

Abstract
Highly diverse antibody (Fab or scFv) libraries have become vital sources to select antibodies with high affinity and novel properties. Combinatorial strategies provide efficient ways of creating antibody libraries containing a large number of individual clones. These strategies include the reassembly of naturally occurring genes encoding the heavy and light chains from either immune or nonimmune B-cell sources, or introduction of synthetic diversity to either the framework regions (FRs) or the complementarity-determining regions (CDRs) of the variable domains of antibodies. In the late 1980s, the smallest known antigen-binding fragment was identified when a murine VH repertoire was screened for binding to lysozyme. This fragment (~15 kDa), called a “domain antibody”, or “dAb”, is approximately four times smaller than an Fab and half the size of an scFv. Here, we describe the construction of a phage-displayed VH library and an approach to introduce genetic diversity in this library, where both diverse human CDRs and synthetic CDRs are combined into a single domain (VH) framework.
Keywords: library construction, phage display, domain antibody, human, VH, CDRs, grafting, diversity
1. Introduction
Monoclonal antibodies are now well established therapeutics - more than twenty antibodies have been approved by the US Food and Drug Administration against various diseases and more antibodies have been in clinical trials in the past decade (1). Antibody fragments which are significantly smaller than full-size antibodies (~150 kDa) e.g. Fabs (~60 kDa) or scFvs (20~30 kDa) have been also widely used especially as components of antibody-based candidate therapeutics and imaging reagents. Recently, an even smaller antigen-binding fragment, dAb (~15 kDa), was isolated from murine VH repertoires (2,3). Because of their smaller size, dAbs are typically better suited for phage display, which has been proved to be one of the most successful technologies developed to construct antibody libraries (4). It has been shown that the use of monoclonal antibodies derived from nonhuman species such as mouse or rabbit results in immune responses to the foreign immunoglobulin epitopes in humans, severely limiting the long-term use of these reagents (5). It is, therefore, desirable to have human monoclonal dAbs.
In this article, we describe a dAb library design based on the introduction of naturally occurring CDR2s, CDR3s, and synthetic CDR1s (random mutation of 4 putative solvent-accessible residues to A/D/S/Y) into a human VH single domain framework. Donation of human blood and immune tissues for research, and commercial availability of the B cells and their RNA or cDNA provide straightforward sources to harvest natural CDR repertoires (6). PCR amplification using randomized primers covering CDRs can readily introduce synthetic diversity (7). It has been already demonstrated that using overlapping oligonucleotides FRs and CDRs can be precisely assembled into the entire VHs (8). The products can be subsequently cloned into phagemid vectors, which are also commercially available. Transformation of highly efficient competent bacteria cells with the phagemid constructs can result in a library containing billions of individual clones.
2. Materials
2.1. Master VH genes and phagemid vectors
Figure 1.
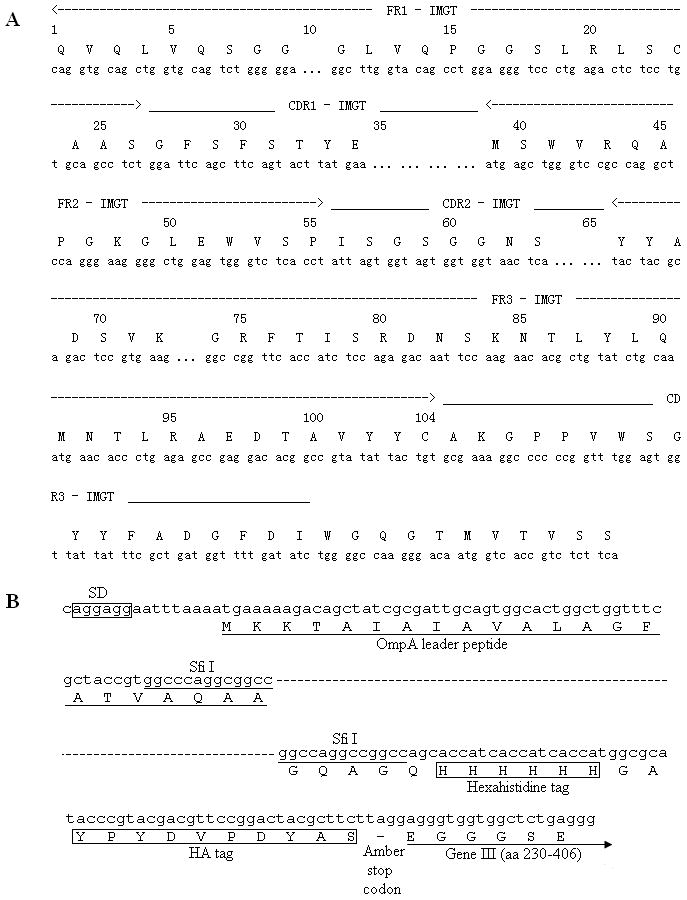
Schematic representations of the m0 master framework and the cloning region of pComb3X. (A) m0 master framework is analyzed using IMGT/V-QUEST tool provided by IMGT immunoglobulin database (http://imgt.cines.fr/IMGT_vquest/vquest?livret=0&Option=humanIg). The FRs and CDRs regions of the master gene are indicated according to the database. (B) Brief description of the cloning region of phagemid pComb3X. SfiI restriction sites are frequently used for cloning of genes. Hexahistidine tag and HA tag are included for purification and detection of protein products. This vector system has been widely used for display of a wide variety of proteins.
