

Abstract
Fluconazole-loaded ethyl cellulose microspheres were prepared by alginate facilitated (water-in-oil)-in-water emulsion technology and the effects of various processing variables on the properties of microspheres were investigated. Scanning electron microscopy revealed spherical nature and smooth surface morphology of the microspheres except those prepared at higher concentration of emulsifiers and higher stirring speeds. The size of microspheres varied between 228 and 592 μm, and as high as 80% drug entrapment efficiency was obtained depending upon the processing variables. When compared up to 2 h, the drug release in pH 1.2 HCl solution was slower than in pH 7.4 phosphate buffer saline solution. However, this trend was reversed at high shear conditions. The microspheres provided extended drug release in alkaline dissolution medium and the drug release was found to be controlled by Fickian-diffusion mechanism. However, the mechanism shifted to anomalous diffusion at high shear rates and emulsifier concentrations. The aging of microspheres did not influence the drug release kinetics. However, the physical interaction between drug and excipients affected the drug dissolution behaviors. X-ray diffractometry (X-RD) and differential scanning calorimetry (DSC) analysis revealed amorphous nature of drug in the microspheres. Fourier transform infrared (FTIR) spectroscopy indicated stable character of fluconazole in the microspheres. The stability testing data also supported the stable nature of fluconazole in the microspheres. The fluconazole extracted from 80% drug-loaded formulation showed good in vitro antifungal activity against Candida albicans. Thus, proper control of the processing variables involved in this modified multiple emulsion technology could allow effective incorporation of slightly water soluble drugs into ethyl cellulose microspheres without affecting drug stability.
Key words: entrapment efficiency, ethyl cellulose, fluconazole, microspheres, water-in-oil-in-water (w/o/w) multiple emulsion
INTRODUCTION
The administration of drugs in the form of microspheres has received much attention because they avoid the vagaries of gastric emptying and different transit rates through the gastrointestinal tract, thereby release drugs more uniformly (1). Moreover, a multiunit system spreads over a large area of the absorbing mucosa and prevents exposure to a high drug concentration, when compared to single unit dosage form on chronic dosing (2). This fact coupled with their ability to prolong the release of drugs has given impetus to the development of oral microparticulate systems for drug delivery. The microencapsulation process in which the removal of the hydrophobic polymer solvent is achieved by evaporation has widely been reported in recent years. The technique of emulsion solvent evaporation offers a versatile, easy, and practical method for the manufacture of microspheres because it requires only mild conditions such as ambient temperature and constant stirring. Moreover, this technique makes possible the entrapment of a wide range of drugs having different physical properties and solubility characteristics (3). Although the solvent evaporation method seems to involve a relatively simple process, final product characteristics depends mainly on the formulation and process variables. In order to obtain batches of microspheres with reproducible sizes, surface/core distribution of drugs and release profiles, manufacturing factors such as emulsifier concentration, stirring rate, and organic phase volume should be under control (4). The water-in-oil (w/o) emulsification process has been developed for the encapsulation of highly water soluble drugs. However, an important drawback of using an oil external phase is cleaning up the final product (5). Unlike w/o emulsification method, water-in-oil-in-water (w/o/w) emulsion solvent evaporation technique proves much more effective when the aqueous solubility of drug is high (>900 mg/ml) and partitioning into the organic phase is disfavorable (6). Accordingly, microspheres containing water soluble drugs have been prepared using different polymers such as poly(lactide-co-glycolide) (PLGA) (7), eudragit RS (8), polymethyl methacrylate (9). In addition, modified procedures of this technique have been proposed to obtain desired properties of the microspheres, loaded with the water soluble active principles. The modifications include the use of enteric wall material in oil phase (10,11), addition of nonsolvent into external aqueous phase (12), incorporation of solid lyophilized powder during primary emulsification (13), and the addition of electrolytes into external aqueous phase (14). Simultaneously, the research efforts have been made to incorporate slightly water soluble drugs into polymeric microspheres by the conventional w/o/w emulsion solvent evaporation technique. Recently, it has been stated that the poly(ε-caprolactone) microparticles resulted in poor encapsulation efficiency (2.89 ± 0.20 μg/mg) for melarsoprol (15). An encapsulation efficiency of only 5–17% has also been reported for zidovudine into PLGA microparticles (16). To the best of our knowledge, no reports are available that describe the modification of w/o/w emulsion solvent evaporation technique for the effective/successful incorporation of slightly water soluble medicaments into polymeric microspheres. Ethyl cellulose, a nonbiodegradable and biocompatible polymer, is an extensively studied encapsulating material for the controlled release of pharmaceuticals. However, it has been little investigated by either conventional or modified w/o/w emulsion technology. Thus, the objective of our present investigation was to improve the trapping efficiency of slightly water soluble drug into ethyl cellulose microspheres by alginate facilitated w/o/w emulsion solvent evaporation technique and to study systematically the effect of processing variables which are likely to influence the properties of microspheres such as percentage yield, particle size distribution, surface morphology, drug entrapment efficiency, and in vitro drug release behaviors. Fluconazole, used for the treatment of oral, esophageal, and vaginal candidiasis (17), has been selected as a model drug in this investigation. The compatibility of drug in ethyl cellulose microspheres was evaluated through X-RD, DSC, and FTIR spectroscopy. The optimized formulation was also evaluated for its in vitro antifungal activity.
