136.
Figure 9.

Cross talk between H2S, CO, NO, O2•−, and H2O2 in vascular angiogenic signaling. Binding of an angiogenic factor to its cell surface receptor results in receptor tyrosine phosphorylation and downstream kinase activation of HO-1 (producing CO), eNOS (producing NO), and NADPH oxidase (producing O2•−). Receptor activation can also increase intracellular calcium levels activating CSE (producing H2S). O2•− is converted to H2O2 by SOD and this is capable of either direct or indirect inhibition of PTPs that normally attenuate receptor downstream signaling. H2O2 provides a reinforcement of the angiogenic signal. O2•− can also react with and consume NO making OONO− decreasing the signaling capacity of both NO and O2•−. NO through sGC is a primary regulator of angiogenesis, blood pressure, vascular permeability, and hemostasis. Likewise CO can bind sGC to regulate the same processes, but with a much lower affinity (pM vs. mM). However, CO also regulates angiogenic signaling in a sGC independent manner. H2S is an inhibitor of ACE with associated effects on blood pressure and has an ill-defined role in angiogenesis. Increase in CO by HO-1 induction could also decrease the levels of H2S derived from CBS.
Efforts to define the roles of other members of this family in angiogenesis are at a much earlier stages than for NO. Indeed, to fully understand and pharmacologically control pathologic angiogenesis we may need to both define their individual targets and integrate all of these molecules into a signaling network. This review has mentioned a few such connections, and it may be instructive for future research efforts to highlight what is known about cross-talk between their signaling pathways (Fig. 9). At physiological concentrations of NO relevant to stimulating angiogenesis, the primary target of NO is clearly sGC. O2•− plays a primary role in pro-angiogenic signaling by providing positive feedback through PTP inactivation to prolong signaling through Tyr-kinase receptors, which in turn prolongs the synthesis of NO via eNOS. Although NO and O2•− can react to neutralize each other, it is unclear that they achieve sufficient concentrations or colocalization in vascular cells for this to provide significant negative cross talk. Although it is clear that CO plays an important role in angiogenesis, the mechanism is less clear. Its direct activation of sGC is weak, so other undefined targets may play a dominant role in angiogenic signaling. One potential indirect pathway is via its inhibition of H2S synthesis via CBS. Physiological CO levels are consistent with this mechanism. Yet, current data suggests that CSE rather than CBS is the major source of H2S synthesis in the vasculature. H2S in turn can clearly inhibit ACE at physiological concentrations, and the resulting loss of Ang II could account for the effects of H2S on blood pressure. It is unclear whether the same pathway mediates the reported effects of H2S on angiogenesis or whether another target needs to be identified for this function. Inhibition of ACE would alter Ang II, which is known to control NO synthesis via the AT2 receptor. Thus, angiogenic signaling of H2S could be mediated by NO.
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, NCI, Center for Cancer Research (D.D.R.) and NIH grant K22 CA128616 (J.S.I.).
11. Abbreviations
- Ang
angiopoietin
- Ang II
Angiotensin II
- CBS
cystathionine β-synthase
- CSE
cystathionine γ-lyase
- cGK
cGMP-dependent protein kinase
- CORM
CO-releasing molecule
- CSE
cystathionine γ-lyase
- EGFR
epidermal growth factor receptor
- EPC
endothelial progenitor cells
- FGF2
Fibroblast growth factor-2, also known as basic fibroblast growth factor
- HO-1
heme oxygenase-1
- Hsp90
heat shock protein-90
- HUVEC
human umbilical vein endothelial cells
- I/R
ischemia/reperfusion
- LDL
low density lipoprotein
- MLC2
myosin light chain 2
- MMP
matrix metalloproteinase
- NOS
nitric oxide synthase
- NOX
NADPH oxidase
- PDE
phosphodiesterase
- PI3K
phosphatidylinositol 3-kinase
- PlGF
placental growth factor
- PPARγ
peroxisome proliferator-activated receptor-γ
- PPIX
protoporphyrin-IX
- Prx
peroxiredoxin
- PTP
protein tyrosine phosphatase
- ROS
reactive oxygen species
- S1P
sphingosine 1-phosphate
- SDF-1
stromal cell-derived growth factor 1
- sGC
soluble guanylate cyclase
- TNFα
tumor necrosis factor α
- TSP
thrombospondin
- VEGF
vascular endothelial growth factor
- VSMC
vascular smooth muscle cells
Biographies
Thomas W. Miller was born in Tacoma Washington in 1978. He received his B.S. in Chemistry from the University of Pittsburgh (2000), M.S. in Chemistry (2003), and Ph.D. in Chemistry (2007) from the University of California, Los Angeles under the guidance of Professor Jon Fukuto studying nitrogen oxide signal transduction. He received a cancer research training award from the National Cancer Institute at the National Institutes of Health and is a postdoctoral research fellow in the laboratory of David D. Roberts. His current research interests include the molecular mechanisms of thrombospondin1 - control of nitric oxide signaling and the modulation of extracellular matrix signaling by changes in the cellular redox environment.
Jeff S. Isenberg completed his undergraduate studies at the University of Pennsylvania and his medical training at Tulane University School of Medicine, where he simultaneously earned a Masters of Public Health. Following general surgery residency he specialized in reconstructive, hand and microsurgery training at Yale University and the University of Southern California. He then completed a post-doctoral research fellowship in the Laboratory of Pathology of the NCI, NIH funded by a Cancer Research Fellow Training Award. He is currently Associate Professor of Medicine and a principal investigator of the Hemostasis and Vascular Biology Research Institute, University of Pittsburgh School of Medicine.
David D. Roberts is Chief of the Biochemical Pathology Section, Laboratory of Pathology in the Center for Cancer Research, NCI in Bethesda. He received his B.S. in Chemistry in 1976 from the Massachusetts Institute of Technology and his Ph.D. in Biological Chemistry in 1983 from The University of Michigan with Irwin Goldstein. After postdoctoral training at Michigan and in the Laboratory of Biochemical Pharmacology of the National Institute of Diabetes and Digestive and Kidney Diseases with Victor Ginsburg, he became a Research Chemist there in 1987 and moved to the NCI in 1988. His research has focused on the role of thrombospondins in regulating vascular cell behavior, tumor immunity and angiogenesis, the biochemistry of cell surface carbohydrates, and host-pathogen interactions.
References
- 1.Black WC, Welch HG. N Engl J Med. 1993;328:1237. doi: 10.1056/NEJM199304293281706. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. N Engl J Med. 1971;285:1182. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 3.Naumov GN, Folkman J, Straume O, Akslen LA, editors. APMIS. Vol. 116. 2008. p. 569. [DOI] [PubMed] [Google Scholar]
- 4.Beecken WD, Engl T, Ringel EM, Camphausen K, Michaelis M, Jonas D, Folkman J, Shing Y, Blaheta RA. Ann Surg Oncol. 2006;13:1241. doi: 10.1245/s10434-006-9009-9. [DOI] [PubMed] [Google Scholar]
- 5.Lien S, Lowman HB. Handb Exp Pharmacol. 2008;181:131. doi: 10.1007/978-3-540-73259-4_6. [DOI] [PubMed] [Google Scholar]
- 6.Kenny PA, Lee GY, Bissell MJ. Front Biosci. 2007;12:3468. doi: 10.2741/2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cabebe E, Wakelee H. Drugs Today (Barc) 2006;42:387. doi: 10.1358/dot.2006.42.6.985633. [DOI] [PubMed] [Google Scholar]
- 8.Boehm T, Folkman J, Browder T, O’Reilly MS. Nature. 1997;390:404. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- 9.O’Reilly MS, Holmgren L, Chen C, Folkman J. Nat Med. 1996;2:689. doi: 10.1038/nm0696-689. [DOI] [PubMed] [Google Scholar]
- 10.Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, Bumgarner R, Goodlett DR, Aebersold R, Hood L. Science. 2001;292:929. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- 11.Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. J Clin Invest. 1997;100:3131. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Nature. 1999;399:597. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Nature. 1999;399:601. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 14.Nieder C, Wiedenmann N, Andratschke NH, Astner ST, Molls M. Rev Recent Clin Trials. 2007;2:163. doi: 10.2174/157488707781662733. [DOI] [PubMed] [Google Scholar]
- 15.Czirok A, Zamir EA, Szabo A, Little CD. Curr Top Dev Biol. 2008;81:269. doi: 10.1016/S0070-2153(07)81009-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heil M, Eitenmuller I, Schmitz-Rixen T, Schaper W. J Cell Mol Med. 2006;10:45. doi: 10.1111/j.1582-4934.2006.tb00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinstein BM. Cell. 2005;120:299. doi: 10.1016/j.cell.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 18.Polykandriotis E, Tjiawi J, Euler S, Arkudas A, Hess A, Brune K, Greil P, Lametschwandtner A, Horch RE, Kneser U. Microvasc Res. 2008;75:25. doi: 10.1016/j.mvr.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Blebea J, Vu JH, Assadnia S, McLaughlin PJ, Atnip RG, Zagon IS. J Vasc Surg. 2002;35:532. doi: 10.1067/mva.2002.120042. [DOI] [PubMed] [Google Scholar]