2.2. Lymphocyte isolation
Defibrinated or anticoagulant-treated human peripheral blood stored at 4 °C and used as soon as possible (see Note 1).
Ficoll-Paque Plus regents (Amersham Bioscience, Piscataway, NJ).
Solution A: 0.1% (w/v) anhydrous D-glucose, 0.05 mM CaCl2, 0.98 mM MgCl2, 5.4 mM KCl, and 145 mM Tris. Dissolve in approximately 950 ml double distilled water (ddH2O) and add 10 N HCl until pH is 7.6. Adjust the volume to 1 L with ddH2O.
Solution B: 140 mM NaCl in ddH2O.
Balanced salt solution (ready to use): Mix 1 volume Solution A with 9 volumes solution B (see Note 2).
Eppendorf centrifuge 5804R (Eppendorf, Westbury, NY), or similar refrigerated centrifuge producing up to at least 400 g and maintaining temperature of 18–20 °C.
BD Falcon™ Conical Tubes (BD Biosciences, San Jose, CA), or others with volume ~15 ml and internal diameter ~1.3 cm.
Pasteur pipettes, 3 ml.
Hemacytometer (Sigma, St. Louis, MO)
0.4% trypan blue stain (Sigma, St. Louis, MO)
2.3. Total RNA extraction and cDNA synthesis
RNeasy Mini Kit (Qiagen, Valencia, CA).
QIAshredder (Qiagen, Valencia, CA).
SuperScript. III First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA).
Corning® PCR tubes, free of RNase and DNase (Sigma, St. Louis, MO).
1.5 ml Eppendorf tubes, treated with distilled water containing 0.05% (v/v) DEPC at 37 °C overnight, dried in an oven, and then autoclaved.
Ultra pure water (Quality Biologicals, Gaithersburg, MD), free of RNase and DNase.
Eppendorf centrifuge 5417R (Eppendorf, Westbury, NY), or other refrigerated centrifuges with adapters for 1.5 ml Eppendorf centrifugal tubes.
Bio-Rad PTC-100 thermal cycler (Bio-Rad, Hercules, CA), or others with hot bonnet heated lid.
2.4. PCR amplification of CDRs and FRs, and assembly of entire VHs
High Fidelity PCR Master (Roche, Indianapolis, IN), or other high-fidelity PCR systems may be used.
-
Primers for PCR amplification of CDRs (see Note 3)
Primers for CDR1:
H1-F: 5′-GAG GAG GAG GAG GAG GAG GCG GGG CCC AGG CGG CCC AGG TGC AGC TGG TGC-3′
H1-R: 5′-GCG GAC CCA GCT CAT TTC ATA AKM AKM GAA AKM GAA AKM AGA GGC TGC ACA GGA GAG -3′
Primers for CDR2:
H2-F1: 5′-GAA ATG AGC TGG GTC CGC CAG GCT CCA GGA CAA SGS CTT GAG TGG-3′
H2-F2: 5′-GAA ATG AGC TGG GTC CGC CAG GCT CCA GGG AAG GCC CTG GAG TGG-3′
H2-F3: 5′-GAA ATG AGC TGG GTC CGC CAG GCT CCA GGG AAG GGN CTR GAG TGG-3′
H2-R1: 5′-ATT GTC TCT GGA GAT GGT GAC CCT KYC CTG RAA CTY-3′
H2-R2: 5′-ATT GTC TCT GGA GAT GGT GAA TCG GCC CTT CAC NGA -3′
H2-R3: 5′-ATT GTC TCT GGA GAT GGT GAC TMG ACT CTT GAG GGA-3′
H2-R4: 5′-ATT GTC TCT GGA GAT GGT GAC STG GCC TTG GAA GGA-3′
H2-R5: 5′-ATT GTC TCT GGA GAT GGT AAA CCG TCC TGT GAA GCC-3′
Primers for CDR3:
H3-F1: 5′-ACC CTG AGA GCC GAG GAC ACR GCY TTR TAT TAC TGT-3′
H3-F2: 5′-ACC CTG AGA GCC GAG GAC ACA GCC AYR TAT TAC TGT-3′
H3-F3: 5′-ACC CTG AGA GCC GAG GAC ACR GCY GTR TAT TAC TGT-3′
H3-R: 5′-GTG GCC GGC CTG GCC ACT TGA GGA GAC GGT GAC C-3′
-
Primers for PCR amplification of FR3 (see Note 4)
FR3-F: 5′-ACC ATC TCC AGA GAC AAT TCC-3′
FR3-R: 5′-GTC CTC GGC TCT CAG GGT G -3′
-
Primers for extension PCR (see Note 5)
HISR: 5′-GTC GCC GTG GTG GTG GTG GTG GTG GCC GGC CTG GCC ACT TG-3′
2.5. Digestion of VHs and ligation of VHs with phagemids
Restriction enzymes SfiI, 20000 units/ml (BioLabs, Ipswich, MA).