MATERIALS AND METHODS
Materials
Fluconazole was a gift from East India Pharmaceutical Works Ltd., Kolkata, India. Ethyl cellulose (18–22 cps viscosity grade), sodium alginate (0.1% w/v aqueous solution exhibits a viscosity of 1.15 cp at 25°C), and Span 80 were purchased from Loba Chemie Pvt. Ltd., Mumbai, India. Dichloromethane and Tween 80 were supplied by Merck Ltd., Mumbai, India. d-Glucose, peptone (granular), and agar (granular) were of bacteriological grade and were obtained from Qualigens Fine Chemicals, Mumbai, India. All other reagents obtained commercially were of analytical grade and used as received.
Preparation of Microspheres by Conventional o/w and w/o/w Emulsion Solvent Evaporation Technique
Fluconazole (60 mg) and ethyl cellulose (500 mg) were dissolved in 25 ml of dichloromethane. The resultant polymeric solution was then added slowly into 140 ml water containing 0.6% (w/v) Tween 80 with continuous agitation to form o/w type emulsion. The agitation was continued at 700 rpm for 2 h to evaporate the organic solvent. Finally, the microspheres were filtered off, washed with cold double distilled water (3 × 100 ml), and dried to constant weight.
Accurately weighed, 60 mg of fluconazole was dispersed in 12 ml water and emulsified in 25 ml 2% (w/v) organic solution of ethyl cellulose containing 0.5% (w/v) Span 80 using a homogenizer. The resulting w/o emulsion was then poured into 140 ml water containing 0.6% (w/v) Tween 80 with continuous mechanical agitation at 700 rpm to form w/o/w type emulsion. This process was continued for 1.5 h to evaporate the organic solvent at room temperature. The microspheres were separated by filtration, washed with cold double distilled water (3 × 100 ml), and dried to constant weight.
Preparation of Microspheres by Modified Multiple Emulsion Technique
The microspheres were prepared by alginate facilitated w/o/w emulsion solvent evaporation technique. Accurately weighed amount of fluconazole (5 mg/ml of internal aqueous phase volume) was uniformly dispersed in 8 ml of 2% (w/v) aqueous sodium alginate solution (pH 6.0). The dispersion was added slowly (0.15 ml/s) into 25 ml 2% (w/v) solution of ethyl cellulose in dichloromethane containing 0.5% (w/v) Span 80 and emulsified using a rotor–stator homogenizer (Model RQ-127A, Remi Motors Ltd., Mumbai, India) for 5 min at 1,200 rpm. The resulting water-in-oil (w/o) emulsion was then transferred (2.5 ml/s) into 100 ml of water containing 0.6% (w/v) Tween 80 with continuous mechanical stirring at 700 rpm to form w/o/w type emulsion at room temperature. The stirring was continued with a three-blade propeller (Type-BL-433, Bio-Lab Instrument Mfg. Co., Mumbai, India) for a period of 1.5 h to allow evaporation of the organic solvent. The resulting microspheres were separated by filtration. Most of the nonencapsulated fluconazole remained in the external aqueous phase and was removed in the filtrate. The remaining loose drug on the surface of the microspheres was removed by washing with cold double distilled water (3 × 100 ml) and finally oven-dried at 40–50°C for 24 h. The following processing variables were investigated:
Concentration of ethyl cellulose solution: 1.0%, 1.5%, and 2.0% (w/v)
Concentration of Span 80 in oil phase: 0.0%, 0.5%, and 1.5% (w/v)
Stirring speed during secondary emulsification: 700, 1,000, 1,300 rpm
Internal aqueous alginate phase volume: 4, 8, and 12 ml
Concentration of Tween 80 in external aqueous phase: While the internal aqueous phase volume was 12 ml, the concentrations of Tween 80 varied (0.3%, 0.6%, and 1.0% w/v).
