

Abstract
Doxorubicin (DOX) is a potent anti-tumor agent. DOX can also induce cardiotoxicity, and high cumulative doses are associated with recalcitrant heart failure. Children are particularly sensitive to DOX-induced heart failure. The ability to genetically modify mice makes them an ideal experimental system to study the molecular basis of DOX-induced cardiotoxicity. However, most mouse DOX studies rely on acute drug administration in adult animals, which typically are analyzed within one week. Here we describe a juvenile mouse model of chronic DOX-induced cardiac dysfunction. DOX treatment was initiated at 2 weeks of age and continued for a period of 5 weeks (25 mg/kg cumulative dose). This resulted in a decline in cardiac systolic function, which was accompanied by marked atrophy of the heart, low levels of cardiomyocyte apoptosis, and decreased growth velocity. Other animals were allowed to recover for 13 weeks following the final DOX injection. Cardiac systolic function improved during this recovery period but remained depressed as compared to the saline injected controls, despite the reversal of cardiac atrophy. Interestingly, increased levels of cardiomyocyte apoptosis and concomitant myocardial fibrosis were observed following DOX withdrawal. These data suggest that different mechanisms contribute to cardiac dysfunction during the treatment and recovery phases.
Keywords: heart failure, chemotherapy, genetically modified mice
Etiologies of childhood cardiomyopathy are diverse, however, one of the most common is chemotherapy, such as anthracycline-induced cardiotoxicity (1). The anthracyclines, primarily doxorubicin (DOX) but also daunomycin, epirubicin and idarubicin, are among the most widely used and successful chemotherapeutics for childhood cancers. About half of the young adult survivors of childhood cancer have received anthracyclines at some point in their treatment. Unfortunately, these drugs are also cardiotoxic. The acute responses to anthracyclines include hypotension, tachycardia, arrhythmia and transient depression of left ventricular function (2–4). Higher cumulative doses are associated with late-onset cardiomyopathy that is refractory to standard treatment and thus limits the total amount of DOX, which may be administered (5, 6). Children and adolescents are particularly susceptible to the cardiotoxic effects of anthracycline chemotherapy, and a significant portion of children treated with DOX develop cardiomyopathy a year or more after cessation of chemotherapy (5, 7–9).
Although the precise mechanism underlying DOX-induced cardiotoxicity is uncertain (10, 11), most studies indicate that free radical-induced oxidative stress plays a central role. Anthracyclines undergo one electron reduction by complex I in mitochondria (12) and in the process generate superoxide and hydroxyl radical (13). Reactive oxygen species-induced mitochondrial damage is thought to contribute to DOX-induced cardiotoxicity (14). DOX is also known to promote DNA damage and inhibit DNA and protein synthesis (15–17), promote myofiber degeneration (18), inhibit expression of cardiomyocyte specific genes (19), and induce cardiomyocyte apoptosis via a caspase 3-dependent mechanism (20–23). Thus, DOX-induced cardiotoxicity is complex and multifaceted.
Animal models have been widely utilized to study the molecular basis underlying DOX cardiotoxicity, and to develop strategies aimed at evoking therapeutic cardioprotection (24). Given the ease of generating specific gain- and loss-of-function genetic models, mice are particularly useful for DOX cardiotoxicity studies. Since children can be expected to survive for decades after being cured of their malignancy, albeit with a propensity for subsequently developing heart failure, an important component of any animal model should be persistent cardiac dysfunction after anthracycline withdrawal. Here we describe a DOX-induced cardiotoxicity model using juvenile mice. DOX administration for 5 weeks resulted in a decline in cardiac systolic function, which was accompanied by a marked atrophy of the cardiac muscle, low levels of cardiomyocyte apoptosis, and myofiber disarray. Reversal of cardiac atrophy and normalization of myofiber organization were observed after a 13-week recovery period. Despite this, cardiac systolic function remained suppressed and was accompanied by an increased level of cardiomyocyte apoptosis and concomitant myocardial fibrosis. The implications and potential utility of this model are discussed.
Methods
DOX treatment
Juvenile DBA/2J mice received a total of 25 mg/kg of DOX (5 intra-peritoneal injections of 5 mg/kg in saline, given at 7-day intervals beginning at 14 days of age, Sigma, St. Louis, MO; control mice received saline injections). To avoid local tissue damage, a different region of the peritoneal cavity was injected at each time point, and no overt inflammation/tissue damage was apparent. One cohort of mice was sacrificed 1 week after the final DOX injection to study acute cardiotoxicity (treatment phase). A second cohort was sacrificed 13 weeks after the final DOX injection to study the chronic effects of the drug (recovery phase). Mortality with this protocol was 10%. All animal protocols were approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee.
Echocardiography
Transthoracic echocardiography was performed immediately before each DOX injection during the treatment phase, and then monthly during the recovery phase, as previously described (25). Mice were lightly anesthetized with 1.5% isofluorane until the heart rate stabilized at 400 to 500 beats per minute. Two-dimensional short-axis images were obtained using a high resolution Micro-Ultrasound system (Vevo 770, VisualSonics Inc., Toronto, Canada, 40-MHz probe). Left ventricular internal diameter during diastole (LVIDD), left ventricular internal diameter during systole (LVIDS), and fractional shortening (FS) were calculated using the Vevo Analysis software (version 2.2.3) as described (25).