T4 DNA Ligase, 400000 units/ml (BioLabs, Ipswich, MA).
2.6. Concentration and desalting of ligations
Centrifugal filter: Amicon Ultra-4 with a cutoff of 3000 MW (Millipore, Billerica, MA).
2.7. Electroporations
TG1 electroporation-competent cells (Stratagene, La Jolla, CA).
Gene Pulser/MicroPulser Cuvettes (Bio-Rad, Hercules, CA).
Gene Pulser (Bio-Rad, Hercules, CA)
2.8. Preparation of library
2YT medium: 0.5% (w/v) NaCl, 1% (w/v) yeast extract, 1.6% (w/v) tryptone in distilled water. Autoclave and store at room temperature.
20% (w/v) glucose in distilled water. Sterilize using 0.22 μm pore size filter (Nalgene, Rochester, NY).
M13KO7 helper phage (BioLabs, Ipswich, MA).
Antibiotics: 100 mg/ml ampicillin and 100 mg/ml kanamycin.
3. Methods
To construct a high-quality (high diversity, low mutation rate, and very few of reading frame shifts) antibody library, it is important to optimize each step before next step can be performed.
3.1. Lymphocyte isolation by Ficoll-Paque Plus regents (see Note 6)
To a 15 ml BD Falcon tube, add 2 ml of defibrinated or anticoagulant-treated blood and 2 ml of balanced salt solution (see Note 7). Mix by drawing the blood and buffer in and out of a Pasteur pipette.
Invert the Ficoll-Paque Plus bottle several times to ensure thorough mixing. Pipette 3 ml of the reagents into a new 15 ml BD Falcon tube. Carefully layer the diluted blood sample (4 ml) onto the Ficoll-Paque Plus. When layering the sample do not mix the regents and the diluted blood sample.
Centrifuge at 400 g for 30–40 minutes at 18–20 °C (see Note 8). After centrifugation, generally four layers can be clearly observed including plasma, lymphocyte, Ficoll-Paque Plus, and granulocyte/erythrocyte layer from top to bottom, respectively. Draw off the upper layer of plasma using a clean Pasteur pipette, leaving the lymphocyte layer undisturbed at the interface.
Transfer the lymphocyte layer to a clean 15 ml BD Falcon tube using a clean Pasteur pipette. It is critical to remove the entire interface but with a minimum amount of Ficoll-Paque Plus and plasma. Removing excess plasma causes contamination by platelets and plasma proteins. Removing excess Ficoll-Paque Plus results in unnecessary granulocyte contamination.
Add at least 3 volumes of balanced salt solution to the lymphocytes. Suspend the cells by gently drawing them in and out of a Pasteur pipette.
Centrifuge at 400 g for 10 minutes at 18–20 °C. Remove the supernatant and resuspend the lymphocytes in 6–8 ml balanced salt solution by pipetting them gently in and out.
Determine the number of living cells by using hemacytometer: Mix 50 μl cell suspension with 50 μl trypan blue stain, load 20 μl of the mixture to the hemacytometer, count the total number of living cells (i.e., the unstained cells, since only the live cells have intact membrane that is not permeable for the dye), and calculate the total cell quantity according to the hematocytometer instructions.
Centrifuge the cell suspension at 400 g for 10 minutes at 18–20 °C. Remove the supernatant. The lymphocyte pellet can be used immediately for RNA extraction or stored at −80 °C for later use.
3.2. Extraction of total RNA from lymphocytes
To extract the total RNA we used RNeasy Mini Kit from Qiagen (see Note 9) following the basic protocol provided by the manufacturer. Below we introduce and describe few modifications in this protocol that improved the yield and quality of the extracted RNA.
Thaw the lymphocyte pellet at room temperature if it is stored at −80 °C (see Note 10). Gently tap the bottom of the tube containing the lymphocyte pellet on the bench to loosen the cells.
Disrupt cells (up to 5 × 106) by addition of 350 μl of Buffer RLT from RNeasy Mini Kit (see Note 11). Vortex or pipet to mix.
Pipet the lysate directly onto a QIAshredder spin column placed in a 2 ml collection tube, and centrifuge for 2 min at 12000 × rpm.
Add 1 volume of 70% ethanol to the homogenized lysate and mix well by pipetting. Do not centrifuge.