External aqueous phase volume: 60, 100, and 140 ml; the microspheres were prepared using an internal aqueous phase volume of 12 ml.
Identification of Alginate Facilitated w/o/w Emulsion Structure
Amaranth, a water soluble dye, was used to identify the multiple emulsion. The internal aqueous phase of sodium alginate solution and the external aqueous phase were colored with the dye. Following the preparation, a drop of multiple emulsion was put on a microscopic slide and observed under an optical microscope (Olympus, Model HB, India). An optical combination of 10× eyepiece and 10× objective was used for the detection of emulsion structure. The multiple emulsion structure was captured with a 2-megapixel digital camera.
Yield of Microspheres
The yield of the microspheres was calculated as a percentage of the total amounts of polymers and drug employed during the preparation.
Particle Size Analysis
Particle size analysis of the fluconazole-loaded ethyl cellulose microspheres was done by sieving method. Several British standard sieves ranging 16 meshes to 120 meshes were arranged in a nest with the coarsest at the top. A weighed amount of samples were placed on the top and the sieve set was shaken with a mechanical shaker (EGG80432, Geologists’ Syndicate Pvt. Ltd., Kolkata, India) for 10 min. The samples retained on each sieve were collected and weighed. The arithmetic mean diameter was calculated following the method reported earlier (18).
Scanning Electron Microscopy
The shape and surface morphology of the fluconazole-loaded ethyl cellulose microspheres were investigated using scanning electron microscope (Jeol, JSM-5200, Japan). Prior to examinations, samples were mounted onto stubs using double-sided dried carbon tape and vacuum coated with gold-palladium film (thickness 2 nm) using sputter coater (Edward S-150, UK) to render them electrically conductive.
Determination of Drug Entrapment Efficiency
Accurately weighed, 10 mg of fluconazole-loaded ethyl cellulose microspheres was dissolved in 2 ml dichloromethane; 30 ml of pH 7.4 phosphate buffer saline (PBS) solution was added and stirred for 30 min with a magnetic stirrer. The mixture was heated at 50–55°C for 45 min in a thermostatic water bath to remove dichloromethane. After that, the volume was adjusted to 50 ml with fresh PBS solution (pH 7.4) heated at 50–55°C. The solution was cooled, filtered, and an aliquot, after suitable dilution, was analyzed spectrophotometrically at 261 nm. Each experiment was carried out in triplicate. Reliability of the method was judged by conducting recovery analyses using known amounts of fluconazole with or without polymer, and recovery averaged 98.17 ± 0.61%.
In Vitro Drug Dissolution Study
The in vitro release of fluconazole from ethyl cellulose microspheres was carried out in enzyme free, gastric, and intestinal fluids using a digital USP type II dissolution rate test apparatus (VDA-6D, Veego Instrument Corporation, Mumbai, India). About 50 mg of dried microspheres, accurately weighed, were suspended in 500 ml of either pH 1.2 HCl solution or pH 7.4 PBS solution. The release studies were conducted using the same size fractions of microspheres (those retained on 60 mesh sieve) to get comparable results. The paddle was rotated at 50 rpm and the temperature was set at 37±0.5°C. At predetermined times, 5 ml sample was withdrawn and replenished with fresh buffer solution. The aliquots were analyzed using a double beam spectrophotometer (UV1, Thermo Spectronic, Great Britain) at 261 nm. Cumulative percentage of fluconazole released was plotted as a function of time. The dissolubility of free drug and that incorporated in the physical mixture was studied in acidic as well as alkaline dissolution medium under similar conditions for 30 min. The physical mixture was prepared by triturating drug, sodium alginate, and ethyl cellulose (1:4:13.33 weight ratios) for about 10 min in a porcelain mortar to obtain the same weight ratios as the microspheres.