Tissue and Serum Analyses
Tissues were harvested and processed for cryo or paraffin sectioning using standard techniques (26). Hematoxylin and Eosin staining was performed according to the manufacturer's protocols (Sigma Diagnostics, St. Louis, MO). Minimal cardiomyocyte fiber diameter was calculated as described (27). At least 400 randomly selected cardiomyocytes from each animal were analyzed. Cardiac alpha-actinin (antibody EA-53, Sigma Diagnostics, St. Louis, MO) immune reactivity was performed on permeiabilized, post-fixed sections (rhodamine conjugated secondary antibody). Activated caspase-3 immune reactivity (antibody #G7481, Promega, Madison, WI) was performed on post-fixed sections (horseradish peroxidase-conjugated secondary antibody, diaminobenzidine reaction) as described previously (28). Four transverse sections from each heart, sampled from the midpoint between the apex and base, were analyzed. TUNEL analyses were performed on adjacent sections using the ApopTag Apoptosis Detection kit according to the manufacturer's procedures (Chemicon International, Billerica, MA). Sirius red-Fast green staining was performed and quantitated on sections post-fixed in Bouin's solution as described previously (29, 30). Hematocrit and hematologic analyses were performed using standard analyses. Serum Troponin I levels were determined using a commercial ELISA kit (Life Diagnostics, Inc., West Chester, PA).
Western blot analyses
Protein was isolated in NP40 buffer and subjected to Western analysis as previously described (31, 32). Antibodies used recognized myosin heavy chain (H-300, #SC-20641, Santa Cruz Biotechnologies, Santa Cruz, CA), phosphorylated GSK3β (#9331, Cell Signaling Technology, Danvers, MA), total GSK3b, (#9315,Cell Signaling Technology), phosphorylated Akt (#9271, Cell Signaling Technology) and total Akt (#9272, Cell Signaling Technology). Signal was visualized by the ECL method according to the manufacturer's protocol (Amersham).
Statistic analysis
All values are presented as mean ± SEM. Statistical significance (p<0.01) was determined by Student's t-test (for groups of two), or by one-way ANOVA with Bonferroni adjustment (for groups of three or more).
Results
Juvenile DBA/2J mice received weekly injections of saline or DOX, beginning at 2 weeks of age and continuing for a period of 5 weeks. Echocardiography was used to measure fractional shortening (FS) of the short axis of the left ventricle. Base line FS was similar before the initiation of saline or DOX treatment (Figure 1A, Table 1). Cardiac function in the saline-injected mice remained constant over the 5-week treatment phase with an FS of approximately 60%. In contrast, FS was suppressed 1 week following the first DOX-injection, and remained suppressed with a value of approximately 45% at the end of the treatment phase (Figure 1A, Table 1). These animals were sacrificed for cell and molecular analyses (see below). Importantly, DOX-treatment had the anticipated impact on hematologic parameters, and serum cardiac Troponin I ELISA analyses indicated the presence of myocardial damage (Table 2).
Figure 1.

Cardiac function in saline- and DOX-injected juvenile mice. (A) Fractional Shortening (FS [%]) in saline-injected (diamonds) and DOX-injected (squares) mice during the treatment and recovery phases. X-axis indicates the age of the mice at analysis; vertical arrows indicate the age of injection. '*' indicates p<0.01 for DOX-injected vs. saline-injected mice. (B) Representative short axis echocardiograms from saline- and DOX injected mice at the end of the treatment and recovery phases.
Table 1.
Cardiac function in saline- and DOX-injected juvenile mice (Mean ± SEM).
| Age (weeks) | 2 | 3 | 4 | 5 | 6 | 7 | 11 | 15 | 19 |
|---|---|---|---|---|---|---|---|---|---|
| Saline | n=10 |
n=11 |
|||||||
| HR (bpm) | 451 ±9 | 452 ±12 | 458±10 | 494±14 | 479±12 | 473±11 | 480±13 | 470±8 | 468±13 |
| LVIDD (mm) | 2.2±0.1 | 2.4±0.1 | 2.6±0.1 | 2.6±0.1 | 2.6±0.1 | 2.7±0.10 | 2.7±0.1 | 2.8±0.2 | 2.9±0.3 |
| LVIDS (mm) | 0.9±0.0 | 1.0±0.1 | 1.0±0.0 | 1.0±0.0 | 1.1 ±0.0 | 1.1 ±0.0 | 1.1 ±0.0 | 1.1 ±0.0 | 1.2±0.1 |
| FS (%) | 60±0.7 | 60±0.7 | 61 ±0.9 | 62±1.2 | 59±0.9 | 59±0.7 | 58±1.9 | 60±1.2 | 59±2.6 |
| DOX | n=11 |
n=17 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| HR (bpm) | 426±8 | 445±14 | 426±14 | 454±11 | 444±7 | 469±13 | 452±15 | 476±13 | 478±11 |
| LVIDD (mm) | 2.2±0.1 | 2.6±0.1 | 2.6±0.1 | 2.8±0.1 | 2.6±0.1 | 2.8±0.1 | 2.7±0.1 | 2.9±0.1 | 2.9±0.1 |
| LVIDS (mm) | 0.9±0.0 | 1.2±0.0* | 1.4±0.1* | 1.5±0.1* | 1.4±0.0* | 1.5±0.1* | 1.4±0.1* | 1.4±0.1* | 1.4±0.1 |
| FS (%) | 59±0.7 | 52±0.9* | 48±1.1* | 47±1.5* | 45±1.3* | 45±1.3* | 47±2.2* | 51±1.2* | 51±1.8* |
p<0.05 vs. Saline-treated mice
Table 2.