- 20.von Tell D, Armulik A, Betsholtz C. Exp Cell Res. 2006;312:623. doi: 10.1016/j.yexcr.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 21.Liersch R, Detmar M. Thromb Haemost. 2007;98:304. [PubMed] [Google Scholar]
- 22.Kopp HG, Ramos CA, Rafii S. Curr Opin Hematol. 2006;13:175. doi: 10.1097/01.moh.0000219664.26528.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Science. 2008;319:195. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- 24.Goon PK, Lip GY, Boos CJ, Stonelake PS, Blann AD. Neoplasia. 2006;8:79. doi: 10.1593/neo.05592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rinderknecht M, Detmar M. J Cell Physiol. 2008;216:347. doi: 10.1002/jcp.21494. [DOI] [PubMed] [Google Scholar]
- 26.Shibuya M. J Biochem Mol Biol. 2006;39:469. doi: 10.5483/bmbrep.2006.39.5.469. [DOI] [PubMed] [Google Scholar]
- 27.Wagner PD, Olfert IM, Tang K, Breen EC. Respir Physiol Neurobiol. 2006;151:159. doi: 10.1016/j.resp.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 28.Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE. N Engl J Med. 2008;358:1129. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Cell. 2007;130:691. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grothey A, Ellis LM. Cancer J. 2008;14:170. doi: 10.1097/PPO.0b013e318178d9de. [DOI] [PubMed] [Google Scholar]
- 31.Dejana E, Orsenigo F, Lampugnani MG. J Cell Sci. 2008;121:2115. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- 32.Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Angiogenesis. 2008;11:109. doi: 10.1007/s10456-008-9099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong H, Bowen JP. Curr Top Med Chem. 2007;7:1379. doi: 10.2174/156802607781696855. [DOI] [PubMed] [Google Scholar]
- 34.Li X, Tjwa M, Van Hove I, Enholm B, Neven E, Paavonen K, Jeltsch M, Juan TD, Sievers RE, Chorianopoulos E, Wada H, Vanwildemeersch M, Noel A, Foidart JM, Springer ML, von Degenfeld G, Dewerchin M, Blau HM, Alitalo K, Eriksson U, Carmeliet P, Moons L. Arterioscler Thromb Vasc Biol. 2008;28:1614. doi: 10.1161/ATVBAHA.107.158725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M, De Mol M, Autiero M, Wyns S, Plaisance S, Moons L, van Rooijen N, Giacca M, Stassen JM, Dewerchin M, Collen D, Carmeliet P. Cell. 2007;131:463. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 36.Carmeliet P. Nat Med. 2002;8:14. doi: 10.1038/nm0102-14. [DOI] [PubMed] [Google Scholar]
- 37.Dumont DJ, Jussila L, Taipale J, Lymboussaki A, Mustonen T, Pajusola K, Breitman M, Alitalo K. Science. 1998;282:946. doi: 10.1126/science.282.5390.946. [DOI] [PubMed] [Google Scholar]
- 38.Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, Betsholtz C, Alitalo K. Nat Immunol. 2004;5:74. doi: 10.1038/ni1013. [DOI] [PubMed] [Google Scholar]
- 39.He Y, Rajantie I, Ilmonen M, Makinen T, Karkkainen MJ, Haiko P, Salven P, Alitalo K. Cancer Res. 2004;64:3737. doi: 10.1158/0008-5472.CAN-04-0088. [DOI] [PubMed] [Google Scholar]
- 40.Haiko P, Makinen T, Keskitalo S, Taipale J, Karkkainen MJ, Baldwin ME, Stacker SA, Achen MG, Alitalo K. Mol Cell Biol. 2008;28:4843. doi: 10.1128/MCB.02214-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, Yin M, Coffin JD, Kong L, Kranias EG, Luo W, Boivin GP, Duffy JJ, Pawlowski SA, Doetschman T. Nat Med. 1998;4:201. doi: 10.1038/nm0298-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Cytokine Growth Factor Rev. 2005;16:159. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Ribatti D, Vacca A, Rusnati M, Presta M. Cytokine Growth Factor Rev. 2007;18:327. doi: 10.1016/j.cytogfr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 44.Rusnati M, Presta M. Curr Pharm Des. 2007;13:2025. doi: 10.2174/138161207781039689. [DOI] [PubMed] [Google Scholar]
- 45.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Cell. 1996;87:1171. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 46.Shimoda H, Bernas MJ, Witte MH, Gale NW, Yancopoulos GD, Kato S. Cell Tissue Res. 2007;328:329. doi: 10.1007/s00441-006-0360-8. [DOI] [PubMed] [Google Scholar]
- 47.Dellinger M, Hunter R, Bernas M, Gale N, Yancopoulos G, Erickson R, Witte M. Dev Biol. 2008;319:309. doi: 10.1016/j.ydbio.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tressel SL, Kim H, Ni CW, Chang K, Velasquez-Castano JC, Taylor WR, Yoon YS, Jo H. Arterioscler Thromb Vasc Biol. 2008;4:4. doi: 10.1161/ATVBAHA.108.175463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brat DJ, Bellail AC, Van Meir EG. Neuro-oncol. 2005;7:122. doi: 10.1215/S1152851704001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abdel-Malak NA, Srikant CB, Kristof AS, Magder SA, Di Battista JA, Hussain SN. Blood. 2008;111:4145. doi: 10.1182/blood-2007-08-110338. [DOI] [PubMed] [Google Scholar]
- 51.Hato T, Tabata M, Oike Y. Trends Cardiovasc Med. 2008;18:6. doi: 10.1016/j.tcm.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 52.Nakamura M, Han B, Nunobiki O, Kakudo K. Curr Cancer Drug Targets. 2006;6:635. doi: 10.2174/156800906778742442. [DOI] [PubMed] [Google Scholar]
- 53.Nikitenko LL, Fox SB, Kehoe S, Rees MC, Bicknell R. Br J Cancer. 2006;94:1. doi: 10.1038/sj.bjc.6602832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shindo T, Kurihara Y, Nishimatsu H, Moriyama N, Kakoki M, Wang Y, Imai Y, Ebihara A, Kuwaki T, Ju KH, Minamino N, Kangawa K, Ishikawa T, Fukuda M, Akimoto Y, Kawakami H, Imai T, Morita H, Yazaki Y, Nagai R, Hirata Y, Kurihara H. Circulation. 2001;104:1964. doi: 10.1161/hc4101.097111. [DOI] [PubMed] [Google Scholar]
- 55.Simoncini T, Mannella P, Fornari L, Caruso A, Varone G, Genazzani AR. Steroids. 2004;69:537. doi: 10.1016/j.steroids.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 56.Caulin-Glaser T, Garcia-Cardena G, Sarrel P, Sessa WC, Bender JR. Circ Res. 1997;81:885. doi: 10.1161/01.res.81.5.885. [DOI] [PubMed] [Google Scholar]
- 57.Johnson ML, Grazul-Bilska AT, Redmer DA, Reynolds LP. Endocrine. 2006;30:333. doi: 10.1007/s12020-006-0012-5. [DOI] [PubMed] [Google Scholar]
- 58.Sutherland TE, Anderson RL, Hughes RA, Altmann E, Schuliga M, Ziogas J, Stewart AG. Drug Discov Today. 2007;12:577. doi: 10.1016/j.drudis.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 59.Bergers G, Hanahan D. Nat Rev Cancer. 2008;8:592. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reed MJ, Edelberg JM. Sci Aging Knowl Environ. 2004;2004:pe7. doi: 10.1126/sageke.2004.7.pe7. [DOI] [PubMed] [Google Scholar]
- 61.Fleischer R, Weston GC, Vollenhoven BJ, Rogers PA. Best Pract Res Clin Obstet Gynaecol. 2008;22:603. doi: 10.1016/j.bpobgyn.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 62.Ong CT, Khoo YT, Tan EK, Mukhopadhyay A, Do DV, Han HC, Lim IJ, Phan TT. J Pathol. 2007;211:95. doi: 10.1002/path.2081. [DOI] [PubMed] [Google Scholar]
- 63.Gira AK, Brown LF, Washington CV, Cohen C, Arbiser JL. J Am Acad Dermatol. 2004;50:850. doi: 10.1016/j.jaad.2003.11.061. [DOI] [PubMed] [Google Scholar]
- 64.Good DJ, Polverini PJ, Rastinejad F, Le BM, Lemons RS, Frazier WA, Bouck NP. Proc Natl Acad Sci U S A. 1990;87:6624. doi: 10.1073/pnas.87.17.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Taraboletti G, Roberts D, Liotta LA, Giavazzi R. J Cell Biol. 1990;111:765. doi: 10.1083/jcb.111.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bagavandoss P, Wilks JW. Biochem Biophys Res Commun. 1990;170:867. doi: 10.1016/0006-291x(90)92171-u. [DOI] [PubMed] [Google Scholar]
- 67.Iruela-Arispe ML, Vazquez F, Ortega MA. Ann N Y Acad Sci. 1999;886:58. doi: 10.1111/j.1749-6632.1999.tb09400.x. [DOI] [PubMed] [Google Scholar]
- 68.Weinstat-Saslow DL, Zabrenetzky VS, VanHoutte K, Frazier WA, Roberts DD, Steeg PS. Cancer Res. 1994;54:6504. [PubMed] [Google Scholar]
- 69.Sheibani N, Frazier WA. Proc Natl Acad Sci U S A. 1995;92:6788. doi: 10.1073/pnas.92.15.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hawighorst T, Oura H, Streit M, Janes L, Nguyen L, Brown LF, Oliver G, Jackson DG, Detmar M. Oncogene. 2002;21:7945. doi: 10.1038/sj.onc.1205956. [DOI] [PubMed] [Google Scholar]
- 71.Gutierrez LS, Suckow M, Lawler J, Ploplis VA, Castellino FJ. Carcinogenesis. 2003;24:199. doi: 10.1093/carcin/24.2.199. [DOI] [PubMed] [Google Scholar]
- 72.Naumov GN, Bender E, Zurakowski D, Kang SY, Sampson D, Flynn E, Watnick RS, Straume O, Akslen LA, Folkman J, Almog N. J Natl Cancer Inst. 2006;98:316. doi: 10.1093/jnci/djj068. [DOI] [PubMed] [Google Scholar]
- 73.Volpert OV, Tolsma SS, Pellerin S, Feige JJ, Chen H, Mosher DF, Bouck N. Biochem Biophys Res Commun. 1995;217:326. doi: 10.1006/bbrc.1995.2780. [DOI] [PubMed] [Google Scholar]
- 74.Streit M, Stephen AE, Hawighorst T, Matsuda K, Lange-Asschenfeldt B, Brown LF, Vacanti JP, Detmar M. Cancer Res. 2002;62:2004. [PubMed] [Google Scholar]