Drug Release Mechanism
The mechanism of in vitro fluconazole release from freshly prepared and aged ethyl cellulose microspheres was determined from the values of diffusional exponent (n) obtained by modeling the first 60% of the drug release into Korsmeyer–Peppas equation (19): Mt/M∞ = ktn, where Mt/M∞ is the fractional solute release at time t and k is a constant which incorporates the structural and geometric characteristics of the device. The mechanism of drug release from spherical polymeric devices may be Fickian diffusion when the value of n = 0.43 or less, anomalous (non-Fickian) transport when the value of n lies between 0.43 and 0.85, and case II transport when n = 0.85. An exponent value of n greater than 0.85 signifies super case II transport mechanism (20).
Flow Property
The flow property of microspheres was evaluated by measuring Carr’s index (C) and Hausner ratio (H). The bulk and tap densities were measured in a 100-ml graduated measuring cylinder. The sample contained in the measuring cylinder was placed in a USP-I tap density test apparatus (Vtap/Matic-II, Mumbai, India) and tapped mechanically by means of constant velocity rotating cam. At first, 500 taps were applied and tapped volume was noted. The tapped volume was measured once again following additional 750 taps. From the initial bulk volume and final tapped volume, respective densities were calculated. Carr’s index was determined by the following formula:
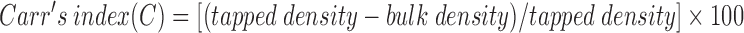 |
A Carr’s compressibility index greater than 25% was considered to be an indication of poor flowability and below 15% of good flowability. The Hausner ratio (H) is a number that is correlated to the flowability of a granular material. It was calculated by the formula: H = 100/(100 − C). A Hausner ratio greater than 1.25 was considered to be an indication of poor flowability.
Swelling Behaviors
The swelling ratio of blank ethyl cellulose microspheres was measured by immersing dried preweighed samples (20 mg) into 100 ml pH 1.2 HCl solution for 2 h and subsequently transferring into pH 7.4 PBS solution for 3 h. At specific time intervals, microspheres were removed from the swelling medium, blotted with a piece of tissue paper to absorb excess surface water, and weighed. The swelling ratios (Q) of the microspheres were calculated from the equation: Q = (w2 − w1)/w1, where w1 is the weight of the dried sample and w2 is the weight of the swollen sample. The swelling study was also conducted in pH 7.4 PBS solution for 3.5 h.
Fourier Transform Infrared Spectroscopy
FTIR spectra of finely powdered pure fluconazole, fluconazole-loaded microspheres, and blank ethyl cellulose microspheres were recorded in Perkin Elmer FT-IR Spectrometer (Spectrux RX 1, UK) from 4,000 to 400 cm−1 at a resolution of 4 cm−1 using KBr pellets. The pellets were made by applying a pressure of 10 tons for 15 min in a hydraulic pellet press (Type-KP, Kimaya Engineers, India).
Qualitative X-Ray Diffractometry
Grinded samples of pure fluconazole, physical mixture of fluconazole and excipients, and fluconazole-loaded ethyl cellulose microspheres were scanned from 5° to 50° 2θ, using a wide angle X-ray diffractometer (Rigaku, Miniflex, Japan), under the following measurement conditions: source, Cu-Kα radiation; voltage, 30 kV; current, 15 mA; and scan speed, 1° per min.
Differential Scanning Calorimetry
DSC thermograms of pure fluconazole, fluconazole-loaded microspheres, and blank ethyl cellulose microspheres were recorded using Perkin-Elmer instrument (Pyris-Diamond TG/DTA, Singapore). Each sample (3–7 mg) was accurately weighed into a 40-μl aluminum pan in a hermetically sealed condition. The measurements were performed in an atmosphere of nitrogen between 30°C and 250°C at a heating rate of 10°C/min.
In Vitro Antifungal Activity
The isolates of Candida albicans were subcultured on sterilized liquid Sabouraud Dextrose Agar (SDA) media and incubated at 35°C for 24 h. Following dissolution of fluconazole-loaded microspheres in either pH 7.4 PBS solution or pH 1.2 HCl solution, 5 ml samples were collected at different time intervals and were sterilized by membrane filtration. The liquid culture (100 μl) was transferred into each test tube aseptically. One milliliter of this liquid culture was added in a SDA plate by spread plate method, incubated at 35°C for 24 h, and the colony of fungi was counted. The initial colony was counted with respect to control and the percentage inhibition of colony forming unit as a function of in vitro fluconazole concentration was plotted.