Blood cell counts and cTnI concentration measurements (Mean ± SEM).
| Leukocytes | WBC (K/uL) | NE (K/uL) | LY (K/uL) | MO (K/uL) | EO (K/uL) | BA (K/uL) |
|---|---|---|---|---|---|---|
| Saline (n=12) | 12.71±1.27 | 4.08±0.48 | 7.23±0.79 | 0.59±0.10 | 0.65±0.24 | 0.16±0.05 |
| DOX (n=16) | 6.92±0.96* | 4.13±0.72 | 2.04±0.21* | 0.41 ±0.10 | 0.27±0.09 | 0.08±0.03 |
| Erythrocytes | RBC (M/uL) | HB (g/dL) | HCT (%) | MCV (fL) | MCH (pg) | MCHC (g/dL) | RDW (%) |
|---|---|---|---|---|---|---|---|
| Saline (n=12) | 10.08±0.26 | 13.51±0.33 | 51.96±1.25 | 51.62±0.57 | 13.42±0.11 | 26.02±0.29 | 22.57±0.28 |
| DOX (n=16) | 7.30±0.65* | 9.79±0.92* | 39.45±3.61* | 53.54±0.68* | 13.22±0.24 | 24.66±0.24* | 24.62±0.56* |
| cTnI | ng/mL |
|---|---|
| Saline (n=12) | 0.31±0.20 |
| DOX (n=16) | 4.56±1.19* |
WBC, white blood cells; NE, neutrophils; LY, lymphocytes; MO, monocytes; EO, eosiniohils; BA, basophils; K/uL, 1000 cells per microliter; RBC, red blood cells; HB, hemoglobin; HCT, hematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular hemaglobin; MCHC, mean corpuscular hemoglobin content; RDW, red blood cell distribution width.
p<0.05 vs. Saline-treated mice.
A second group of mice subjected to the same regimen of saline or DOX injection was allowed to recover for 13 weeks. DOX treatment resulted in a similar decrease in cardiac function (not shown). Cardiac function remained suppressed in the DOX-injected animals throughout the entire recovery phase as compared to the saline-injected controls (Figure 1A). Decreased left ventricular contraction was apparent in representative echocardiograms from DOX-injected mice at the end of the treatment and recovery phases, as compared to the saline-injected controls (Figure 1B). Of interest, mechanical alternans were occasionally observed in the DOX-injected group.
Growth velocity was markedly lower in the DOX-injected animals during the treatment phase as compared to the saline-injected mice. Although growth velocity was similar in the two groups during the recovery phase, absolute body weight was less in the DOX treated animals due to growth inhibition during the treatment phase (Figure 2A). A reduction in heart weight was noted at the end of the treatment phase in DOX-injected animals as compared to saline-injected control animals. However, by the end of the recovery phase heart weight in DOX-injected mice was largely restored, approaching values observed in control animals (Figure 2B). A similar result was obtained when heart weight was normalized to tibia length (Figure 2C).
Figure 2.

Growth parameters and histochemical analysis of saline- and DOX-injected juvenile mice. (A) Growth velocity (GV [g/wk]) during the treatment and recovery phases. (B) Heart weight in milligrams (HW [mg]) at the end of the treatment and recovery phases. (C) Heart weight / tibia length (HW/TL) measurements at the end of the treatment and recovery phases. (D) Representative survey and high power micrographs of heart sections from mice at the end of the treatment and recovery phases. Note the thin left ventricular wall in the heart from the DOX-injected mouse at the end of the treatment phase. Magnification bar in the survey micrographs = 2 mm; magnification bar in the high power micrographs = 20 microns. (E) Cardiomyocyte minimal fiber diameter (MFD) measurements at the end of the treatment and recovery phases. For all panels, '*' indicates p<0.01 for DOX-injected vs. saline-injected mice.
Hearts harvested at the end of the treatment and recovery phases were processed for histochemical analysis. Survey micrographs from Hematoxylin and Eosin (H&E) stained sections suggested a reduction in left ventricular wall thickness in DOX-injected animals at the end of the treatment phase, which appeared to return to normal by the end of the recovery period (Figure 2D). Quantitative morphometric analyses were performed to validate these gross observations. Previous studies have shown that minimal fiber diameter (MFD) measurements can be used as an index for cardiomyocyte hypertrophy or atrophy in histology sections (33). A reduction in cardiomyocyte MFD was observed at the end of the treatment phase in hearts from DOX-injected mice (Figure 2E). This suggests that the DOX-induced decrease in heart weight resulted at least in part from cardiomyocyte atrophy. Interestingly, cardiomyocyte MFD in hearts from DOX-injected mice returned to control values during the recovery phase.