- 75.Hawighorst T, Velasco P, Streit M, Hong YK, Kyriakides TR, Brown LF, Bornstein P, Detmar M. EMBO J. 2001;20:2631. doi: 10.1093/emboj/20.11.2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J. Cell. 1994;79:315. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 77.Cao Y, Chen A, An SS, Ji RW, Davidson D, Llinas M. J Biol Chem. 1997;272:22924. doi: 10.1074/jbc.272.36.22924. [DOI] [PubMed] [Google Scholar]
- 78.O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Cell. 1997;88:277. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 79.Wickstrom SA, Alitalo K, Keski-Oja J. Adv Cancer Res. 2005;94:197. doi: 10.1016/S0065-230X(05)94005-0. [DOI] [PubMed] [Google Scholar]
- 80.Deininger MH, Wybranietz WA, Graepler FT, Lauer UM, Meyermann R, Schluesener HJ. FASEB J. 2003;17:1267. doi: 10.1096/fj.02-1118com. [DOI] [PubMed] [Google Scholar]
- 81.Mundel TM, Kalluri R. Microvascular research. 2007;74:85. doi: 10.1016/j.mvr.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Woodall BP, Nystrom A, Iozzo RA, Eble JA, Niland S, Krieg T, Eckes B, Pozzi A, Iozzo RV. J Biol Chem. 2008;283:2335. doi: 10.1074/jbc.M708364200. [DOI] [PubMed] [Google Scholar]
- 83.Gerwins P, Skoldenberg E, Claesson-Welsh L. Crit Rev Oncol Hematol. 2000;34:185. doi: 10.1016/s1040-8428(00)00062-7. [DOI] [PubMed] [Google Scholar]
- 84.Geretti E, Shimizu A, Klagsbrun M. Angiogenesis. 2008;11:31. doi: 10.1007/s10456-008-9097-1. [DOI] [PubMed] [Google Scholar]
- 85.Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. J Cell Biol. 2006;174:593. doi: 10.1083/jcb.200602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blanes MG, Oubaha M, Rautureau Y, Gratton JP. J Biol Chem. 2007;282:10660. doi: 10.1074/jbc.M609048200. [DOI] [PubMed] [Google Scholar]
- 87.Gelinas DS, Bernatchez PN, Rollin S, Bazan NG, Sirois MG. Br J Pharmacol. 2002;137:1021. doi: 10.1038/sj.bjp.0704956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reihill JA, Ewart MA, Hardie DG, Salt IP. Biochem Biophys Res Commun. 2007;354:1084. doi: 10.1016/j.bbrc.2007.01.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Duval M, Le Boeuf F, Huot J, Gratton JP. Mol Biol Cell. 2007;18:4659. doi: 10.1091/mbc.E07-05-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fulton D, Ruan L, Sood SG, Li C, Zhang Q, Venema RC. Circ Res. 2008;102:497. doi: 10.1161/CIRCRESAHA.107.162933. [DOI] [PubMed] [Google Scholar]
- 91.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL, Jain RK. Proc Natl Acad Sci U S A. 2001;98:2604. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jones MK, Tsugawa K, Tarnawski AS, Baatar D. Biochem Biophys Res Commun. 2004;318:520. doi: 10.1016/j.bbrc.2004.04.055. [DOI] [PubMed] [Google Scholar]
- 93.Thomas DD, Miranda KM, Espey MG, Citrin D, Jourd’heuil D, Paolocci N, Hewett SJ, Colton CA, Grisham MB, Feelisch M, Wink DA. Methods Enzymol. 2002;359:84. doi: 10.1016/s0076-6879(02)59174-6. [DOI] [PubMed] [Google Scholar]
- 94.Ridnour LA, Thomas DD, Donzelli S, Espey MG, Roberts DD, Wink DA, Isenberg JS. Antioxid Redox Signal. 2006;8:1329. doi: 10.1089/ars.2006.8.1329. [DOI] [PubMed] [Google Scholar]
- 95.Ridnour LA, Thomas DD, Switzer C, Flores-Santana W, Isenberg JS, Ambs S, Roberts DD, Wink DA. Nitric Oxide. 2008;19:73. doi: 10.1016/j.niox.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dudzinski DM, Michel T. Cardiovasc Res. 2007;75:247. doi: 10.1016/j.cardiores.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Babaei S, Teichert-Kuliszewska K, Zhang Q, Jones N, Dumont DJ, Stewart DJ. Am J Pathol. 2003;162:1927. doi: 10.1016/S0002-9440(10)64326-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Urano T, Ito Y, Akao M, Sawa T, Miyata K, Tabata M, Morisada T, Hato T, Yano M, Kadomatsu T, Yasunaga K, Shibata R, Murohara T, Akaike T, Tanihara H, Suda T, Oike Y. Arterioscler Thromb Vasc Biol. 2008;28:827. doi: 10.1161/ATVBAHA.107.149674. [DOI] [PubMed] [Google Scholar]
- 99.Shindo T, Kurihara H, Kuno K, Yokoyama H, Wada T, Kurihara Y, Imai T, Wang Y, Ogata M, Nishimatsu H, Moriyama N, Oh-hashi Y, Morita H, Ishikawa T, Nagai R, Yazaki Y, Matsushima K. J Clin Invest. 2000;105:1345. doi: 10.1172/JCI8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rikitake Y, Hirata K, Kawashima S, Ozaki M, Takahashi T, Ogawa W, Inoue N, Yokoyama M. Arterioscler Thromb Vasc Biol. 2002;22:108. doi: 10.1161/hq0102.101843. [DOI] [PubMed] [Google Scholar]
- 101.Babaei S, Teichert-Kuliszewska K, Monge JC, Mohamed F, Bendeck MP, Stewart DJ. Circ Res. 1998;82:1007. doi: 10.1161/01.res.82.9.1007. [DOI] [PubMed] [Google Scholar]
- 102.Kim KH, Moriarty K, Bender JR. Steroids. 2008;73:864. doi: 10.1016/j.steroids.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee YM, Bae MH, Lee OH, Moon EJ, Moon CK, Kim WH, Kim KW. Oncol Rep. 2004;12:843. [PubMed] [Google Scholar]
- 104.Rabinovsky ED, Draghia-Akli R. Mol Ther. 2004;9:46. doi: 10.1016/j.ymthe.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 105.Zhao X, Lu X, Feng Q. Am J Physiol Heart Circ Physiol. 2002;283:H2371. doi: 10.1152/ajpheart.00383.2002. [DOI] [PubMed] [Google Scholar]
- 106.Morishita T, Tsutsui M, Shimokawa H, Horiuchi M, Tanimoto A, Suda O, Tasaki H, Huang PL, Sasaguri Y, Yanagihara N, Nakashima Y. FASEB J. 2002;16:1994. doi: 10.1096/fj.02-0155fje. [DOI] [PubMed] [Google Scholar]
- 107.Lundberg JO, Weitzberg E. Am J Physiol Heart Circ Physiol. 2008;295:H477. doi: 10.1152/ajpheart.00611.2008. [DOI] [PubMed] [Google Scholar]
- 108.Williams JM, White CR, Chang MM, Injeti ER, Zhang L, Pearce WJ. J Appl Physiol. 2006;100:1857. doi: 10.1152/japplphysiol.00662.2005. [DOI] [PubMed] [Google Scholar]
- 109.Fukumura D, Kashiwagi S, Jain RK. Nat Rev Cancer. 2006;6:521. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 110.Jadeski LC, Lala PK. Am J Pathol. 1999;155:1381. doi: 10.1016/S0002-9440(10)65240-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kashiwagi S, Izumi Y, Gohongi T, Demou ZN, Xu L, Huang PL, Buerk DG, Munn LL, Jain RK, Fukumura D. J Clin Invest. 2005;115:1816. doi: 10.1172/JCI24015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee YC, Huang CH, Wang CJ, Liu CC, Wu WJ, Chang LL, Lin HH. BJU Int. 2007;100:1116. doi: 10.1111/j.1464-410X.2007.07110.x. [DOI] [PubMed] [Google Scholar]
- 113.Yang Q, Tian Y, Liu S, Zeine R, Chlenski A, Salwen HR, Henkin J, Cohn SL. Cancer Res. 2007;67:1716. doi: 10.1158/0008-5472.CAN-06-2595. [DOI] [PubMed] [Google Scholar]
- 114.Hefler LA, Tempfer CB, Bashford MT, Unfried G, Zeillinger R, Schneeberger C, Koelbl H, Nagele F, Huber JC. Mol Hum Reprod. 2002;8:95. doi: 10.1093/molehr/8.1.95. [DOI] [PubMed] [Google Scholar]
- 115.Riener EK, Hefler LA, Grimm C, Galid A, Zeillinger R, Tong-Cacsire D, Gitsch G, Leodolter S, Tempfer CB. Gynecol Oncol. 2004;93:686. doi: 10.1016/j.ygyno.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 116.Ghilardi G, Biondi ML, Cecchini F, DeMonti M, Guagnellini E, Scorza R. Nitric Oxide. 2003;9:118. doi: 10.1016/j.niox.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 117.Medeiros RM, Morais A, Vasconcelos A, Costa S, Pinto D, Oliveira J, Ferreira P, Lopes C. Clin Cancer Res. 2002;8:3433. [PubMed] [Google Scholar]
- 118.Marangoni K, Araujo TG, Neves AF, Goulart LR. BMC Cancer. 2008;8:273. doi: 10.1186/1471-2407-8-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Murad F, Mittal CK, Arnold WP, Katsuki S, Kimura H. Adv Cyclic Nucleotide Res. 1978;9:145. [PubMed] [Google Scholar]
- 120.Gruetter CA, Barry BK, McNamara DB, Gruetter DY, Kadowitz PJ, Ignarro L. J Cyclic Nucleotide Res. 1979;5:211. [PubMed] [Google Scholar]
- 121.Stuehr DJ, Fasehun OA, Kwon NS, Gross SS, Gonzalez JA, Levi R, Nathan CF. FASEB J. 1991;5:98. doi: 10.1096/fasebj.5.1.1703974. [DOI] [PubMed] [Google Scholar]
- 122.Palmer RM, Ashton DS, Moncada S. Nature. 1988;333:664. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- 123.Bredt DS, Snyder SH. Proc Natl Acad Sci U S A. 1990;87:682. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Burstyn JN, Yu AE, Dierks EA, Hawkins BK, Dawson JH. Biochemistry. 1995;34:5896. doi: 10.1021/bi00017a019. [DOI] [PubMed] [Google Scholar]
- 125.Martin E, Berka V, Bogatenkova E, Murad F, Tsai AL. J Biol Chem. 2006;281:27836. doi: 10.1074/jbc.M601078200. [DOI] [PubMed] [Google Scholar]
- 126.Stone JR, Marletta MA. Biochemistry. 1994;33:5636. doi: 10.1021/bi00184a036. [DOI] [PubMed] [Google Scholar]
- 127.Ignarro LJ. J Physiol Pharmacol. 2002;53:503. [PubMed] [Google Scholar]
- 128.Lancaster JR, Jr, Langrehr JM, Bergonia HA, Murase N, Simmons RL, Hoffman RA. J Biol Chem. 1992;267:10994. [PubMed] [Google Scholar]
- 129.Guazzi M, Samaja M. Curr Med Chem. 2007;14:2181. doi: 10.2174/092986707781389619. [DOI] [PubMed] [Google Scholar]
- 130.Ghiadoni L, Versari D, Taddei S. Curr Hypertens Rep. 2008;10:52. doi: 10.1007/s11906-008-0011-4. [DOI] [PubMed] [Google Scholar]
- 131.Dony E, Lai YJ, Dumitrascu R, Pullamsetti SS, Savai R, Ghofrani HA, Weissmann N, Schudt C, Flockerzi D, Seeger W, Grimminger F, Schermuly RT. Eur Respir J. 2008;31:599. doi: 10.1183/09031936.00002007. [DOI] [PubMed] [Google Scholar]
- 132.Pande A, Lombardo J, Spangenthal E, Javle M. Anticancer Res. 2007;27:3465. [PubMed] [Google Scholar]