Apply all sample, including any precipitate that may have formed, to an RNeasy mini column placed in a 2 ml collection tube. Centrifuge for 15 s at 10000 × rpm. Discard the flow-through.
Add 700 μl Buffer RW1 to the RNeasy column. Centrifuge for 15 s at 10000 × rpm. Discard the flow-through and collection tube.
Transfer the RNeasy column into a new 2 ml collection tube. Pipet 500 μl Buffer RPE onto the RNeasy column. Centrifuge for 15 s at 10000 × rpm. Discard the flow-through.
Add another 500 μl Buffer RPE to the RNeasy column. Centrifuge for 2 min at 10000 × rpm to dry the membrane in the column.
Transfer the RNeasy column to a new 1.5 ml collection tube. Pipet 30–50 μl RNase-free water onto the membrane in the column. Centrifuge for 1 min at 10000 × rpm to elute. Store the RNA product at −80 °C and use it as soon as possible.
3.3. Retro-transcription of RNAs to cDNAs
These instructions assume the use of SuperScript™ III First-Strand Synthesis System from Invitrogen. The following procedure is designed to convert total RNA (5 pg to 25 μg) or mRNA (5 pg to 2.5 μg) into first-strand cDNA.
Mix and briefly centrifuge each component in the kit before use.
-
Prepare two RNA/primer mixtures, one with oligo (dT)20 and the other with random hexamers, in 0.2 ml Corning® PCR tubes by combining the following:
Total RNA x μl (up to 25 μg) 50 μM oligo (dT)20 or 50 ng/μl random hexamers 5 μl 10 mM dNTP mix 5 μl DEPC-treated water 40-x μl Total 50 μl Incubate at 65 °C for 5 min, then place on ice for at least 1 min.
- During the incubation, set up two tubes containing the same cDNA synthesis mixtures by adding each component in the indicated order:
10 × RT buffer 10 μl 2.5 mM MgCl2 20 μl 0.1 M DTT 10 μl RNase OUT™ (40 u/μl) 5 μl Superscript ™ III RT (200 u/μl) 5 μl Total 50 μl -
Add 50 μl cDNA synthesis mixtures to the 50 μl RNA/primer mixtures, mix gently, and collect by brief centrifugation. Incubate as follows:
For oligo (dT)20 primer: 50 min at 50 °C.
For random hexamer primer: 10 min at 25 °C, followed by 50 min at 50 °C.
Terminate the reactions at 85 °C for 5 min. Chill on ice.
Collect the reactions by brief centrifugation add 5 μl of RNase H to each reaction, and incubate for 20 min at 37 °C
Run a 0.8% (w/v) agarose gel using 2 μl of the reaction to simply check the amount and length distribution of cDNA. An example of the results produced is shown in Fig. 2. The cDNA reactions are combined and stored at −20 °C for further use.
Figure 2.
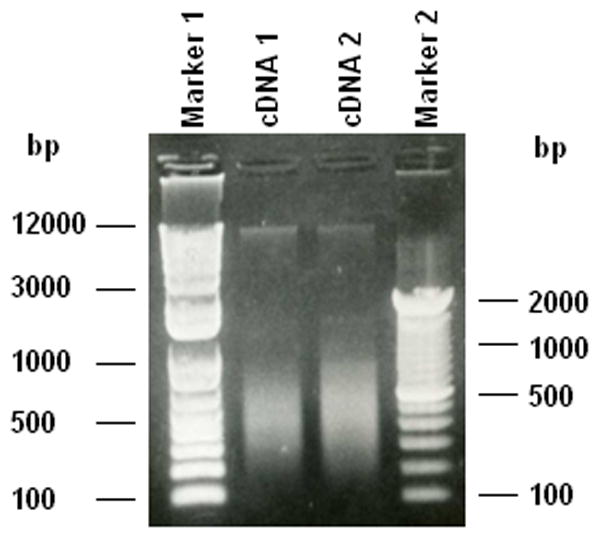
Preparation of cDNA by reverse transcription of total RNA from human peripheral blood mononuclear cells. Total RNA was extracted from human mononuclear cells with RNeasy Mini Kit (Qiagen, Cat. #74104) as described in Methods 3.2. Using a SuperScript. III First-Strand Synthesis System (Invitrogen, Cat. #18080-051) containing oligo (dT)20 primers and random hexamers, total RNA was then reverse transcribed into cDNA (Methods 3.3). The cDNA products were separated on a 0.8% (w/v) agarose gel. The cDNA 1 and cDNA 2 are from reactions using oligo (dT)20 primers and random hexamers, respectively. Two molecular weight DNA mass markers, Marker 1 (Invitrogen Cat. #15628-019) and Marker 2 (Invitrogen Cat. #10787-018) were included.