Anti-cardiac alpha-actinin immune reactivity was monitored to ascertain the impact of DOX on myofiber structure (Figure 3A). Well organized sarcomeric structure was apparent in hearts from saline-injected animals during the treatment and recovery phases. In contrast, myofiber structure was disrupted in hearts from DOX-injected animals during the treatment phase, as evidenced by the widespread reduction of sarcomeric staining. Sarcomeric structure was reorganized by the end of the recovery phase in hearts from DOX-injected animals.
Figure 3.
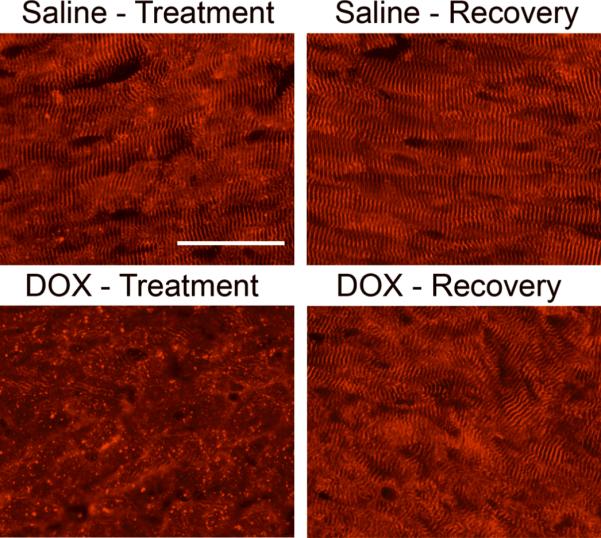
Myocardial alterations induced by DOX treatment. Cardiac alpha-actinin immune reactivity in hearts from saline- and DOX-injected mice during the treatment and recovery phases (rhodamine-conjugated secondary antibody, red signal). Magnification bar = 50 microns.
Anti-activated caspase 3 immune histology was used to quantitate cardiomyocyte apoptosis in hearts from saline- and DOX-injected mice. Since activated caspase 3 is distributed in the cytoplasm, cardiomyocytes at early stages of apoptosis were easily identified by the presence of immune reactivity in rod-shaped cells (28). Figure 4A shows a representative image of an activated caspase 3 immune reactive cardiomyocyte from a DOX-treated heart. An approximately 5-fold increase in the number of activated caspase 3 immune reactive cardiomyocytes per mm2 myocardium was observed in DOX-injected mice at the end of the treatment phase as compared to saline-injected controls (Figure 4B). Interestingly, the level of cardiomyocyte apoptosis was higher in the DOX treated animals at the end of the recovery phase (i.e., 13 weeks after the last drug injection, see Figure 4B). A similar increase in the number of apoptotic cells was obtained using TUNEL staining as an index for apoptosis, with a 4.5 +/−0.3-fold increase in DOX-injected mice at the end of the treatment phase (n=5), and a 19.5 +/- 2.5-fold increase at the end of the recovery phase (n=6), as compared to the respective saline-injected controls (n=7).
Figure 4.
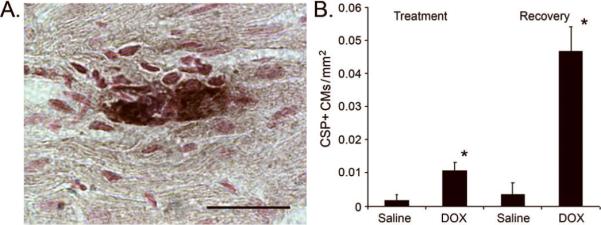
Cardiomyocyte activated caspase 3 immune reactivity in saline- and DOX-injected juvenile mice. (A) Representative image of an anti-activated caspase 3 immune-reactive cardiomyocyte from a DOX-injected heart harvested at the end of the treatment phase. Magnification bar = 20 microns. (B) Number of activated caspase 3 immune-reactive cardiomyocytes (CSP+) per mm2 at the end of the treatment and recovery phases. '*' indicates p<0.01 for DOX-injected vs. saline-injected mice.
Sirius red-Fast green-staining was performed to monitor the level of myocardial collagen deposition, which is indicative of fibrosis. Representative images shown in Figure 5A indicate that collagen deposition (red signal) was minimal at the end of the treatment phase in hearts from DOX-injected mice, but become prevalent at the end of the recovery phase. Quantitative image analyses confirmed this observation, consistent with the presence of DOX-induced myocardial fibrosis (Figure 5B). Finally, Western blot analyses were performed to monitor the expression and/or activity of several myocardial proteins, which are known to be modulated by DOX (Figure 6). Lower levels of myosin heavy chain (MHC), phosphorylated glycogen synthase kinase (GSK)3β, and phosphorylated Akt were apparent in hearts from DOX-injected animals at the end of the treatment phase, as compared to the saline-injected controls. DOX treatment had no effect on total GSK3β or Akt levels. At the end of the recovery phase, expression of these proteins in hearts from DOX-injected mice returned those seen in the saline-injected animals.
Figure 5.

Myocardial fibrosis in saline- and DOX-injected juvenile mice. (A) Representative images of Sirius red-Fast green stained sections from hearts harvested at the end of the treatment and recovery phases. Red signal indicates collagen deposition. Magnification bar = 40 microns. (B) Collagen (COL) content at the end of the treatment and recovery phases. '*' indicates p<0.01 for DOX-injected vs. saline-injected mice.
Figure 6.