Statistical Analysis
The differences in drug release rate from the microspheres were evaluated by one-way analysis of variance: single factor, using Microsoft Excel 2002 software. Differences were considered significant when p < 0.05. The drug release rate before and after aging was evaluated by t test: paired two samples for means, using Microsoft Excel 2002 software. The aging effect was considered significant when the t stat value was greater than the t critical two-tail value at alpha level of 0.05 (p two-tail <0.05). The difference between drug entrapment efficiencies of the microspheres before and after stability testing was also analyzed statistically by paired t test.
RESULTS AND DISCUSSION
The present work was aimed to prepare ethyl cellulose microspheres with high entrapment levels of fluconazole by alginate facilitated w/o/w emulsion solvent evaporation technique. This approach to replace more simple process technologies, such as o/w or w/o/w emulsion solvent evaporation, for the model drug was justified primarily on the basis of their drug entrapment efficiencies. While the drug entrapment efficiency (%) of the ethyl cellulose microspheres, prepared by conventional o/w emulsion solvent evaporation technique, was 24.52 ± 2.13, the same was found to be 10.93 ± 1.23 when prepared by conventional w/o/w emulsion solvent evaporation technique. Hence, a high level of fluconazole entrapment into ethyl cellulose microspheres was not possible by using these simple process technologies. On the other hand, the use of 2% (w/v) sodium alginate in the internal aqueous phase of this conventional multiple emulsion technique improved the drug entrapment efficiency (55.55 ± 4.30) of ethyl cellulose microspheres under similar processing conditions. The formation of w/o/w emulsion structure by this modified technology has been represented in Fig. 1. As several processing parameters were involved in this modified technique, the properties of microspheres could be affected. Thus, the focus of the present work was directed toward the systematic investigation of these parameters which could influence the properties of microspheres.
Fig. 1.
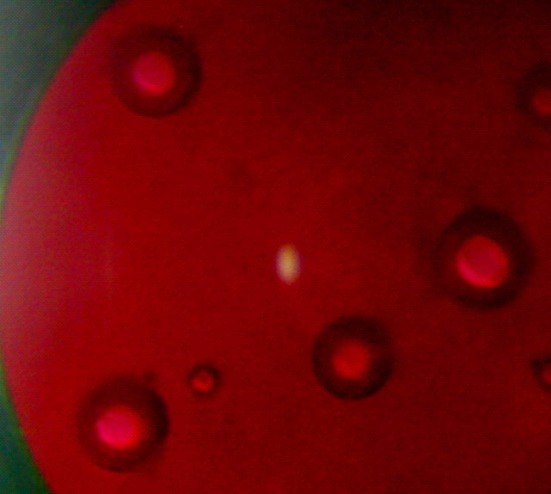
A photograph showing the presence of alginate facilitated (water-in-oil)-in-water emulsion structure; water phase (red); organic phase (black)
Effect of Polymer Concentration
Fluconazole-loaded microspheres were prepared using different concentration of ethyl cellulose (1.0%, 1.5%, and 2.0% w/v) to investigate the eventual modifications of the percentage yield, particle size, surface topography, drug entrapment efficiency, and release behavior in different dissolution media. The yield of microspheres was improved from 88.43% to 96.15% when the concentration of polymer was increased (Table I). Increasing the weight of polymer in a fixed volume of organic solvent resulted in an increase in mean particle diameter from 321 to 331 μm (Table I). It could be postulated that higher concentration of polymer in the sample led to an increased frequency of collisions, resulted in fusion of semiformed particles, and finally increased the size of the microspheres (21). Nevertheless, 72 to 85 weight percentages retained on British standard sieve (no. 60) which was indicative of monomodal size distribution of the microspheres. When observed under scanning electron microscope, all the microspheres appeared spherical having smooth surfaces. A representative micrograph has been displayed in Fig. 2a. Higher polymer concentration in the emulsion droplets led to an enhancement of the efficiency of fluconazole entrapment from 27.80% to 72.79% (Table I). Probably, the