- 133.Wu S, Chen JJ, Kudelka A, Lu J, Zhu X. Lancet Oncol. 2008;9:117. doi: 10.1016/S1470-2045(08)70003-2. [DOI] [PubMed] [Google Scholar]
- 134.van Heeckeren WJ, Sanborn SL, Narayan A, Cooney MM, McCrae KR, Schmaier AH, Remick SC. Curr Opin Hematol. 2007;14:468. doi: 10.1097/MOH.0b013e3282a6457f. [DOI] [PubMed] [Google Scholar]
- 135.Ku DD, Zaleski JK, Liu S, Brock TA. Am J Physiol. 1993;265:H586. doi: 10.1152/ajpheart.1993.265.2.H586. [DOI] [PubMed] [Google Scholar]
- 136.Curwen JO, Musgrove HL, Kendrew J, Richmond GH, Ogilvie DJ, Wedge SR. Clin Cancer Res. 2008;14:3124. doi: 10.1158/1078-0432.CCR-07-4783. [DOI] [PubMed] [Google Scholar]
- 137.Osol G, Celia G, Gokina N, Barron C, Chien E, Mandala M, Luksha L, Kublickiene K. Am J Physiol Heart Circ Physiol. 2008;294:H1381. doi: 10.1152/ajpheart.00922.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.De Matteo R, May CN. Br J Pharmacol. 2003;140:1414. doi: 10.1038/sj.bjp.0705572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, Roberts DD. Proc Natl Acad Sci U S A. 2005;102:13141. doi: 10.1073/pnas.0502977102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Isenberg JS, Wink DA, Roberts DD. Cardiovasc Res. 2006;71:785. doi: 10.1016/j.cardiores.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 141.Patel MK, Lymn JS, Clunn GF, Hughes AD. Arterioscler Thromb Vasc Biol. 1997;17:2107. doi: 10.1161/01.atv.17.10.2107. [DOI] [PubMed] [Google Scholar]
- 142.Yabkowitz R, Mansfield PJ, Ryan US, Suchard SJ. J Cell Physiol. 1993;157:24. doi: 10.1002/jcp.1041570104. [DOI] [PubMed] [Google Scholar]
- 143.Isenberg J, Calzada M, Zhou L, Guo N, Lawler J, Wang X, Frazier W, Roberts D. Matrix Biol. 2005;24:110. doi: 10.1016/j.matbio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 144.Baenziger NL, Brodie GN, Majerus PW. Proc Natl Acad Sci U S A. 1971;68:240. doi: 10.1073/pnas.68.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Baenziger NL, Brodie GN, Majerus PW. J Biol Chem. 1972;247:2723. [PubMed] [Google Scholar]
- 146.Vogel T, Guo NH, Krutzsch HC, Blake DA, Hartman J, Mendelovitz S, Panet A, Roberts DD. J Cell Biochem. 1993;53:74. doi: 10.1002/jcb.240530109. [DOI] [PubMed] [Google Scholar]
- 147.Chandrasekaran L, He C-Z, Al-Barazi HO, Krutzsch HC, Iruela-Arispe ML, Roberts DD. Mol Biol Cell. 2000;11:2885. doi: 10.1091/mbc.11.9.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Calzada MJ, Sipes JM, Krutzsch HC, Yurchenco PD, Annis DS, Mosher DF, Roberts DD. J Biol Chem. 2003;278:40679. doi: 10.1074/jbc.M302014200. [DOI] [PubMed] [Google Scholar]
- 149.Calzada MJ, Annis DS, Zeng B, Marcinkiewicz C, Banas B, Lawler J, Mosher DF, Roberts DD. J Biol Chem. 2004;279:41734. doi: 10.1074/jbc.M406267200. [DOI] [PubMed] [Google Scholar]
- 150.Dawson DW, Pearce SF, Zhong R, Silverstein RL, Frazier WA, Bouck NP. J Cell Biol. 1997;138:707. doi: 10.1083/jcb.138.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Nat Med. 2000;6:41. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 152.Iruela-Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD. Circulation. 1999;100:1423. doi: 10.1161/01.cir.100.13.1423. [DOI] [PubMed] [Google Scholar]
- 153.Isenberg JS, Ridnour LA, Dimitry J, Frazier WA, Wink DA, Roberts DD. J Biol Chem. 2006 doi: 10.1074/jbc.M605040200. [DOI] [PubMed] [Google Scholar]
- 154.Isenberg JS, Romeo MJ, Maxhimer JB, Smedley J, Frazier WA, Roberts DD. Ann Surg. 2008;247:860. doi: 10.1097/SLA.0b013e31816c4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gao AG, Lindberg FP, Finn MB, Blystone SD, Brown EJ, Frazier WA. J Biol Chem. 1996;271:21. doi: 10.1074/jbc.271.1.21. [DOI] [PubMed] [Google Scholar]
- 156.Radomski MW, Palmer RM, Moncada S. Proc Natl Acad Sci U S A. 1990;87:5193. doi: 10.1073/pnas.87.13.5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Isenberg JS, Romeo MJ, Yu C, Yu CK, Nghiem K, Monsale J, Rick ME, Wink DA, Frazier WA, Roberts DD. Blood. 2008;111:613. doi: 10.1182/blood-2007-06-098392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhu W, Smart EJ. J Biol Chem. 2005;280:29543. doi: 10.1074/jbc.M501238200. [DOI] [PubMed] [Google Scholar]
- 159.Febbraio M, Hajjar DP, Silverstein RL. J Clin Invest. 2001;108:785. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Isenberg JS, Jia Y, Fukuyama J, Switzer CH, Wink DA, Roberts DD. J Biol Chem. 2007;282:15404. doi: 10.1074/jbc.M701638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Denninger JW, Marletta MA. Biochim Biophys Acta. 1999;1411:334. doi: 10.1016/s0005-2728(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 162.Llorens S, Jordan J, Nava E. J Physiol Biochem. 2002;58:179. doi: 10.1007/BF03179855. [DOI] [PubMed] [Google Scholar]
- 163.Murphy RA, Rembold CM. Can J Physiol Pharmacol. 2005;83:857. doi: 10.1139/y05-090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tang DD, Anfinogenova Y. J Cardiovasc Pharmacol Ther. 2008;13:130. doi: 10.1177/1074248407313737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Isenberg JS, Hyodo F, Matsumoto K, Romeo MJ, Abu-Asab M, Tsokos M, Kuppusamy P, Wink DA, Krishna MC, Roberts DD. Blood. 2007;109:1945. doi: 10.1182/blood-2006-08-041368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Isenberg JS, Romeo MJ, Abu-Asab M, Tsokos M, Oldenborg A, Pappan L, Wink DA, Frazier WA, Roberts DD. Circ Res. 2007;100:712. doi: 10.1161/01.RES.0000259579.35787.4e. [DOI] [PubMed] [Google Scholar]
- 167.Lien YH, Lai LW, Silva AL. Life Sci. 2003;74:543. doi: 10.1016/j.lfs.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 168.Inglott FS, Mathie RT. Hepatogastroenterology. 2000;47:1722. [PubMed] [Google Scholar]
- 169.Isenberg JS, Maxhimer JB, Powers P, Tsokos M, Frazier WA, Roberts DD. Surgery. 2008;144:752. doi: 10.1016/j.surg.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Isenberg JS, Pappan LK, Romeo MJ, Abu-Asab M, Tsokos M, Wink DA, Frazier WA, Roberts DD. Ann Surg. 2008;247:180. doi: 10.1097/SLA.0b013e31815685dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Moeller BJ, Richardson RA, Dewhirst MW. Cancer Metastasis Rev. 2007;26:241. doi: 10.1007/s10555-007-9056-0. [DOI] [PubMed] [Google Scholar]
- 172.Isenberg JS, Hyodo F, Ridnour LA, Shannon CS, Wink DA, Krishna MC, Roberts DD. Neoplasia. 2008;10:886. doi: 10.1593/neo.08264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jain RK. Int J Radiat Biol. 1991;60:85. doi: 10.1080/09553009114551621. [DOI] [PubMed] [Google Scholar]
- 174.Baluk P, Hashizume H, McDonald DM. Curr Opin Genet Dev. 2005;15:102. doi: 10.1016/j.gde.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 175.Jirtle RL. Int J Hyperthermia. 1988;4:355. doi: 10.3109/02656738809016490. [DOI] [PubMed] [Google Scholar]
- 176.Yamashita Y, Kurohiji T, Tuszynski GP, Sakai T, Shirakusa T. Cancer. 1998;82:632. doi: 10.1002/(sici)1097-0142(19980215)82:4<632::aid-cncr3>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 177.Nathan FE, Hernandez E, Dunton CJ, Treat J, Switalska HI, Joseph RR, Tuszynski GP. Cancer. 1994;73:2853. doi: 10.1002/1097-0142(19940601)73:11<2853::aid-cncr2820731131>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 178.Liebmann J, DeLuca AM, Coffin D, Keefer LK, Venzon D, Wink DA, Mitchell JB. Cancer Res. 1994;54:3365. [PubMed] [Google Scholar]
- 179.Chen Y, Stanford A, Simmons RL, Ford HR, Hoffman RA. Cell Immunol. 2001;214:72. doi: 10.1006/cimm.2001.1880. [DOI] [PubMed] [Google Scholar]
- 180.Shi S, Wang G, Wang Y, Zhang L. Nitric Oxide. 2005;13:1. doi: 10.1016/j.niox.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 181.Isenberg JS, Maxhimer JB, Hyodo F, Pendrak ML, Ridnour LA, DeGraff WG, Tsokos M, Wink DA, Roberts DD. Am J Pathol. 2008;173:1100. doi: 10.2353/ajpath.2008.080237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Gonzalez C, Corbacho AM, Eiserich JP, Garcia C, Lopez-Barrera F, Morales-Tlalpan V, Barajas-Espinosa A, Diaz-Munoz M, Rubio R, Lin SH, Martinez de la Escalera G, Clapp C. Endocrinology. 2004;145:5714. doi: 10.1210/en.2004-0647. [DOI] [PubMed] [Google Scholar]
- 183.Garcia C, Aranda J, Arnold E, Thebault S, Macotela Y, Lopez-Casillas F, Mendoza V, Quiroz-Mercado H, Hernandez-Montiel HL, Lin SH, de la Escalera GM, Clapp C. J Clin Invest. 2008;118:2291. doi: 10.1172/JCI34508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lee SH, Nishino M, Mazumdar T, Garcia GE, Galfione M, Lee FL, Lee CL, Liang A, Kim J, Feng L, Eissa NT, Lin SH, Yu-Lee LY. Cancer Res. 2005;65:7984. doi: 10.1158/0008-5472.CAN-05-0631. [DOI] [PubMed] [Google Scholar]
- 185.Urbich C, Reissner A, Chavakis E, Dernbach E, Haendeler J, Fleming I, Zeiher AM, Kaszkin M, Dimmeler S. FASEB J. 2002;16:706. doi: 10.1096/fj.01-0637fje. [DOI] [PubMed] [Google Scholar]
- 186.Schmidt A, Wenzel D, Thorey I, Werner S, Fleischmann BK, Bloch W. Endothelium. 2005;12:251. doi: 10.1080/10623320500476690. [DOI] [PubMed] [Google Scholar]
- 187.Chavakis E, Dernbach E, Hermann C, Mondorf UF, Zeiher AM, Dimmeler S. Circulation. 2001;103:2102. doi: 10.1161/01.cir.103.16.2102. [DOI] [PubMed] [Google Scholar]