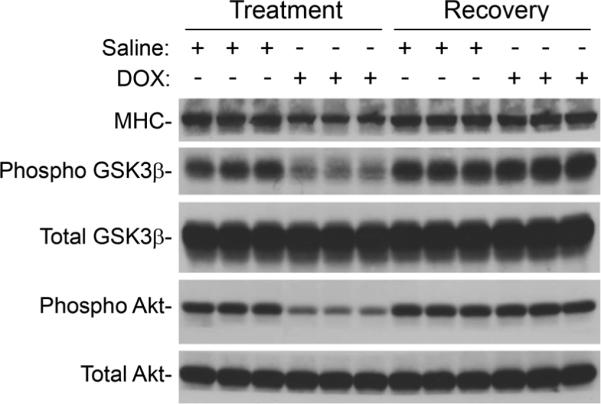
Western blot analysis of protein expression in hearts from saline- and DOX-injected mice during the treatment and recovery phases. Equal protein loading between samples was confirmed by Napthol blue staining of the membrane before hybridization (not shown).
Discussion
The results presented here demonstrate that treatment of juvenile mice with DOX resulted in cardiotoxicity manifested by decreased cardiac systolic function, as well as heart and cardiomyocyte atrophy, myofiber disarray, low levels of cardiomyocyte apoptosis, and altered expression of structural and regulatory proteins. Cardiac systolic dysfunction persisted for as long as 13 weeks after the termination of DOX treatment (the latest time-point analyzed) despite the normalization of organ mass, cell mass, myofiber structure and protein expression. Cardiac dysfunction in the recovery phase was accompanied by increased levels of cardiomyocyte apoptosis and myocardial fibrosis.
In the vast majority of mouse DOX cardiotoxicity studies, animals typically received a single injection of drug and were analyzed within 1 week. None utilized juvenile mice. Nonetheless, several published studies share methodological aspects with the current work, and as such provide an interesting comparison. For example, adult mice receiving cumulative DOX doses of 15 mg/kg (34, 35) to 24 mg/kg (36) and analyzed at 2 (34), 4 (36) or 8 (35) weeks after the last drug injection all exhibited sustained depression of cardiac function. Somewhat surprisingly, the two studies using a 15 mg/kg cumulative DOX dose differed markedly with respect to the level of cardiomyocyte apoptosis, with no apoptosis detected at 2 weeks post drug delivery in one study (34) and consistently elevated levels of apoptosis at 0, 2, 4 and 8 weeks after the last drug injection in the other study (35). In contrast, the levels of cardiomyocyte apoptosis observed in our study were more dynamic. Difficulties encountered when identifying cardiomyocyte nuclei in histology sections (37) may explain the discrepant levels of apoptosis reported in the previous studies, as the TUNEL and ISEL analyses employed label both cardiomyocyte and non-myocyte nuclei. The use of activated caspase 3 immune reactivity in the current study (which permitted cardiomyocyte identification via morphologic criteria) circumvented this problem.
Although it is well known that acute DOX cardiotoxicity is associated with myofiber disarray and cardiomyocyte shrinkage (38), the marked reduction in heart weight and cardiomyocyte size observed at the end of the treatment period was somewhat unexpected. Several recent studies observed a similar impact on heart weight and/or cardiomyocyte minimal fiber diameter (39–42) following a single injection of DOX (15 mg/kg) in adult mice. Myofiber disarray was also apparent in adult mice receiving a single or multiple injections of DOX (12–15 mg/kg cumulative dose) for as long as 2 weeks after drug delivery (39, 41, 43). Myofiber disarray appears to result at least in part from calpain-induced degradation of titin (18). Similar to our results, Yoda and colleagues observed reversal of myofiber disarray 56 days after a single injection of DOX at 14 mg/kg (44).
Several studies have described DOX-induced changes in protein expression similar to that observed in the current study (19, 38, 45, 46). This occurs at least in part via phospho-regulation of both ubiquitous and cardiac-restricted transcriptional co-factors (47, 48). In addition, recent studies using a reporter transgene, which targets cardiac-restricted expression of enhanced green fluorescent protein engineered to carry ubiquination signal sequence demonstrated that DOX treatment results in a marked induction of 26-proteosome activity (49). Collectively, these transcriptional and proteolytic processes likely contribute to the cardiomyocyte atrophy and myofiber disarray observed during the treatment phase of the current study.
Different mechanisms may contribute to the cardiac systolic dysfunction observed in the treatment versus recovery phases in the model presented here. Cardiomyocyte atrophy, loss of myofiber sarcomeric structure, and altered protein expression are likely major contributors to cardiac dysfunction during the DOX treatment phase. Given the relatively low levels of cardiomyocyte apoptosis and the absence of reactive fibrosis, these processes may have little or no impact on cardiac function during the treatment phase of the model. Although the decreased growth velocity and cardiac systolic function observed in the DOX-treated animals, as well as the absence of overt histopathology in the kidney and gut (not shown), is consistent with the presence of heart failure, the lack of standardized definition for heart failure in mice makes it somewhat tenuous to apply this designation. Unfortunately, many of the ancillary analyses, which would assist in diagnosing heart failure, cannot be performed on mice at the ages studied during the treatment phase.