3.4. PCR amplification of CDRs and FRs
-
First round of PCR to get CDR2s
To amplify the CDR2s from cDNA samples, perform eight amplifications. Set up one PCR tube for each primer combination as the following:ddH2O 23-x μl 2 × High Fidelity PCR Master 25 μl Forward primer (25 μM) 1 μl Reverse primer (25 μM) 1 μl cDNA x μl (~1 μg) Total 50 μl Primer recombinations:
H2-F1/H2-R1; H2-F1/H2-R2; H2-F1/H2-R5; H2-F2/H2-R3; H2-F3/H2-R1; H2-F3/H2-R2; H2-F3/H2-R3; H2-F3/H2-R4
Perform the PCR under the following conditions:
Step 1: 4 min at 94 °C for initial denaturation
Step 2: 45 sec at 94 °C; 45 sec at 55 °C; 1 min at 72 °C (30 cycles)
Step 3: 5 min at 72 °C
Run the products separately on a 2% agarose gel to check the specific amplification of CDR2s. An example of the results produced is shown in Fig. 3A. Cut out the correct-sized bands on the gel and purify the DNA using, for example, QIAquick Gel Extraction Kit (Qiagen, Cat. #28706) (see Note 12). Pool the purified DNA and quantify it by reading the optical density (O.D.) at 260 nm (1 O.D. unit = 50 μg/ml). Store the sample at −20 °C for later use.
-
First round of PCR to get CDR3s
Three amplifications are performed to obtain CDR3. Set up the reaction for each primer combination and perform the PCR as it is described above for CDR2 amplification except for the use of different primers.
Primer combinations:
H3-F1/H3R; H3-F2/H3R; H3-F3/H3R
Run the products separately on a 2% agarose gel to check the specific amplification of CDR3s. An example of the results produced is shown in Fig. 3B. Purify the DNA (see Note 13), pool and quantify it by reading the optical density (O.D.) at 260 nm (1 O.D. unit = 50 μg/ml). Store the sample at −20 °C for later use.
-
First round of PCR to get CDR1s
Only one reaction is needed for CDR1 amplification as the following.ddH2O 46-x μl 2 × High Fidelity PCR Master 50 μl H1-F (25 μM) 2 μl H1-R (25 μM) 2 μl m0 x μl (~0.1 μg) Total 100 μl Perform the PCR under the same conditions as for the CDR2 amplification. At least 1 μg of purified DNA is required in order to proceed further.
-
First round of PCR to get FR3
Only one reaction is needed for FR3 amplification as the followingddH2O 46-x μl 2 × High Fidelity PCR Master 50 μl FR3-F (25 μM) 2 μl FR3-R (25 μM) 2 μl m0 x μl (~0.1 μg) Total 100 μl Perform the PCR under the same conditions as for the CDR2 amplification. At least 1 μg of purified DNA is required in order to proceed further.
Figure 3.
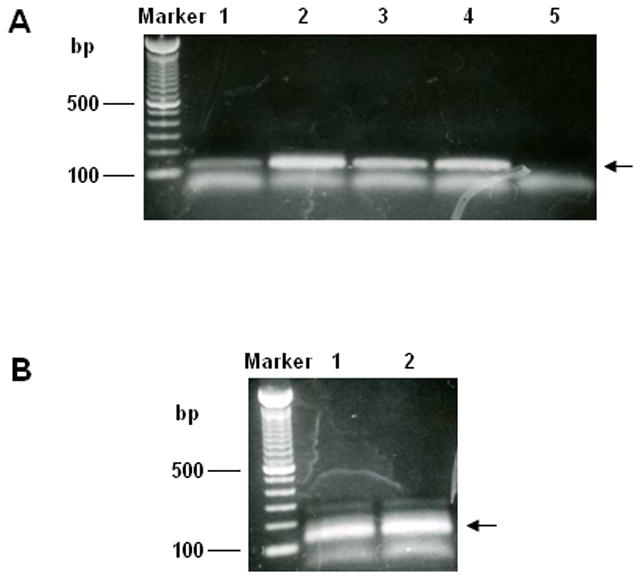
PCR amplification of CDR2 and CDR3 repertoires from cDNA. (A) Eight recombinations of primers were used for CDR2s amplification as described in Methods 3.4. The products of the first five recombinations (H2-F1/H2-R1, H2-F1/H2-R2, H2-F1/H2-R5, H2-F2/H2-R3, H2-F3/H2-R1, and H2-F3/H2-R2) were shown on lane 1 to lane 5, respectively. (B) Three recombinations of primers were used for CDR3 amplification. The products of the first two recombinations (H3-F1/H3R and H3-F2/H3R) were shown on lane 1 and 2, respectively. The correct-sized bands were indicated by arrows.
3.5. Assembly of entire VHs
-
Second round of PCR (overlap extension) to get CDR1s and CDR2s together
The primers in the first round of PCR create identical sequences in the downstream regions of the CDR1s and the upstream regions of CDR2s. These identical sequences serve as the overlap for the second-round extension.