- 188.Fleming I, Mohamed A, Galle J, Turchanowa L, Brandes RP, Fisslthaler B, Busse R. Cardiovasc Res. 2005;65:897. doi: 10.1016/j.cardiores.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 189.Chow SE, Hshu YC, Wang JS, Chen JK. J Appl Physiol. 2007;102:1520. doi: 10.1152/japplphysiol.00881.2006. [DOI] [PubMed] [Google Scholar]
- 190.Thomas DD, Ridnour LA, Espey MG, Donzelli S, Ambs S, Hussain SP, Harris CC, Degraff W, Roberts DD, Mitchell JB, Wink DA. J Biol Chem. 2006 doi: 10.1074/jbc.M602242200. [DOI] [PubMed] [Google Scholar]
- 191.Febbraio M, Silverstein RL. Int J Biochem Cell Biol. 2007;39:2012. doi: 10.1016/j.biocel.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Halliwell B. Haemostasis. 1993;23(Suppl 1):118. doi: 10.1159/000216921. [DOI] [PubMed] [Google Scholar]
- 193.Meier B, Radeke HH, Selle S, Younes M, Sies H, Resch K, Habermehl GG. Biochem J. 1989;263:539. doi: 10.1042/bj2630539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Science. 1995;270:296. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 195.Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. J Biol Chem. 1997;272:217. [PubMed] [Google Scholar]
- 196.Mahadev K, Wu X, Zilbering A, Zhu L, Lawrence JT, Goldstein BJ. J Biol Chem. 2001;276:48662. doi: 10.1074/jbc.M105061200. [DOI] [PubMed] [Google Scholar]
- 197.Stone JR, Yang S. Antioxid Redox Signal. 2006;8:243. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 198.Szatrowski TP, Nathan CF. Cancer Res. 1991;51:794. [PubMed] [Google Scholar]
- 199.Ushio-Fukai M. Cardiovasc Res. 2006;71:226. doi: 10.1016/j.cardiores.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 200.Brauchle M, Funk JO, Kind P, Werner S. J Biol Chem. 1996;271:21793. doi: 10.1074/jbc.271.36.21793. [DOI] [PubMed] [Google Scholar]
- 201.Kuroki M, Voest EE, Amano S, Beerepoot LV, Takashima S, Tolentino M, Kim RY, Rohan RM, Colby KA, Yeo KT, Adamis AP. J Clin Invest. 1996;98:1667. doi: 10.1172/JCI118962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ruef J, Hu ZY, Yin LY, Wu Y, Hanson SR, Kelly AB, Harker LA, Rao GN, Runge MS, Patterson C. Circ Res. 1997;81:24. doi: 10.1161/01.res.81.1.24. [DOI] [PubMed] [Google Scholar]
- 203.Chua CC, Hamdy RC, Chua BH. Free Radic Biol Med. 1998;25:891. doi: 10.1016/s0891-5849(98)00115-4. [DOI] [PubMed] [Google Scholar]
- 204.Kosmidou I, Xagorari A, Roussos C, Papapetropoulos A. Am J Physiol Lung Cell Mol Physiol. 2001;280:L585. doi: 10.1152/ajplung.2001.280.4.L585. [DOI] [PubMed] [Google Scholar]
- 205.Cho M, Hunt TK, Hussain MZ. Am J Physiol Heart Circ Physiol. 2001;280:H2357. doi: 10.1152/ajpheart.2001.280.5.H2357. [DOI] [PubMed] [Google Scholar]
- 206.Arbiser JL, Petros J, Klafter R, Govindajaran B, McLaughlin ER, Brown LF, Cohen C, Moses M, Kilroy S, Arnold RS, Lambeth JD. Proc Natl Acad Sci U S A. 2002;99:715. doi: 10.1073/pnas.022630199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Cao Y, Cao R. Nature. 1999;398:381. doi: 10.1038/18793. [DOI] [PubMed] [Google Scholar]
- 208.Saccani A, Saccani S, Orlando S, Sironi M, Bernasconi S, Ghezzi P, Mantovani A, Sica A. Proc Natl Acad Sci U S A. 2000;97:2761. doi: 10.1073/pnas.97.6.2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Urao N, Inomata H, Razvi M, Kim HW, Wary K, McKinney R, Fukai T, Ushio-Fukai M. Circ Res. 2008;103:212. doi: 10.1161/CIRCRESAHA.108.176230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lambeth JD. Curr Opin Hematol. 2002;9:11. doi: 10.1097/00062752-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 211.Loschen G, Azzi A, Richter C, Flohe L. FEBS Lett. 1974;42:68. doi: 10.1016/0014-5793(74)80281-4. [DOI] [PubMed] [Google Scholar]
- 212.Forman HJ, Kennedy JA. Biochem Biophys Res Commun. 1974;60:1044. doi: 10.1016/0006-291x(74)90418-5. [DOI] [PubMed] [Google Scholar]
- 213.Massey V, Strickland S, Mayhew SG, Howell LG, Engel PC, Matthews RG, Schuman M, Sullivan PA. Biochem Biophys Res Commun. 1969;36:891. doi: 10.1016/0006-291x(69)90287-3. [DOI] [PubMed] [Google Scholar]
- 214.Babior BM. IUBMB Life. 2000;50:267. doi: 10.1080/713803730. [DOI] [PubMed] [Google Scholar]
- 215.DeLeo FR, Renee J, McCormick S, Nakamura M, Apicella M, Weiss JP, Nauseef WM. J Clin Invest. 1998;101:455. doi: 10.1172/JCI949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lambeth JD, Kawahara T, Diebold B. Free Radic Biol Med. 2007;43:319. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Isogai Y, Iizuka T, Shiro Y. J Biol Chem. 1995;270:7853. doi: 10.1074/jbc.270.14.7853. [DOI] [PubMed] [Google Scholar]
- 218.Sumimoto H. FEBS J. 2008;275:3249. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 219.Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, Drain PF, Sasidhar M, Nabel EG, Takahashi T, Lukacs NW, Ryter SW, Morita K, Choi AM. J Exp Med. 2006;203:2377. doi: 10.1084/jem.20060845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Cheng G, Diebold BA, Hughes Y, Lambeth JD. J Biol Chem. 2006;281:17718. doi: 10.1074/jbc.M512751200. [DOI] [PubMed] [Google Scholar]
- 221.Griendling KK, Sorescu D, Ushio-Fukai M. Circ Res. 2000;86:494. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 222.BelAiba RS, Djordjevic T, Petry A, Diemer K, Bonello S, Banfi B, Hess J, Pogrebniak A, Bickel C, Gorlach A. Free Radic Biol Med. 2007;42:446. doi: 10.1016/j.freeradbiomed.2006.10.054. [DOI] [PubMed] [Google Scholar]
- 223.Jones SA, O’Donnell VB, Wood JD, Broughton JP, Hughes EJ, Jones OT. Am J Physiol. 1996;271:H1626. doi: 10.1152/ajpheart.1996.271.4.H1626. [DOI] [PubMed] [Google Scholar]
- 224.Qin Z, Reszka KJ, Fukai T, Weintraub NL. Transl Res. 2008;151:68. doi: 10.1016/j.trsl.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Deisseroth A, Dounce AL. Physiol Rev. 1970;50:319. doi: 10.1152/physrev.1970.50.3.319. [DOI] [PubMed] [Google Scholar]
- 226.Chance B, Sies H, Boveris A. Physiol Rev. 1979;59:527. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 227.Oshino N, Chance B, Sies H, Bucher T. Arch Biochem Biophys. 1973;154:117. doi: 10.1016/0003-9861(73)90040-4. [DOI] [PubMed] [Google Scholar]
- 228.Antunes F, Cadenas E. Free Radic Biol Med. 2001;30:1008. doi: 10.1016/s0891-5849(01)00493-2. [DOI] [PubMed] [Google Scholar]
- 229.Antunes F, Cadenas E. FEBS Lett. 2000;475:121. doi: 10.1016/s0014-5793(00)01638-0. [DOI] [PubMed] [Google Scholar]
- 230.Kulagina NV, Michael AC. Anal Chem. 2003;75:4875. doi: 10.1021/ac034573g. [DOI] [PubMed] [Google Scholar]
- 231.Forman HJ, Fukuto JM, Torres M. Am J Physiol Cell Physiol. 2004;287:C246. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 232.Stone JR. Arch Biochem Biophys. 2004;422:119. doi: 10.1016/j.abb.2003.12.029. [DOI] [PubMed] [Google Scholar]
- 233.Leung-Toung R, Zhao Y, Li W, Tam TF, Karimian K, Spino M. Curr Med Chem. 2006;13:547. doi: 10.2174/092986706776055733. [DOI] [PubMed] [Google Scholar]
- 234.Cho SH, Lee CH, Ahn Y, Kim H, Ahn CY, Yang KS, Lee SR. FEBS Lett. 2004;560:7. doi: 10.1016/s0014-5793(04)00112-7. [DOI] [PubMed] [Google Scholar]
- 235.Denu JM, Dixon JE. Curr Opin Chem Biol. 1998;2:633. doi: 10.1016/s1367-5931(98)80095-1. [DOI] [PubMed] [Google Scholar]
- 236.Dougher-Vermazen M, Hulmes JD, Bohlen P, Terman BI. Biochem Biophys Res Commun. 1994;205:728. doi: 10.1006/bbrc.1994.2726. [DOI] [PubMed] [Google Scholar]
- 237.Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, Quinn MT, Pagano PJ, Johnson C, Alexander RW. Circ Res. 2002;91:1160. doi: 10.1161/01.res.0000046227.65158.f8. [DOI] [PubMed] [Google Scholar]
- 238.Colavitti R, Pani G, Bedogni B, Anzevino R, Borrello S, Waltenberger J, Galeotti T. J Biol Chem. 2002;277:3101. doi: 10.1074/jbc.M107711200. [DOI] [PubMed] [Google Scholar]
- 239.Lin MT, Yen ML, Lin CY, Kuo ML. Mol Pharmacol. 2003;64:1029. doi: 10.1124/mol.64.5.1029. [DOI] [PubMed] [Google Scholar]
- 240.Kim YM, Kim KE, Koh GY, Ho YS, Lee KJ. Cancer Res. 2006;66:6167. doi: 10.1158/0008-5472.CAN-05-3640. [DOI] [PubMed] [Google Scholar]
- 241.Chen JX, Zeng H, Lawrence ML, Blackwell TS, Meyrick B. Am J Physiol Heart Circ Physiol. 2006;291:H1563. doi: 10.1152/ajpheart.01081.2005. [DOI] [PubMed] [Google Scholar]
- 242.Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, Griendling KK. J Biol Chem. 1999;274:22699. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- 243.Mahadev K, Zilbering A, Zhu L, Goldstein BJ. J Biol Chem. 2001;276:21938. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- 244.Cunnick JM, Dorsey JF, Mei L, Wu J. Biochem Mol Biol Int. 1998;45:887. doi: 10.1002/iub.7510450506. [DOI] [PubMed] [Google Scholar]
- 245.Meng TC, Fukada T, Tonks NK. Mol Cell. 2002;9:387. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 246.Peters KG, Davis MG, Howard BW, Pokross M, Rastogi V, Diven C, Greis KD, Eby-Wilkens E, Maier M, Evdokimov A, Soper S, Genbauffe F. J Inorg Biochem. 2003;96:321. doi: 10.1016/s0162-0134(03)00236-8. [DOI] [PubMed] [Google Scholar]
- 247.Weibrecht I, Bohmer SA, Dagnell M, Kappert K, Ostman A, Bohmer FD. Free Radic Biol Med. 2007;43:100. doi: 10.1016/j.freeradbiomed.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 248.Visse R, Nagase H. Circ Res. 2003;92:827. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 249.Page-McCaw A, Ewald AJ, Werb Z. Nat Rev Mol Cell Biol. 2007;8:221. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Newby AC. Physiol Rev. 2005;85:1. doi: 10.1152/physrev.00048.2003. [DOI] [PubMed] [Google Scholar]