In contrast, the DOX-induced changes in cardiomyocyte size, myofiber organization and protein expression returned to control values during the recovery phase, and thus are likely not to have contributed to the sustained cardiac dysfunction. Rather, a marked increase in cardiomyocyte apoptosis and concomitant induction myocardial fibrosis was observed. Increased extracellular matrix content contributes to diastolic stiffness, and ultimately promotes ventricular dysfunction (50). Thus, apoptosis-induced fibrosis may be a major contributor to the depressed cardiac function observed during the recovery phase in this model.
Although a similar mechanism may underlie the appearance of heart failure in children previously treated with DOX, it is important to acknowledge the limitations of the current study. Notably, a decrease in left ventricular wall thickness is observed in the long-term survivors of anthracycline therapy in childhood. In contrast, wall thickness appeared to have normalized during the recovery phase in the mouse model. However, given the presence of sustained cardiomyocyte apoptosis and concomitant myocardial fibrosis at the end of the recovery phase, wall thinning and frank heart failure may very well occur at later time points. Another potential concern is that the limited blood volume in mice precluded clinical chemistry analysis, which would have been helpful to determine the extent to which non-cardiac tissue damage may have contributed to myocardial dysfunction. However, this caveat could be unequivocally addressed by experiments employing mice harboring cardiac-restricted, cardioprotective genetic modifications.
In light of the multifaceted nature of DOX-induced cardiotoxicity (see Introduction), other mechanisms are also likely to contribute to the phenotypes observed during the treatment and recovery phases. Nonetheless, if the interpretation of our data is correct, studies examining cardioprotective interventions in models utilizing a single injection of DOX followed by a short recovery period might benefit from re-evaluation. In particular, reassessment of therapeutics aimed at inducing cardioprotection by blocking apoptosis during acute DOX cardiotoxicity is likely to be warranted (11). Evaluation of genetically modified mice using the DOX-induced model described here could help address this important issue.
Acknowledgements
We thank Dorothy Field for excellent technical assistance, and Drs. Michael Rubart, Mark Soonpaa and Alex Becker for comments on the manuscript.
Financial Support: This work was funded through grants from the National Heart, Lung and Blood Institute, National Institutes of Health.
Abbreviations
- DOX
Doxorubicin
- FS
fractional shortening
Footnotes
Publisher's Disclaimer: Pediatric Research Articles Ahead of Print contains articles in unedited manuscript form that have been peer-reviewed and accepted for publication. As a service to our readers, we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting and review of the resulting proof before it is published in its final definitive form. Please note that during the production process errors may be discovered, which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morrow WR. Cardiomyopathy and heart transplantation in children. Curr Opin Cardiol. 2000;15:216–223. doi: 10.1097/00001573-200007000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Bristow MR, Billingham ME, Mason JW, Daniels JR. Clinical spectrum of anthracycline antibiotic cardiotoxicity. Cancer Treat Rep. 1978;62:873–879. [PubMed] [Google Scholar]
- 3.Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973;32:302–314. doi: 10.1002/1097-0142(197308)32:2<302::aid-cncr2820320205>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 4.Rinehart JJ, Lewis RP, Balcerzak SP. Adriamycin cardiotoxicity in man. Ann Intern Med. 1974;81:475–478. doi: 10.7326/0003-4819-81-4-475. [DOI] [PubMed] [Google Scholar]
- 5.Sorensen K, Levitt GA, Bull C, Dorup I, Sullivan ID. Late anthracycline cardiotoxicity after childhood cancer: a prospective longitudinal study. Cancer. 2003;97:1991–1998. doi: 10.1002/cncr.11274. [DOI] [PubMed] [Google Scholar]
- 6.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 7.Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, Gelber RD, Colan SD. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J Clin Oncol. 2005;23:2629–2636. doi: 10.1200/JCO.2005.12.121. [DOI] [PubMed] [Google Scholar]
- 8.Steinherz LJ, Steinherz PG, Tan C. Cardiac failure and dysrhythmias 6–19 years after anthracycline therapy: a series of 15 patients. Med Pediatr Oncol. 1995;24:352–361. doi: 10.1002/mpo.2950240604. [DOI] [PubMed] [Google Scholar]