Set up a reaction without primers as the following. Pay attention that CDR1s and CDR2s should be added in the same molarities.ddH2O 46-x-y μl 2 × High Fidelity PCR Master 50 μl CDR1s x μl (~100 ng) CDR2s y μl (~120 ng) Total 100 μl Perform the PCR under the following conditions:
Step 1: 4 min at 94 °C for initial denaturation
Step 2: 45 sec at 94 °C; 45 sec at 55 °C; 1 min at 72 °C (7 cycles)
Step 3: 5 min at 72 °C
After the cycling, add primers to the reaction: 2 μl H1-F (25 μM) and 2 μl H2-R1-5 mixture (25 μM). Then perform another 15 cycles of PCR under the same condition (see Note 14).
-
Second round of PCR (overlap extension) to get FR3 and CDR3s together
The procedure is almost the same as above except the use of different gene fragments and primers for overlap. The reaction after addition of primers contains the following:ddH2O 46-x-y μl 2 × High Fidelity PCR Master 50 μl FR3 x μl (~100 ng) CDR3s y μl (~120 ng) FR3-F (25 μM) 2 μl H3-R (25 μM) 2 μl Total 100 μl Run the products on a 2% agarose gel, purify the DNA, and quantify it. At least 2 μg each of the purified CDR1-CDR2 and FR3-CDR3 DNA is required to proceed.
-
Third round of PCR (final overlap extension) to get whole-length VHs.
Also, the procedure is almost the same as above except the use of resultant gene fragments from Step 1 (CDR1-CDR2) and 2 (FR3-CDR3) above, and extension primers for overlap. The reaction after addition of primers contains the following:ddH2O 46-x-y μl 2 × High Fidelity PCR Master 50 μl CDR1-CDR2 x μl (~100 ng) FR3-CDR3 y μl (~100 ng) H1-F (25 μM) 2 μl HISR 2 μl Total 100 μl Run the products on a 2% agarose gel, purify the DNA with gel extraction kit (elute the DNA with ultra pure water in this step instead of elution buffer provided with the kit), and quantify it. At least 50 μg of purified VHs is needed to make a library with a size of up to 1010. If the yields are too low, repeat the final overlap PCR and pool the end products.
3.6. Digestion of VHs and phagemid vector, and ligation between them
-
Digestion of VHs and phagemid vector
The reaction for VH digestion should contain:ddH2O 870-x μl 10 × Buffer 2 100 μl VHs x μl (up to 50 μg) SfiF (20 units/μl) 20 μl BSA 10 μl Total 1000 μl Set up two reactions for phagemid digestion. Each should contain:ddH2O 870-x μl 10 × Buffer 2 100 μl pComb3X x μl (up to 100 μg) SfiF (20 units/μl) 20 μl BSA (100×) 10 μl Total 1000 μl Incubate both digests at 50 °C for 3 hours. Run the digested products on agarose gels (2% for VHs and 1% for phagemids), purify the DNA with gel extraction kit (elute the DNA with ultra pure water), and quantify it (see Note 15).
-
Ligation of VHs with phagemid vector (see Note 16)
Assemble the reaction as the following:ddH2O 1750-x-y μl 10 × T4 ligase buffer 200 μl SfiI-digested VHs x μl (~30 μg) SfiI-digested pCom3bX y μl (~90 μg) T4 ligase (200 units/μl) 50 μl Total 2000 μl Incubate at 16 °C for 72 hours.
3.7. Concentration and desalting of ligated products
Concentrate and desalt the reactions by passing through a 4 ml Amicon Ultra-4 centrifugal filter with a cutoff 3000 MW:
Add all 2000 μl reactions into the filter; centrifuge at 4000 × g for 20 minutes at room temperature, Remove the flow-through to a 15 ml Falcon tube (do not discard the flow-through at this moment just in case most of DNA is lost due to the broken membrane of the filter).
Add 3.5 ml ultra pure water into the filter and centrifuge under the same condition for 30 min, remove the flow-through to a 15 ml Falcon tube.
Repeat Step 2 at least twice (see Note 17). In the last repeat, centrifuge for longer time, make sure that about 50 μl reactions remain in the filter.
Gently pipette the reactions and remove them to a 1.5 ml Eppendorf tube, store at −20 °C for later use.
3.8. Electroporations and preparation of library
Pre-warm 1 L 2YT medium containing 1% glucose (w/v) at 37 °C. Chill 50 gene pulser cuvettes on ice. At the same time thaw, on ice, the desalted ligations and 2 ml of TG1 electroporation-competent cells.
Divide 2 ml of TG1 competent cells into 5 pre-chilled 1.5 ml Eppendorf tubes with 400 μl each. Add 10 μl ligations to each tube and pipet gently to mix. Transfer 41 μl mixtures to each cuvette. Gently tap the cuvette on the bench to make the mixture fill out the bottom of the cuvette.