- 251.Handsley MM, Edwards DR. Int J Cancer. 2005;115:849. doi: 10.1002/ijc.20945. [DOI] [PubMed] [Google Scholar]
- 252.Egeblad M, Werb Z. Nat Rev Cancer. 2002;2:161. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 253.Grote K, Flach I, Luchtefeld M, Akin E, Holland SM, Drexler H, Schieffer B. Circ Res. 2003;92:e80. doi: 10.1161/01.RES.0000077044.60138.7C. [DOI] [PubMed] [Google Scholar]
- 254.Cook-Mills JM. Cell Mol Biol (NoisyLe - -Grand) 2006;52:8. [PMC free article] [PubMed] [Google Scholar]
- 255.Yasuda M, Ohzeki Y, Shimizu S, Naito S, Ohtsuru A, Yamamoto T, Kuroiwa Y. Life Sci. 1999;64:249. doi: 10.1016/s0024-3205(98)00560-8. [DOI] [PubMed] [Google Scholar]
- 256.Shono T, Ono M, Izumi H, Jimi SI, Matsushima K, Okamoto T, Kohno K, Kuwano M. Mol Cell Biol. 1996;16:4231. doi: 10.1128/mcb.16.8.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Brar SS, Kennedy TP, Sturrock AB, Huecksteadt TP, Quinn MT, Murphy TM, Chitano P, Hoidal JR. Am J Physiol Lung Cell Mol Physiol. 2002;282:L782. doi: 10.1152/ajplung.00206.2001. [DOI] [PubMed] [Google Scholar]
- 258.Ni W, Zhan Y, He H, Maynard E, Balschi JA, Oettgen P. Circ Res. 2007;101:985. doi: 10.1161/CIRCRESAHA.107.152439. [DOI] [PubMed] [Google Scholar]
- 259.Lee JH, Jeong MW, Kim W, Choi YH, Kim KT. J Biol Chem. 2008;283:19826. doi: 10.1074/jbc.M706201200. [DOI] [PubMed] [Google Scholar]
- 260.Ushio-Fukai M. Sci STKE. 2006;2006:re8. doi: 10.1126/stke.3492006re8. [DOI] [PubMed] [Google Scholar]
- 261.Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. Cell. 2002;111:471. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- 262.Georgiou G, Masip L. Science. 2003;300:592. doi: 10.1126/science.1084976. [DOI] [PubMed] [Google Scholar]
- 263.Wood ZA, Poole LB, Karplus PA. Science. 2003;300:650. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 264.Young LJ, Caughey WS. Biochemistry. 1986;25:152. doi: 10.1021/bi00349a022. [DOI] [PubMed] [Google Scholar]
- 265.Kreck TC, Shade ED, Lamm WJ, McKinney SE, Hlastala MP. Am J Respir Crit Care Med. 2001;163:458. doi: 10.1164/ajrccm.163.2.2003039. [DOI] [PubMed] [Google Scholar]
- 266.Kaczorowski DJ, Zuckerbraun BS. Curr Med Chem. 2007;14:2720. doi: 10.2174/092986707782023181. [DOI] [PubMed] [Google Scholar]
- 267.Wu L, Wang R. Pharmacol Rev. 2005;57:585. doi: 10.1124/pr.57.4.3. [DOI] [PubMed] [Google Scholar]
- 268.Kharitonov VG, Sharma VS, Magde D, Koesling D. Biochemistry. 1997;36:6814. doi: 10.1021/bi970201o. [DOI] [PubMed] [Google Scholar]
- 269.Kharitonov VG, Russwurm M, Magde D, Sharma VS, Koesling D. Biochem Biophys Res Commun. 1997;239:284. doi: 10.1006/bbrc.1997.7470. [DOI] [PubMed] [Google Scholar]
- 270.Ryter SW, Otterbein LE, Morse D, Choi AM. Mol Cell Biochem. 2002;234–235:249. doi: 10.1023/A:1015957026924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Yoshida T, Migita CT. J Inorg Biochem. 2000;82:33. doi: 10.1016/s0162-0134(00)00156-2. [DOI] [PubMed] [Google Scholar]
- 272.Abraham NG, Kappas A. Pharmacol Rev. 2008;60:79. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- 273.Morse D, Choi AM. Am J Respir Cell Mol Biol. 2002;27:8. doi: 10.1165/ajrcmb.27.1.4862. [DOI] [PubMed] [Google Scholar]
- 274.Hangaishi M, Ishizaka N, Aizawa T, Kurihara Y, Taguchi J, Nagai R, Kimura S, Ohno M. Biochem Biophys Res Commun. 2000;279:582. doi: 10.1006/bbrc.2000.3973. [DOI] [PubMed] [Google Scholar]
- 275.Christou H, Morita T, Hsieh CM, Koike H, Arkonac B, Perrella MA, Kourembanas S. Circ Res. 2000;86:1224. doi: 10.1161/01.res.86.12.1224. [DOI] [PubMed] [Google Scholar]
- 276.Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, Soares MP. J Exp Med. 2000;192:1015. doi: 10.1084/jem.192.7.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Goodman AI, Choudhury M, da Silva JL, Schwartzman ML, Abraham NG. Proc Soc Exp Biol Med. 1997;214:54. doi: 10.3181/00379727-214-44069. [DOI] [PubMed] [Google Scholar]
- 278.Maines MD, Abrahamsson PA. Urology. 1996;47:727. doi: 10.1016/s0090-4295(96)00010-6. [DOI] [PubMed] [Google Scholar]
- 279.Doi K, Akaike T, Fujii S, Tanaka S, Ikebe N, Beppu T, Shibahara S, Ogawa M, Maeda H. Br J Cancer. 1999;80:1945. doi: 10.1038/sj.bjc.6690624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Sahoo SK, Sawa T, Fang J, Tanaka S, Miyamoto Y, Akaike T, Maeda H. Bioconjug Chem. 2002;13:1031. doi: 10.1021/bc020010k. [DOI] [PubMed] [Google Scholar]
- 281.Sunamura M, Duda DG, Ghattas MH, Lozonschi L, Motoi F, Yamauchi J-I, Matsuno S, Shibahara S, Abraham NG. Angiogenesis. 2003;6:15. doi: 10.1023/a:1025803600840. [DOI] [PubMed] [Google Scholar]
- 282.Deramaudt BM, Braunstein S, Remy P, Abraham NG. J Cell Biochem. 1998;68:121. doi: 10.1002/(sici)1097-4644(19980101)68:1<121::aid-jcb12>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 283.Li Volti G, Sacerdoti D, Sangras B, Vanella A, Mezentsev A, Scapagnini G, Falck JR, Abraham NG. Antioxid Redox Signal. 2005;7:704. doi: 10.1089/ars.2005.7.704. [DOI] [PubMed] [Google Scholar]
- 284.Nishie A, Ono M, Shono T, Fukushi J, Otsubo M, Onoue H, Ito Y, Inamura T, Ikezaki K, Fukui M, Iwaki T, Kuwano M. Clin Cancer Res. 1999;5:1107. [PubMed] [Google Scholar]
- 285.Torisu-Itakura H, Furue M, Kuwano M, Ono M. Jpn J Cancer Res. 2000;91:906. doi: 10.1111/j.1349-7006.2000.tb01033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Suzuki M, Iso-o N, Takeshita S, Tsukamoto K, Mori I, Sato T, Ohno M, Nagai R, Ishizaka N. Biochem Biophys Res Commun. 2003;302:138. doi: 10.1016/s0006-291x(03)00114-1. [DOI] [PubMed] [Google Scholar]
- 287.Dulak J, Jozkowicz A, Foresti R, Kasza A, Frick M, Huk I, Green CJ, Pachinger O, Weidinger F, Motterlini R. Antioxid Redox Signal. 2002;4:229. doi: 10.1089/152308602753666280. [DOI] [PubMed] [Google Scholar]
- 288.Abdel-Aziz MT, el-Asmar MF, el-Miligy D, Atta H, Shaker O, Ghattas MH, Hosni H, Kamal N. Int J Biochem Cell Biol. 2003;35:324. doi: 10.1016/s1357-2725(02)00172-3. [DOI] [PubMed] [Google Scholar]
- 289.Jozkowicz A, Huk I, Nigisch A, Weigel G, Dietrich W, Motterlini R, Dulak J. Antioxid Redox Signal. 2003;5:155. doi: 10.1089/152308603764816514. [DOI] [PubMed] [Google Scholar]
- 290.Jozkowicz A, Dulak J, Piatkowska E, Placha W, Dembinska-Kiec A. Acta Biochim Pol. 2000;47:1147. [PubMed] [Google Scholar]
- 291.Yamakawa K, Hosoi M, Koyama H, Tanaka S, Fukumoto S, Morii H, Nishizawa Y. Biochem Biophys Res Commun. 2000;271:571. doi: 10.1006/bbrc.2000.2665. [DOI] [PubMed] [Google Scholar]
- 292.Inoue M, Itoh H, Tanaka T, Chun TH, Doi K, Fukunaga Y, Sawada N, Yamshita J, Masatsugu K, Saito T, Sakaguchi S, Sone M, Yamahara K, Yurugi T, Nakao K. Arterioscler Thromb Vasc Biol. 2001;21:560. doi: 10.1161/01.atv.21.4.560. [DOI] [PubMed] [Google Scholar]
- 293.Bamba H, Ota S, Kato A, Kawamoto C, Fujiwara K. Biochem Biophys Res Commun. 2000;273:485. doi: 10.1006/bbrc.2000.2969. [DOI] [PubMed] [Google Scholar]
- 294.Straus DS, Glass CK. Trends Immunol. 2007;28:551. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 295.Kronke G, Kadl A, Ikonomu E, Bluml S, Furnkranz A, Sarembock IJ, Bochkov VN, Exner M, Binder BR, Leitinger N. Arterioscler Thromb Vasc Biol. 2007;27:1276. doi: 10.1161/ATVBAHA.107.142638. [DOI] [PubMed] [Google Scholar]
- 296.Zingarelli B, Sheehan M, Hake PW, O’Connor M, Denenberg A, Cook JA. J Immunol. 2003;171:6827. doi: 10.4049/jimmunol.171.12.6827. [DOI] [PubMed] [Google Scholar]
- 297.Wright MM, Schopfer FJ, Baker PR, Vidyasagar V, Powell P, Chumley P, Iles KE, Freeman BA, Agarwal A. Proc Natl Acad Sci U S A. 2006;103:4299. doi: 10.1073/pnas.0506541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Alvarez-Maqueda M, El Bekay R, Alba G, Monteseirin J, Chacon P, Vega A, Martin-Nieto J, Bedoya FJ, Pintado E, Sobrino F. J Biol Chem. 2004;279:21929. doi: 10.1074/jbc.M400492200. [DOI] [PubMed] [Google Scholar]
- 299.Liu JD, Tsai SH, Lin SY, Ho YS, Hung LF, Pan S, Ho FM, Lin CM, Liang YC. Life Sci. 2004;74:2451. doi: 10.1016/j.lfs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 300.Gong P, Stewart D, Hu B, Li N, Cook J, Nel A, Alam J. Antioxid Redox Signal. 2002;4:249. doi: 10.1089/152308602753666307. [DOI] [PubMed] [Google Scholar]
- 301.Renedo M, Gayarre J, Garcia-Dominguez CA, Perez-Rodriguez A, Prieto A, Canada FJ, Rojas JM, Perez-Sala D. Biochemistry. 2007;46:6607. doi: 10.1021/bi602389p. [DOI] [PubMed] [Google Scholar]
- 302.Jozkowicz A, Huk I, Nigisch A, Weigel G, Weidinger F, Dulak J. Antioxid Redox Signal. 2002;4:577. doi: 10.1089/15230860260220076. [DOI] [PubMed] [Google Scholar]
- 303.Kim EH, Na HK, Surh YJ. Ann N Y Acad Sci. 2006;1090:375. doi: 10.1196/annals.1378.041. [DOI] [PubMed] [Google Scholar]
- 304.Pae HO, Oh GS, Choi BM, Kim YM, Chung HT. Endocrinology. 2005;146:2229. doi: 10.1210/en.2004-1431. [DOI] [PubMed] [Google Scholar]
- 305.Frank S, Hubner G, Breier G, Longaker MT, Greenhalgh DG, Werner S. J Biol Chem. 1995;270:12607. doi: 10.1074/jbc.270.21.12607. [DOI] [PubMed] [Google Scholar]