- 9.Lipshultz SE, Colan SD, Gelber RD, Perez-Atayde AR, Sallan SE, Sanders SP. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N Engl J Med. 1991;324:808–815. doi: 10.1056/NEJM199103213241205. [DOI] [PubMed] [Google Scholar]
- 10.Singal PK, Li T, Kumar D, Danelisen I, Iliskovic N. Adriamycin-induced heart failure: mechanism and modulation. Mol Cell Biochem. 2000;207:77–86. doi: 10.1023/a:1007094214460. [DOI] [PubMed] [Google Scholar]
- 11.Takemura G, Fujiwara H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis. 2007;49:330–352. doi: 10.1016/j.pcad.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem. 1986;261:3060–3067. [PubMed] [Google Scholar]
- 13.Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem. 1986;261:3068–3074. [PubMed] [Google Scholar]
- 14.Zhou S, Starkov A, Froberg MK, Leino RL, Wallace KB. Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res. 2001;61:771–777. [PubMed] [Google Scholar]
- 15.Arena E, D'Alessandro N, Dusonchet L, Geraci M, Rausa L, Sanguedolce R. Repair kinetics of DNA, RNA and proteins in the tissues of mice treated with doxorubicin. Arzneimittelforschung. 1979;29:901–902. [PubMed] [Google Scholar]
- 16.Billingham ME, Mason JW, Bristow MR, Daniels JR. Anthracycline cardiomyopathy monitored by morphologic changes. Cancer Treat Rep. 1978;62:865–872. [PubMed] [Google Scholar]
- 17.Monti E, Prosperi E, Supino R, Bottiroli G. Free radical-dependent DNA lesions are involved in the delayed cardiotoxicity induced by adriamycin in the rat. Anticancer Res. 1995;15:193–197. [PubMed] [Google Scholar]
- 18.Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, Eppenberger HM, Suter TM, Liao R, Sawyer DB. Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J Biol Chem. 2004;279:8290–8299. doi: 10.1074/jbc.M308033200. [DOI] [PubMed] [Google Scholar]
- 19.Papoian T, Lewis W. Selective alterations in rat cardiac mRNA induced by doxorubicin: possible subcellular mechanisms. Exp Mol Pathol. 1991;54:112–121. doi: 10.1016/0014-4800(91)90024-r. [DOI] [PubMed] [Google Scholar]
- 20.Wang L, Ma W, Markovich R, Chen JW, Wang PH. Regulation of cardiomyocyte apoptotic signaling by insulin-like growth factor I. Circ Res. 1998;83:516–522. doi: 10.1161/01.res.83.5.516. [DOI] [PubMed] [Google Scholar]
- 21.Kitta K, Day RM, Kim Y, Torregroza I, Evans T, Suzuki YJ. Hepatocyte growth factor induces GATA-4 phosphorylation and cell survival in cardiac muscle cells. J Biol Chem. 2003;278:4705–4712. doi: 10.1074/jbc.M211616200. [DOI] [PubMed] [Google Scholar]
- 22.Kim R, Tanabe K, Uchida Y, Emi M, Inoue H, Toge T. Current status of the molecular mechanisms of anticancer drug-induced apoptosis. The contribution of molecular-level analysis to cancer chemotherapy. Cancer Chemother Pharmacol. 2002;50:343–352. doi: 10.1007/s00280-002-0522-7. [DOI] [PubMed] [Google Scholar]
- 23.Ueno M, Kakinuma Y, Yuhki K, Murakoshi N, Iemitsu M, Miyauchi T, Yamaguchi I. Doxorubicin induces apoptosis by activation of caspase-3 in cultured cardiomyocytes in vitro and rat cardiac ventricles in vivo. J Pharmacol Sci. 2006;101:151–158. doi: 10.1254/jphs.fp0050980. [DOI] [PubMed] [Google Scholar]
- 24.Herman EH, Ferrans VJ. Preclinical animal models of cardiac protection from anthracycline-induced cardiotoxicity. Semin Oncol. 1998;25:15–21. [PubMed] [Google Scholar]
- 25.Chen H, Yong W, Ren S, Shen W, He Y, Cox KA, Zhu W, Li W, Soonpaa M, Payne RM, Franco D, Field LJ, Rosen V, Wang Y, Shou W. Overexpression of bone morphogenetic protein 10 in myocardium disrupts cardiac postnatal hypertrophic growth. J Biol Chem. 2006;281:27481–27491. doi: 10.1074/jbc.M604818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bullock GR, Petrusz P. Techniques in immunocytochemistry. Academic Press; London; New York: 1982. [Google Scholar]
- 27.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol. 1997;272:H220–H226. doi: 10.1152/ajpheart.1997.272.1.H220. [DOI] [PubMed] [Google Scholar]
- 28.Nakajima H, Nakajima HO, Tsai SC, Field LJ. Expression of mutant p193 and p53 permits cardiomyocyte cell cycle reentry after myocardial infarction in transgenic mice. Circ Res. 2004;94:1606–1614. doi: 10.1161/01.RES.0000132279.99249.f4. [DOI] [PubMed] [Google Scholar]
- 29.Nakajima H, Nakajima HO, Dembowsky K, Pasumarthi KB, Field LJ. Cardiomyocyte cell cycle activation ameliorates fibrosis in the atrium. Circ Res. 2006;98:141–148. doi: 10.1161/01.RES.0000197783.70106.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pasumarthi KB, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res. 2005;96:110–118. doi: 10.1161/01.RES.0000152326.91223.4F. [DOI] [PubMed] [Google Scholar]
- 31.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 33.Dubowitz V, Sewry CA, Fitzsimons RB. Muscle biopsy: a practical approach. Baillière Tindall; London; Philadelphia: 1985. p. 120. [Google Scholar]