Electroporate at 1.8 kV, 25 μF, and 200 Ω. Flush the cuvette immediately with 1 ml and then twice with 2 ml of pre-warmed 2YT medium and combine the 3 ml in a 2 L flask. After all electroporations are completed, add 850 ml pre-warmed 2YT medium left to the flask.
Shake at 250 rpm for 30 min at 37 °C. Serially dilute 10 μl of the culture in 100 μl of 2YT medium, and spread on LB agar plates containing 2% glucose (w/v) and 100 μg/ml of ampicillin. Incubate the plates overnight at 37 °C. Calculate the total number of transformants by counting the number of colonies, multiplying by the culture volume, and dividing by the plating volume.
Add 1 ml of 100 mg/ml ampicillin to the 1-L culture and shake for additional 2 hours at 37 °C.
Take 1 ml of the culture and measure the cell density by reading O.D.600. Calculate the total number of cells by multiplying the O.D.600 value by 5 × 108 (estimated number of cells in 1 ml culture when O.D.600 reaches 1) and the culture volume (1000 in this case). Add 10 M.O.I. of M13KO7 helper phage to the culture. Incubate at 37 °C for 30 min, shaking for homogenization every 10 min.
Spin down the cells at 5000 rpm for 10 min. Resuspend in 2 L 2YT medium containing 100 μg/ml of ampicillin and 50 μg/ml of kanamycin. Incubate at 250 rpm overnight at 30 °C.
Spin at 5000 rpm for 15 min at 4 °C. Save the bacterial pellet for phagemid preparation using, for example, the Qiagen HiSpeed Plasmid Maxi Kit. For phage precipitation, transfer the supernatant to a clean 2 L flask and add ¼ volume of 20% (w/v) PEG8000 and 2.5 M NaCl solution. Mix well and incubate on ice for at least 1 h.
Spin at 14000 g for 20 min at 4 °C. Discard the supernatant. Resuspend the phage pellet in 50 ml PBS, pH7.4 by pipetting up and down along the side of the centrifuge bottle by using a 10-ml pipette.
Spin at 5000 rpm for 10 min at 4 °C. Transfer the supernatant to a clean 200 ml flask and add volume of 20% (w/v) PEG8000 and 2.5 M NaCl solution. Mix well and incubate on ice for 1 h.
Spin at 14000 g for 20 min. Discard the supernatant. Resuspend the phage pellet in 50 ml PBS, pH7.4.
Spin at 5000 rpm for 10 min at 4 °C. Transfer the supernatant to a clean 200 ml flask.
Add the same volume of autoclaved glycerol and mix well.
Measure the concentration of phage by reading O.D.280 (1 O.D.280 = 2.33 × 1012/ml). Aliquot the phage to make sure that each contains phage particles at least 100 times of the total number of transformants (calculated in step 4). Store the phage at −80 °C. The phage library is now ready for panning.
Acknowledgments
This project was supported by the NIH Intramural AIDS Targeted Antiviral Program (IATAP) and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Total RNA, PolyA+ RNA, and cDNA of human blood and other immune tissues such as bone marrow, spleen, and lymph node are commercially available, e.g. from Clontech.
Prepare the solution fresh each week. Other standard salt solutions like PBS, pH7.4 may be used.
To construct a highly diverse antibody library, it is essential for the primers to be able to cover as many human antibody genes as possible. To design those primers, it is necessary to possess some sequence information and try to understand how human antibody genes are organized. We have designed the primers listed (sequences underlined are homologous to CDRs), which are currently being used in our laboratory and proved to be capable of creating high-quality antibody libraries.
The products of CDR1 primers and CDR2 primers will cover FR1 and FR2, respectively, so that there are only four fragments instead of six (FR1-3 and CDR1-3) for the entire VH assembly. It is highly recommended to reduce the number of fragments which will decrease reading frame shifts.
We found that highly efficient digestion of VH products with restriction enzymes is critical for the construction of a large library. PCR amplification using the extension primers will result in long overhangs at both 5 and 3 ends of VH products so that there is an obvious difference in length change after digestion with restriction enzyme SfiI, which can be observed clearly on agarose gel.
At the beginning, it is important to determine whether the blood sample is collected and stored appropriately.
Tissue culture plasticware or pretreated glassware is required. All glassware which comes in contact with the samples should be siliconized before use. The glassware should be immersed in a 1% silicone solution for 10 seconds, washed with distilled water (6 times) and then dried in an oven.
We have found that it is very important to maintain exactly 18–20 °C temperature in the centrifuge. Lower temperature will result in precipitates in the layer of plasma making the lymphocyte layer unclear. Optimization of the duration of centrifuging is also recommended to yield a clear lymphocyte layer. This can be accomplished through a practice using irrelevant blood samples.