- 306.Jazwa A, Loboda A, Golda S, Cisowski J, Szelag M, Zagorska A, Sroczynska P, Drukala J, Jozkowicz A, Dulak J. Free Radic Biol Med. 2006;40:1250. doi: 10.1016/j.freeradbiomed.2005.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Loboda A, Jazwa A, Wegiel B, Jozkowicz A, Dulak J. Cell Mol Biol (Noisy-Le-Grand) 2005;51:347. [PMC free article] [PubMed] [Google Scholar]
- 308.Malaguarnera L, Pilastro MR, Quan S, Ghattas MH, Yang L, Mezentsev AV, Kushida T, Abraham NG, Kappas A. Int J Mol Med. 2002;10:433. [PubMed] [Google Scholar]
- 309.Ueda E, Ozerdem U, Chen YH, Yao M, Huang KT, Sun H, Martins-Green M, Bartolini P, Walker AM. Endocr Relat Cancer. 2006;13:95. doi: 10.1677/erc.1.01076. [DOI] [PubMed] [Google Scholar]
- 310.Fernandez M, Bonkovsky HL. Br J Pharmacol. 2003;139:634. doi: 10.1038/sj.bjp.0705272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Terry CM, Clikeman JA, Hoidal JR, Callahan KS. Am J Physiol. 1999;276:H1493. doi: 10.1152/ajpheart.1999.276.5.H1493. [DOI] [PubMed] [Google Scholar]
- 312.Morbidelli L, Donnini S, Ziche M. Curr Pharm Des. 2003;9:521. doi: 10.2174/1381612033391405. [DOI] [PubMed] [Google Scholar]
- 313.Dulak J, Jozkowicz A. Antioxid Redox Signal. 2003;5:123. doi: 10.1089/152308603321223612. [DOI] [PubMed] [Google Scholar]
- 314.Pryor WA, Houk KN, Foote CS, Fukuto JM, Ignarro LJ, Squadrito GL, Davies KJ. Am J Physiol Regul Integr Comp Physiol. 2006;291:R491. doi: 10.1152/ajpregu.00614.2005. [DOI] [PubMed] [Google Scholar]
- 315.Fukuto JM, Collins MD. Curr Pharm Des. 2007;13:2952. doi: 10.2174/138161207782110525. [DOI] [PubMed] [Google Scholar]
- 316.Ding Y, McCoubrey WK, Jr, Maines MD. Eur J Biochem. 1999;264:854. doi: 10.1046/j.1432-1327.1999.00677.x. [DOI] [PubMed] [Google Scholar]
- 317.Kim YM, Bergonia H, Lancaster JR., Jr FEBS Lett. 1995;374:228. doi: 10.1016/0014-5793(95)01115-u. [DOI] [PubMed] [Google Scholar]
- 318.Hara E, Takahashi K, Takeda K, Nakayama M, Yoshizawa M, Fujita H, Shirato K, Shibahara S. Biochem Pharmacol. 1999;58:227. doi: 10.1016/s0006-2952(99)00097-0. [DOI] [PubMed] [Google Scholar]
- 319.Tran MD, Neary JT. Proc Natl Acad Sci U S A. 2006;103:9321. doi: 10.1073/pnas.0603146103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Dulak J, Jozkowicz A, Dembinska-Kiec A, Guevara I, Zdzienicka A, Zmudzinska-Grochot D, Florek I, Wojtowicz A, Szuba A, Cooke JP. Arterioscler Thromb Vasc Biol. 2000;20:659. doi: 10.1161/01.atv.20.3.659. [DOI] [PubMed] [Google Scholar]
- 321.Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Li Calzi S, Lach R, Hock TD, Chen B, Hill-Kapturczak N, Siegal GP, Dulak J, Jozkowicz A, Grant MB, Agarwal A. J Exp Med. 2007;204:605. doi: 10.1084/jem.20061609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Lin HH, Chen YH, Chang PF, Lee YT, Yet SF, Chau LY. J Mol Cell Cardiol. 2008;45:44. doi: 10.1016/j.yjmcc.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 323.Kucia M, Jankowski K, Reca R, Wysoczynski M, Bandura L, Allendorf DJ, Zhang J, Ratajczak J, Ratajczak MZ. J Mol Histol. 2004;35:233. doi: 10.1023/b:hijo.0000032355.66152.b8. [DOI] [PubMed] [Google Scholar]
- 324.Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Nature. 1998;393:595. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 325.Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N, Nishikawa S, Kishimoto T, Nagasawa T. Nature. 1998;393:591. doi: 10.1038/31261. [DOI] [PubMed] [Google Scholar]
- 326.Kucia M, Reca R, Miekus K, Wanzeck J, Wojakowski W, Janowska-Wieczorek A, Ratajczak J, Ratajczak MZ. Stem Cells. 2005;23:879. doi: 10.1634/stemcells.2004-0342. [DOI] [PubMed] [Google Scholar]
- 327.Herbrig K, Pistrosch F, Foerster S, Gross P. Kidney Blood Press Res. 2006;29:24. doi: 10.1159/000092484. [DOI] [PubMed] [Google Scholar]
- 328.Bainbridge SA, Smith GN. Free Radic Biol Med. 2005;38:979. doi: 10.1016/j.freeradbiomed.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 329.Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, Sibai BM, Epstein FH, Romero R, Thadhani R, Karumanchi SA. N Engl J Med. 2006;355:992. doi: 10.1056/NEJMoa055352. [DOI] [PubMed] [Google Scholar]
- 330.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. J Clin Invest. 2003;111:649. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Cudmore M, Ahmad S, Al-Ani B, Fujisawa T, Coxall H, Chudasama K, Devey LR, Wigmore SJ, Abbas A, Hewett PW, Ahmed A. Circulation. 2007;115:1789. doi: 10.1161/CIRCULATIONAHA.106.660134. [DOI] [PubMed] [Google Scholar]
- 332.Gabriele Sass PL, Volker Schmitz, Esther Raskopf, Matthias Ocker, Daniel Neureiter, Matthias Meissnitzer, Elena Tasika, Andrea Tannapfel, Gisa Tiegs. Int J Cancer. 2008;123:1269. [Google Scholar]
- 333.Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH. Science. 1993;259:381. doi: 10.1126/science.7678352. [DOI] [PubMed] [Google Scholar]
- 334.Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Nat Med. 2000;6:422. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 335.Fukuto JM, Ignarro LJ. Acc Chem Res. 1997;30:149. [Google Scholar]
- 336.Halliwell B, Zhao K, Whiteman M. Free Radic Res. 1999;31:651. doi: 10.1080/10715769900301221. [DOI] [PubMed] [Google Scholar]
- 337.Turkseven S, Kruger A, Mingone CJ, Kaminski P, Inaba M, Rodella LF, Ikehara S, Wolin MS, Abraham NG. Am J Physiol Heart Circ Physiol. 2005;289:H701. doi: 10.1152/ajpheart.00024.2005. [DOI] [PubMed] [Google Scholar]
- 338.Kruger AL, Peterson S, Turkseven S, Kaminski PM, Zhang FF, Quan S, Wolin MS, Abraham NG. Circulation. 2005;111:3126. doi: 10.1161/CIRCULATIONAHA.104.517102. [DOI] [PubMed] [Google Scholar]
- 339.Zuckerbraun BS, Chin BY, Bilban M, d’Avila JC, Rao J, Billiar TR, Otterbein LE. FASEB J. 2007;21:1099. doi: 10.1096/fj.06-6644com. [DOI] [PubMed] [Google Scholar]
- 340.Cooper CE, Brown GC. J Bioenerg Biomembr. 2008;7:7. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 341.D’Amico G, Lam F, Hagen T, Moncada S. J Cell Sci. 2006;119:2291. doi: 10.1242/jcs.02914. [DOI] [PubMed] [Google Scholar]
- 342.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Proc Natl Acad Sci U S A. 1998;95:11715. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Connor KM, Subbaram S, Regan KJ, Nelson KK, Mazurkiewicz JE, Bartholomew PJ, Aplin AE, Tai YT, Aguirre-Ghiso J, Flores SC, Melendez JA. J Biol Chem. 2005;280:16916. doi: 10.1074/jbc.M410690200. [DOI] [PubMed] [Google Scholar]
- 344.Li L, Moore PK. Trends Pharmacol Sci. 2008;29:84. doi: 10.1016/j.tips.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 345.Pearson RJ, Wilson T, Wang R. Clin Invest Med. 2006;29:146. [PubMed] [Google Scholar]
- 346.Lowicka E, Beltowski J. Pharmacol Rep. 2007;59:4. [PubMed] [Google Scholar]
- 347.Kimura H, Nagai Y, Umemura K, Kimura Y. Antioxid Redox Signal. 2005;7:795. doi: 10.1089/ars.2005.7.795. [DOI] [PubMed] [Google Scholar]
- 348.Kamoun P. Amino Acids. 2004;26:243. doi: 10.1007/s00726-004-0072-x. [DOI] [PubMed] [Google Scholar]
- 349.Kimura H. Mol Neurobiol. 2002;26:13. doi: 10.1385/MN:26:1:013. [DOI] [PubMed] [Google Scholar]
- 350.Taoka S, Banerjee R. J Inorg Biochem. 2001;87:245. doi: 10.1016/s0162-0134(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 351.Wang R. Antioxid Redox Signal. 2003;5:493. doi: 10.1089/152308603768295249. [DOI] [PubMed] [Google Scholar]
- 352.Ishii I, Akahoshi N, Yu XN, Kobayashi Y, Namekata K, Komaki G, Kimura H. Biochem J. 2004;381:113. doi: 10.1042/BJ20040243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Mudd SH, Finkelstein JD, Irreverre F, Laster L. J Biol Chem. 1965;240:4382. [PubMed] [Google Scholar]
- 354.Zhao W, Ndisang JF, Wang R. Can J Physiol Pharmacol. 2003;81:848. doi: 10.1139/y03-077. [DOI] [PubMed] [Google Scholar]
- 355.Zhao W, Zhang J, Lu Y, Wang R. EMBO J. 2001;20:6008. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Warenycia MW, Steele JA, Karpinski E, Reiffenstein RJ. Neurotoxicology. 1989;10:191. [PubMed] [Google Scholar]
- 357.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. Science. 2008;322:587. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC. Cardiovasc Res. 2007;76:29. doi: 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 359.Isenberg JS, Jia Y, Field L, Ridnour LA, Sparatore A, Del Soldato P, Sowers AL, Yeh GC, Moody TW, Wink DA, Ramchandran R, Roberts DD. Br J Pharmacol. 2007;151:63. doi: 10.1038/sj.bjp.0707194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Li JM, Shah AM. J Biol Chem. 2003;278:12094. doi: 10.1074/jbc.M209793200. [DOI] [PubMed] [Google Scholar]
- 361.Cai H, Li Z, Dikalov S, Holland SM, Hwang J, Jo H, Dudley SC, Jr, Harrison DG. J Biol Chem. 2002;277:48311. doi: 10.1074/jbc.M208884200. [DOI] [PubMed] [Google Scholar]
- 362.Cifuentes ME, Rey FE, Carretero OA, Pagano PJ. Am J Physiol Heart Circ Physiol. 2000;279:H2234. doi: 10.1152/ajpheart.2000.279.5.H2234. [DOI] [PubMed] [Google Scholar]
- 363.Grishko V, Pastukh V, Solodushko V, Gillespie M, Azuma J, Schaffer S. Am J Physiol Heart Circ Physiol. 2003;285:H2364. doi: 10.1152/ajpheart.00408.2003. [DOI] [PubMed] [Google Scholar]
- 364.Nakagami H, Takemoto M, Liao JK. J Mol Cell Cardiol. 2003;35:851. doi: 10.1016/s0022-2828(03)00145-7. [DOI] [PubMed] [Google Scholar]