- 34.Kim KH, Oudit GY, Backx PH. Erythropoietin protects against doxorubicin-induced cardiomyopathy via a phosphatidylinositol 3-kinase-dependent pathway. J Pharmacol Exp Ther. 2008;324:160–169. doi: 10.1124/jpet.107.125773. [DOI] [PubMed] [Google Scholar]
- 35.Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. 2005;111:1601–1610. doi: 10.1161/01.CIR.0000160359.49478.C2. [DOI] [PubMed] [Google Scholar]
- 36.Delgado RM, 3rd, Nawar MA, Zewail AM, Kar B, Vaughn WK, Wu KK, Aleksic N, Sivasubramanian N, McKay K, Mann DL, Willerson JT. Cyclooxygenase-2 inhibitor treatment improves left ventricular function and mortality in a murine model of doxorubicin-induced heart failure. Circulation. 2004;109:1428–1433. doi: 10.1161/01.CIR.0000121354.34067.48. [DOI] [PubMed] [Google Scholar]
- 37.Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15–26. doi: 10.1161/01.res.83.1.15. [DOI] [PubMed] [Google Scholar]
- 38.Singal PK, Deally CM, Weinberg LE. Subcellular effects of adriamycin in the heart: a concise review. J Mol Cell Cardiol. 1987;19:817–828. doi: 10.1016/s0022-2828(87)80392-9. [DOI] [PubMed] [Google Scholar]
- 39.Esaki M, Takemura G, Kosai K, Takahashi T, Miyata S, Li L, Goto K, Maruyama R, Okada H, Kanamori H, Ogino A, Ushikoshi H, Minatoguchi S, Fujiwara T, Fujiwara H. Treatment with an adenoviral vector encoding hepatocyte growth factor mitigates established cardiac dysfunction in doxorubicin-induced cardiomyopathy. Am J Physiol Heart Circ Physiol. 2008;294:H1048–H1057. doi: 10.1152/ajpheart.01102.2007. [DOI] [PubMed] [Google Scholar]
- 40.Li K, Sung RY, Huang WZ, Yang M, Pong NH, Lee SM, Chan WY, Zhao H, To MY, Fok TF, Li CK, Wong YO, Ng PC. Thrombopoietin protects against in vitro and in vivo cardiotoxicity induced by doxorubicin. Circulation. 2006;113:2211–2220. doi: 10.1161/CIRCULATIONAHA.105.560250. [DOI] [PubMed] [Google Scholar]
- 41.Li L, Takemura G, Li Y, Miyata S, Esaki M, Okada H, Kanamori H, Khai NC, Maruyama R, Ogino A, Minatoguchi S, Fujiwara T, Fujiwara H. Preventive effect of erythropoietin on cardiac dysfunction in doxorubicin-induced cardiomyopathy. Circulation. 2006;113:535–543. doi: 10.1161/CIRCULATIONAHA.105.568402. [DOI] [PubMed] [Google Scholar]
- 42.Li L, Takemura G, Li Y, Miyata S, Esaki M, Okada H, Kanamori H, Ogino A, Maruyama R, Nakagawa M, Minatoguchi S, Fujiwara T, Fujiwara H. Granulocyte colony-stimulating factor improves left ventricular function of doxorubicin-induced cardiomyopathy. Lab Invest. 2007;87:440–455. doi: 10.1038/labinvest.3700530. [DOI] [PubMed] [Google Scholar]
- 43.Alderton PM, Gross J, Green MD. Comparative study of doxorubicin, mitoxantrone, and epirubicin in combination with ICRF-187 (ADR-529) in a chronic cardiotoxicity animal model. Cancer Res. 1992;52:194–201. [PubMed] [Google Scholar]
- 44.Yoda Y, Nakazawa M, Abe T, Kawakami Z. Prevention of doxorubicin myocardial toxicity in mice by reduced glutathione. Cancer Res. 1986;46:2551–2556. [PubMed] [Google Scholar]
- 45.Esaki M, Takemura G, Kosai K, Takahashi T, Miyata S, Li L, Goto K, Maruyama R, Okada H, Kanamori H, Ogino A, Ushikoshi H, Minatoguchi S, Fujiwara T, Fujiwara H. Treatment with an adenoviral vector encoding hepatocyte growth factor mitigates established cardiac dysfunction in doxorubicin-induced cardiomyopathy. Am J Physiol Heart Circ Physiol. 2008;294:H1048–H1057. doi: 10.1152/ajpheart.01102.2007. [DOI] [PubMed] [Google Scholar]
- 46.Negoro S, Oh H, Tone E, Kunisada K, Fujio Y, Walsh K, Kishimoto T, Yamauchi-Takihara K. Glycoprotein 130 regulates cardiac myocyte survival in doxorubicin-induced apoptosis through phosphatidylinositol 3-kinase/Akt phosphorylation and Bcl-xL/caspase-3 interaction. Circulation. 2001;103:555–561. doi: 10.1161/01.cir.103.4.555. [DOI] [PubMed] [Google Scholar]
- 47.Jeyaseelan R, Poizat C, Baker RK, Abdishoo S, Isterabadi LB, Lyons GE, Kedes L. A novel cardiac-restricted target for doxorubicin. CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. J Biol Chem. 1997;272:22800–22808. doi: 10.1074/jbc.272.36.22800. [DOI] [PubMed] [Google Scholar]
- 48.Poizat C, Puri PL, Bai Y, Kedes L. Phosphorylation-dependent degradation of p300 by doxorubicin-activated p38 mitogen-activated protein kinase in cardiac cells. Mol Cell Biol. 2005;25:2673–2687. doi: 10.1128/MCB.25.7.2673-2687.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumarapeli AR, Horak KM, Glasford JW, Li J, Chen Q, Liu J, Zheng H, Wang X. A novel transgenic mouse model reveals deregulation of the ubiquitinproteasome system in the heart by doxorubicin. FASEB J. 2005;19:2051–2053. doi: 10.1096/fj.05-3973fje. [DOI] [PubMed] [Google Scholar]
- 50.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]