We recommend this kit because it provides enrichment for mRNA by eliminating most RNAs shorter than 200 nucleotides such as 5.8S rRNA, 5S rRNA, and tRNAs. It, therefore, may be more efficient for the products to be retro-transcribed to cDNA.
It is suggested that people don’t freeze the lymphocytes but go to RNA extraction directly. Also it will be better for RNA products to be retro-transcribed to cDNA without freeze-thaw cycle. Just don’t hold RNA for long time. Finish these steps within one day.
Since there are limitations with the capacity of the buffer to lyse the cells and the binding capacity of the column, it is essential to use appropriate number of cells in order to obtain optimal RNA yield and purity.
Running the eight products separately on the gel is strongly recommended to make observation. We have found that, when different templates are used, products may not be observed on the gel for one or two primer combinations. This should be due to very limited number of templates for the specific primer combinations so it is not necessary to repeat the reaction. However, it is recommended to cut out the correct-sized gel and purify the limited number of DNA for these primer combinations as is done to those with clear bands. This essentially helps maintain the diversity of the repertoires.
When running the CDR3 products on 2% agarose gel, the bands will not be so sharp due to their highly diverse lengths. Thus, cut as wide a gel as you can to make sure it covers those CDR3s with long or short lengths.
Other than the major correct-sized fragments, this procedure could generate minor nonspecific products for some reasons. Optimization is needed to minimize nonspecific amplification. We found that increasing the annealing temperature from 55 °C to 60 °C and reducing the number of cycles from 7 to 5, 15 to 12 before and after addition of primers, respectively, helped.
In some cases the digestion of phagemid vectors may not be complete due to bad quality of DNA. To address this problem, additional treatment is needed to further purify the phagemids before digestion, or use more SfiI to digest for longer time, for example, overnight.
Before large-scale ligation can be performed, it is highly recommended to take two ligation tests. One is to assess the suitability of the vector and inserts for high-efficiency ligation and transformation. This can be accomplished through assembling small reactions either with vector only (test for vector self-ligation) or with both vector and insert, and transforming chemical competent cells like DH5α. The other is to determine the optimal ratio between insert and vector for the highest efficiency of ligation. This can be accomplished through assembling small reactions with insert and vector in different morality ratios such as 3:1, 2:1, and 1:1, and transforming chemical competent cells. We found that the highest ligation efficiency may be obtained by using insert in two-fold molar excess.
The desalting of DNA samples is a key step to the success of electroporations. High concentration of ions in the DNA solution will result in a long and intense pulse in electroporations, which causes cell damage or rupture. We found that at least 1000-time dilution of DNA solution was needed to generate time constants of 4.6–5.0 s in electroporations that generally gave the highest efficiency.
References
- 1.Zhang Q, Chen G, Liu X, Qian Q. Monoclonal antibodies as therapeutic agents in oncology and antibody gene therapy. Cell Res. 2007;17:89–99. doi: 10.1038/sj.cr.7310143. [DOI] [PubMed] [Google Scholar]
- 2.Ward ES, Güssow D, Griffiths AD, Jones PT, Winter G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature. 1989;341:544–546. doi: 10.1038/341544a0. [DOI] [PubMed] [Google Scholar]
- 3.Muyldermans S, Cambillau C, Wyns D. Recognition of antigens by single domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem Sci. 2001;26:230–235. doi: 10.1016/s0968-0004(01)01790-x. [DOI] [PubMed] [Google Scholar]
- 4.Holt LJ, Herring C, Jespers LS, Woolven BP, Tomlinson IM. Domain antibodies: proteins for therapy. Trends Biotechnol. 2003;21:484–490. doi: 10.1016/j.tibtech.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Schellekens H. Immunogenicity of therapeutic proteins: clinical implications and future prospects. Clin Ther. 2002;24:1720–1740. doi: 10.1016/s0149-2918(02)80075-3. [DOI] [PubMed] [Google Scholar]
- 6.Söderlind E, Strandberg L, Jirholt P, Kobayashi N, Alexeiva1 V, Åberg AM, et al. Recombining germline-derived CDR sequences for creating diverse singleframework antibody libraries. Nature Biotechnol. 2000;18:852–856. doi: 10.1038/78458. [DOI] [PubMed] [Google Scholar]
- 7.Reiter Y, Schuck P, Boyd LF, Plaksin D. An antibody single-domain phage display library of a native heavy chain variable region: isolation of functional single-domain VH molecules with a unique interface. J Mol Biol. 1999;290:685–698. doi: 10.1006/jmbi.1999.2923. [DOI] [PubMed] [Google Scholar]
- 8.Jirholt P, Ohlin M, Borrebaeck CAK, Söderlind E. Exploiting sequence space: shuffling in vivo formed complementarity determining regions into a master framework. Gene. 1998;215:471–476. doi: 10.1016/s0378-1119(98)00317-5. [DOI] [PubMed] [Google Scholar]