high viscosity of organic phase tended to restrict migration of the inner aqueous/drug phase to the external water phase and enhanced the drug entrapment efficiency (22,23). The variation in polymer concentration affected the drug release significantly (p < 0.05) as evident from t50% values (time required for the release of 50% encapsulated drug). The in vitro drug release profile of the microspheres prepared with the lowest polymer concentration ran always higher than those prepared with higher concentrations in either pH 1.2 HCl solution or pH 7.4 PBS solution (Fig. 3a). The microspheres of 1% (w/v) ethyl cellulose showed an initial burst drug release in PBS solution (releasing 50.59% drug in 1 h). However, the burst effect was suppressed by 28.94% and 59.30% with gradual increase in polymer concentration to 2% (w/v). A similar observation with protein-loaded PLGA microcapsules has been reported (24). It was realized that an increase in amount of polymer led to a dense less porous polymeric phase and inhibited the burst effect. The microspheres released almost half of the entrapped drug in acidic dissolution medium within 2 h at the lowest polymer concentration. The same were found 39.08% and 20.49% with increasing polymer concentrations. Indirectly, it can be said that a polymer concentration of 1% (w/v) was insufficient to form a uniform film around the microspheres which could prevent the drug release in either dissolution fluids. Although the drug release was dependent on the polymer concentrations, the drug release was predominated by Fickian-diffusion controlled mechanism (Table I).
Table I.
Effect of Process Variables on the Properties of Fluconazole-Loaded Ethyl Cellulose Microspheres
| Processing variables | %Yield | Entrapment efficiencya (%) | Mean size (μm) | t 50% value (h) in PBS solution | Korsmeyer–Peppas model | ||
|---|---|---|---|---|---|---|---|
| r 2 | k | n | |||||
| EC (% w/v) | |||||||
| 1.0 | 88.43 | 27.80 ± 3.64 | 321.68 | 0.93 ± 0.05 | 0.9498 | 0.5179 | 0.3019 |
| 1.5 | 91.04 | 43.25 ± 2.91 | 328.00 | 2.83 ± 0.25 | 0.9945 | 0.3619 | 0.3121 |
| 2.0 | 96.15 | 72.79 ± 2.11 | 331.62 | 7.90 ± 0.10 | 0.9754 | 0.2184 | 0.3194 |
| Span 80 (% w/v) | |||||||
| 0.0 | 92.00 | 26.27 ± 4.37 | 361.01 | 1.10 ± 0.26 | 0.9157 | 0.4727 | 0.4022 |
| 0.5 | 96.15 | 72.79 ± 2.11 | 331.62 | 7.90 ± 0.10 | 0.9754 | 0.2184 | 0.3194 |
| 1.5 | 95.00 | 28.07 ± 0.38 | – | 2.13 ± 0.21 | 0.9914 | 0.3418 | 0.4505 |
| Stirring speed (rpm) | |||||||
| 700 | 96.15 | 72.79 ± 2.11 | 331.62 | 7.90 ± 0.10 | 0.9754 | 0.2184 | 0.3194 |
| 1,000 | 85.43 | 49.80 ± 5.57 | 248.11 | 6.33 ± 0.15 | 0.9262 | 0.2091 | 0.4893 |
| 1,300 | 78.60 | 31.04 ± 1.85 | 228.56 | 2.70 ± 0.40 | 0.8189 | 0.3545 | 0.5004 |
| Internal aqueous phase volume (ml) | |||||||
| 4 | 98.33 | 28.17 ± 4.10 | 329.36 | 8.57 ± 0.12 | 0.9637 | 0.1503 | 0.4986 |
| 8 | 96.15 | 72.79 ± 2.11 | 331.62 | 7.90 ± 0.10 | 0.9754 | 0.2184 | 0.3194 |
| 12 | 81.25 | 77.26 ± 2.08 | 333.02 | 6.37 ± 0.51 | 0.9802 | 0.2797 | 0.2989 |
| Tween 80 (% w/v) | |||||||
| 0.3 | 78.00 | 74.39 ± 4.32 | 351.84 | 4.30 ± 0.10 | 0.9782 | 0.3419 | 0.2818 |
| 0.6 | 81.25 | 77.26 ± 2.08 | 333.02 | 6.37 ± 0.51 | 0.9802 | 0.2797 | 0.2989 |
| 1.0 | 93.00 | 69.68 ± 1.63 | 302.89 | 8.43 ± 0.25 | 0.9862 | 0.1591 | 0.4824 |
| External aqueous phase volume (ml) | |||||||
| 60 | 80.00 | 80.03 ± 1.63 | 287.62 | 4.87 ± 0.15 | 0.9761 | 0.3206 | 0.2847 |
| 100 | 81.25 | 77.26 ± 2.08 | 333.02 | 6.37 ± 0.51 | 0.9802 | 0.2797 | 0.2989 |
| 140 | 93.00 | 55.55 ± 4.30 | 592.03 | 8.67 ± 0.21 | 0.9778 | 0.2545 | 0.2817 |
PBS phosphate buffered saline, EC ethyl cellulose
aData are the mean ± SD, n = 3
Fig. 2.