- 365.Escobar E, Rodriguez-Reyna TS, Arrieta O, Sotelo J. Curr Vasc Pharmacol. 2004;2:385. doi: 10.2174/1570161043385556. [DOI] [PubMed] [Google Scholar]
- 366.Laggner H, Hermann M, Esterbauer H, Muellner MK, Exner M, Gmeiner BM, Kapiotis S. J Hypertens. 2007;25:2100. doi: 10.1097/HJH.0b013e32829b8fd0. [DOI] [PubMed] [Google Scholar]
- 367.Yusuf M, Kwong Huat BT, Hsu A, Whiteman M, Bhatia M, Moore PK. Biochem Biophys Res Commun. 2005;333:1146. doi: 10.1016/j.bbrc.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 368.Chadefaux B, Rethore MO, Raoul O, Ceballos I, Poissonnier M, Gilgenkranz S, Allard D. Biochem Biophys Res Commun. 1985;128:40. doi: 10.1016/0006-291x(85)91641-9. [DOI] [PubMed] [Google Scholar]
- 369.Kamoun P, Belardinelli MC, Chabli A, Lallouchi K, Chadefaux-Vekemans B. Am J Med Genet A. 2003;116A:310. doi: 10.1002/ajmg.a.10847. [DOI] [PubMed] [Google Scholar]
- 370.Kamoun P. Med Hypotheses. 2001;57:389. doi: 10.1054/mehy.2001.1377. [DOI] [PubMed] [Google Scholar]
- 371.Greene AK, Kim S, Rogers GF, Fishman SJ, Olsen BR, Mulliken JB. Pediatrics. 2008;121:e135. doi: 10.1542/peds.2007-1316. [DOI] [PubMed] [Google Scholar]
- 372.Zorick TS, Mustacchi Z, Bando SY, Zatz M, Moreira-Filho CA, Olsen B, Passos-Bueno MR. Eur J Hum Genet. 2001;9:811. doi: 10.1038/sj.ejhg.5200721. [DOI] [PubMed] [Google Scholar]
- 373.Distrutti E, Mencarelli A, Santucci L, Renga B, Orlandi S, Donini A, Shah V, Fiorucci S. Hepatology. 2008;47:659. doi: 10.1002/hep.22037. [DOI] [PubMed] [Google Scholar]
- 374.Meng QH, Yang G, Yang W, Jiang B, Wu L, Wang R. Am J Pathol. 2007;170:1406. doi: 10.2353/ajpath.2007.060939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Atta HM, El-Rehani MA, Raheim SA, Galal AM. J Surg Res. 2008;146:202. doi: 10.1016/j.jss.2007.04.038. [DOI] [PubMed] [Google Scholar]
- 376.Oh GS, Pae HO, Lee BS, Kim BN, Kim JM, Kim HR, Jeon SB, Jeon WK, Chae HJ, Chung HT. Free Radic Biol Med. 2006;41:106. doi: 10.1016/j.freeradbiomed.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 377.Hosoki R, Matsuki N, Kimura H. Biochem Biophys Res Commun. 1997;237:527. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- 378.Irvine JC, Ritchie RH, Favaloro JL, Andrews KL, Widdop RE, Kemp-Harper BK. Trends Pharmacol Sci. 2008;1:1. doi: 10.1016/j.tips.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 379.Paolocci N, Jackson MI, Lopez BE, Miranda K, Tocchetti CG, Wink DA, Hobbs AJ, Fukuto JM. Pharmacol Ther. 2007;113:442. doi: 10.1016/j.pharmthera.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Irvine JC, Favaloro JL, Kemp-Harper BK. Hypertension. 2003;41:1301. doi: 10.1161/01.HYP.0000072010.54901.DE. [DOI] [PubMed] [Google Scholar]
- 381.Paolocci N, Saavedra WF, Miranda KM, Martignani C, Isoda T, Hare JM, Espey MG, Fukuto JM, Feelisch M, Wink DA, Kass DA. Proc Natl Acad Sci U S A. 2001;98:10463. doi: 10.1073/pnas.181191198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Cheong E, Tumbev V, Abramson J, Salama G, Stoyanovsky DA. Cell Calcium. 2005;37:87. doi: 10.1016/j.ceca.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 383.Miranda KM, Nims RW, Thomas DD, Espey MG, Citrin D, Bartberger MD, Paolocci N, Fukuto JM, Feelisch M, Wink DA. J Inorg Biochem. 2003;93:52. doi: 10.1016/s0162-0134(02)00498-1. [DOI] [PubMed] [Google Scholar]
- 384.Donzelli S, Espey MG, Thomas DD, Mancardi D, Tocchetti CG, Ridnour LA, Paolocci N, King SB, Miranda KM, Lazzarino G, Fukuto JM, Wink DA. Free Radic Biol Med. 2006;40:1056. doi: 10.1016/j.freeradbiomed.2005.10.058. [DOI] [PubMed] [Google Scholar]
- 385.Miranda KM, Yamada K-i, Espey MG, Thomas DD, DeGraff W, Mitchell JB, Krishna MC, Colton CA, Wink DA. Arch Biochem Biophys. 2002;401:134. doi: 10.1016/S0003-9861(02)00031-0. [DOI] [PubMed] [Google Scholar]
- 386.Fukuto JM, Jackson MI, Kaludercic N, Paolocci N, Enrique C, Lester P. Methods Enzymol. Vol. 440. Academic Press; 2008. [DOI] [PubMed] [Google Scholar]
- 387.Norris AJ, Sartippour MR, Lu M, Park T, Rao JY, Jackson MI, Fukuto JM, Brooks MN. Int J Cancer. 2008;122:1905. doi: 10.1002/ijc.23305. [DOI] [PubMed] [Google Scholar]
- 388.Jain M, Townsend RR. Curr Hypertens Rep. 2007;9:320. doi: 10.1007/s11906-007-0058-7. [DOI] [PubMed] [Google Scholar]
- 389.Isenberg JS, Hyodo F, Ridnour LA, Shannon CS, Wink DA, Krishna MC, Roberts DD. Neoplasia. 2008;10:886. doi: 10.1593/neo.08264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Saharinen P, Petrova TV. Ann N Y Acad Sci. 2004;1014:76. doi: 10.1196/annals.1294.008. [DOI] [PubMed] [Google Scholar]
- 391.Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F, Devy L, Beck H, Scholz D, Acker T, DiPalma T, Dewerchin M, Noel A, Stalmans I, Barra A, Blacher S, Vandendriessche T, Ponten A, Eriksson U, Plate KH, Foidart JM, Schaper W, Charnock-Jones DS, Hicklin DJ, Herbert JM, Collen D, Persico MG. Nat Med. 2001;7:575. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- 392.Fiedler U, Augustin HG. Trends Immunol. 2006;27:552. doi: 10.1016/j.it.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 393.Caron KM, Smithies O. Proc Natl Acad Sci U S A. 2001;98:615. doi: 10.1073/pnas.021548898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Pietras K, Pahler J, Bergers G, Hanahan D. PLoS Med. 2008;5:e19. doi: 10.1371/journal.pmed.0050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Kanda S, Shono T, Tomasini-Johansson B, Klint P, Saito Y. Exp Cell Res. 1999;252:262. doi: 10.1006/excr.1999.4622. [DOI] [PubMed] [Google Scholar]
- 396.Simantov R, Febbraio M, Silverstein RL. Matrix Biol. 2005;24:27. doi: 10.1016/j.matbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 397.Bikfalvi A. Biochem Pharmacol. 2004;68:1017. doi: 10.1016/j.bcp.2004.05.030. [DOI] [PubMed] [Google Scholar]
- 398.Sidky YA, Borden EC. Cancer Res. 1987;47:5155. [PubMed] [Google Scholar]
- 399.McCarty MF, Bielenberg D, Donawho C, Bucana CD, Fidler IJ. Clin Exp Metastasis. 2002;19:609. doi: 10.1023/a:1020923326441. [DOI] [PubMed] [Google Scholar]
- 400.Nakamura T, Matsumoto K. Biochem Biophys Res Commun. 2005;333:289. doi: 10.1016/j.bbrc.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 401.Dawson DW, Volpert OV, Gillis P, Crawford SE, Xu H, Benedict W, Bouck NP. Science. 1999;285:245. doi: 10.1126/science.285.5425.245. [DOI] [PubMed] [Google Scholar]
- 402.Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, Wei BY, Stetler-Stevenson WG. Cell. 2003;114:171. doi: 10.1016/s0092-8674(03)00551-8. [DOI] [PubMed] [Google Scholar]
- 403.Moser TL, Kenan DJ, Ashley TA, Roy JA, Goodman MD, Misra UK, Cheek DJ, Pizzo SV. Proc Natl Acad Sci U S A. 2001;98:6656. doi: 10.1073/pnas.131067798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Shi H, Huang Y, Zhou H, Song X, Yuan S, Fu Y, Luo Y. Blood. 2007;110:2899. doi: 10.1182/blood-2007-01-064428. [DOI] [PubMed] [Google Scholar]
- 405.Clapp C, Aranda J, Gonzalez C, Jeziorski MC, Martinez de la Escalera G. Trends Endocrinol Metab. 2006;17:301. doi: 10.1016/j.tem.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 406.Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, Nakhasi H, Teruya-Feldstein J, Wirth P, Gupta G, Tosato G. J Exp Med. 1998;188:2349. doi: 10.1084/jem.188.12.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Merkulova-Rainon T, England P, Ding S, Demerens C, Tobelem G. J Biol Chem. 2003;278:37400. doi: 10.1074/jbc.M212768200. [DOI] [PubMed] [Google Scholar]
- 408.Ludwig RJ, Schon MP, Boehncke WH. Expert Opin Ther Targets. 2007;11:1103. doi: 10.1517/14728222.11.8.1103. [DOI] [PubMed] [Google Scholar]
- 409.Volpert OV, Lawler J, Bouck NP. Proc Natl Acad Sci U S A. 1998;95:6343. doi: 10.1073/pnas.95.11.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Isenberg JS, Ridnour LA, Thomas DD, Wink DA, Roberts DD, Espey MG. Free Radic Biol Med. 2006;40:1028. doi: 10.1016/j.freeradbiomed.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 411.Goldstein S, Czapski G. Free Radic Biol Med. 1995;19:505. doi: 10.1016/0891-5849(95)00034-u. [DOI] [PubMed] [Google Scholar]
- 412.Flint DH, Tuminello JF, Emptage MH. J Biol Chem. 1993;268:22369. [PubMed] [Google Scholar]
- 413.Fridovich I. Annu Rev Biochem. 1975;44:147. doi: 10.1146/annurev.bi.44.070175.001051. [DOI] [PubMed] [Google Scholar]
- 414.Chance B, Greenstein DS, Roughton FJ. Arch Biochem Biophys. 1952;37:301. doi: 10.1016/0003-9861(52)90194-x. [DOI] [PubMed] [Google Scholar]
- 415.Takebe G, Yarimizu J, Saito Y, Hayashi T, Nakamura H, Yodoi J, Nagasawa S, Takahashi K. J Biol Chem. 2002;277:41254. doi: 10.1074/jbc.M202773200. [DOI] [PubMed] [Google Scholar]
- 416.Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. J Biol Chem. 2007;282:11885. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 417.Goldman R, Stoyanovsky DA, Day BW, Kagan VE. Biochemistry. 1995;34:4765. doi: 10.1021/bi00014a034. [DOI] [PubMed] [Google Scholar]
- 418.Denu JM, Tanner KG. Biochemistry. 1998;37:5633. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 419.Winterbourn CC, Metodiewa D. Free Radic Biol Med. 1999;27:322. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]