Scanning electron micrographs of the fluconazole-loaded microspheres: a 2% ethyl cellulose; b 1.5% Span 80; c 1,300 rpm; d 1% Tween 80; e 60 ml external phase volume; f surface before dissolution (60 ml external aqueous phase); g surface after dissolution in pH 1.2 HCl solution; h surface after dissolution in pH 7.4 PBS solution
Fig. 3.

Release profiles of fluconazole-loaded ethyl cellulose microspheres: a variation of ethyl cellulose; b Span 80; c rpm; d internal phase volume; e Tween 80; f external phase volume. Release study in pH 7.4 PBS solution: closed square lowest value of the variables, closed triangle intermediate value of the variables, closed circle highest value of the variables. Release study in pH 1.2 HCl solution: open square lowest value of the variables, open triangle intermediate value of the variables, open circle highest value of the variables
Effect of Span 80 in Oil Phase
Span 80, a representative of the nonionic dispersing agent, was used to stabilize the w/o primary emulsion. It has hydrophilic–lipophilic balance value of 4.3 and is expected to have a high disparity for the present emulsion system by reducing the surface tension at the interface. One formulation was made without Span 80 since ethyl cellulose is reported to have an additional property of stabilizing w/o primary emulsion (25). Two formulations were also made using different concentrations of Span 80 (0.5% and 1.5% w/v). The microspheres prepared without surfactant in the oil phase became elongated; however, at the intermediate concentration, the microspheres were spherical and individual in nature (Fig. 2a). Further increase in concentration of Span 80 lost their individuality and existed as aggregates (Fig. 2b). Hence, it was very difficult to determine the size of microspheres by sieving method at 1.5% (w/v) of Span 80. While the microspheres showed monomodal size distribution at 0.5% (w/v) of Span 80, the distribution shifted to bimodal pattern when prepared without Span 80 (data not shown). The mean diameter of the microspheres decreased from 361 to 331 μm with the use of Span 80 (0.5% w/v). Drug trapping efficiency of the microspheres was higher at the intermediate concentration of emulsifier than those observed at other concentrations (Table I). Since the particle size and drug encapsulation efficiency are greatly related to the stability of primary emulsion, this could be explained by the tensioactive properties of Span 80 which stabilized the first emulsion and prevented the fast coalescence of the droplets (26,27). Figure 3b represents the influence of Span 80 on the release characteristics of drug-loaded microspheres in both acidic and alkaline dissolution media. The microspheres prepared with 1.5% Span 80 or without stabilizer showed faster drug release in both media. Comparatively, a slower drug release profile was achieved at the intermediate concentration of Span 80. However, the drug release from all the microspheres became slower in acidic dissolution medium than in alkaline medium when compared up to 2 h. A statistically significant difference (p < 0.05) was noted in t50% values (Table I) and suggested that the emulsifier concentration in oil phase was influential in controlling drug release from the microspheres. Probably, the absence of surfactant hindered the division of aqueous inner phase into finer droplets, caused slower precipitation of the polymer, created possibilities for the drug to be present over the surface of semiformed microspheres, and thus led to quicker drug release. On contrary, the particles may be smaller at higher concentration of Span 80 (although not measurable by sieving method) and may increase the effective surface area for faster drug dissolution. The drug release from the microspheres deviated from Fickian to non-Fickian/anomalous transport mechanism at 1.5% (w/v) of Span 80 (Table I). This suggested that in addition to diffusion, polymer chain relaxation process was also involved in controlling the drug release at higher concentration of Span 80.
Effect of Stirring Speed During Secondary Emulsification
The impact of agitation during final emulsification on the production of drug-loaded microspheres was studied. The change in agitation speed from 700 to 1,300 rpm was associated with decrease in percentage yield of the microspheres (Table I). This trend continued in drug entrapment efficiency as well as in mean diameter of the microspheres (Table I). The increased stress generated in the emulsion with an increase in speed of mechanical agitation tended to divide the droplets of the emulsion and finally resulted in small particles. The formation of smaller emulsion droplets facilitated drug diffusion out of the microspheres before they harden and ensured lower drug entrapment efficiency (9). When the agitation speed was set at 1,300 rpm, a little change in sphericity was observed. A surface structure with some troughs was seen scattered over the microspheres (Fig. 2c). However, the microspheres maintained their spherical shape and smooth surface characteristics at lower speeds. The variation in mechanical speed affected the drug release behaviors in both acidic and alkaline dissolution media (Fig. 3c). Irrespective of the dissolution media, the drug release rates were always faster from the microspheres prepared at higher shear rates. In acidic medium, the microspheres released about 55.51% of entrapped drug in 2 h when prepared at 1,300 rpm. The same were found 39.31% and 20.49% at the intermediate and lowest speeds, respectively. In pH 7.4 PBS solution, the microspheres prepared at 1,300 rpm exhibited an initial booster release phase (releasing 39.09% drug in 1 h) followed by a slower one. Further, at high shear conditions, the drug release mechanism deviated to non-Fickian transport (Table I).
Effect of Internal Aqueous Phase Volume
An increase in volume of the internal phase of primary emulsion produced a lower yield of the microspheres. Parikh et al. (7) obtained a similar result with 5-flurouracil-loaded PLGA microspheres. Increase in volume of internal aqueous phase caused only a negligible increase in mean diameter of the microspheres (Table I). The droplet size of the primary emulsion may increase with aqueous phase volume of the primary emulsion, which in turn slightly increased the mean particle diameter. Similar observations have been reported (28,29). Scanning electron microscopy revealed that the variation in internal aqueous phase volume did not influence the spherical shape of the microspheres (micrographs not shown). An increasing tendency in drug entrapment efficiency (from 28.17% to 77.26%) was noted as a function of internal aqueous phase volume (Table I). However, this finding was just opposite to the earlier reports (26,30,31). The microspheres prepared with higher internal phase volume released its content at a much faster rate than those prepared with lower internal phase volume in both the dissolution media (Fig. 3d). Crotts and Gwan Park (32) reported that with an increase in volume of inner aqueous phase in the primary emulsion, the porosity of the microspheres increased and contributed to the faster drug release rates associated with the use of higher internal aqueous phase volume. The volume of internal phase of primary emulsion had no apparent effect on the release mechanism. The values of diffusional exponent (Table I) indicated that the drug release from all the microspheres could be explained by Fickian diffusion mechanism.
Effect of Tween 80 in External Aqueous Phase
The use of Tween 80 as an emulsion stabilizer in the external aqueous phase allowed higher fluconazole entrapment (77.26%) at a concentration of 0.6%. The concentrations below and above 0.6% did not improve drug entrapment efficiency of the microspheres. The production yield was gradually increased from 78% to 93% with increasing concentration of Tween 80 (Table I). Particle mean diameter also considerably altered as a function of surfactant concentration in the external phase. When the concentration was increased, a significant reduction in particle size was noticed (Table I) and the size distribution was shifted from bimodal to unimodal pattern (data not shown). At higher surfactant concentration, the rate at which the emulsion stabilizer molecules diffused at the primary emulsion droplets/external aqueous phase interface was probably higher and localized at the surface of emulsion droplets. This could provide a better protection of droplets from coalescence and ultimately resulted in formation of smaller emulsion droplets at higher surfactant concentrations. As the solvents evaporated form the system, these droplets hardened to form the microspheres. Therefore, the size of the finally obtained microspheres became dependent upon the size and stability of emulsion droplets formed during the agitation process (33). At low surfactant concentration, small emulsion droplets were not stable and the resulting microspheres were larger in size than those prepared with higher surfactant concentration. While the microspheres retained their spherical shape at a concentration well below 1%, however, a slight change in their sphericity and surface characteristics was observed at 1% (Fig. 2d). Figure 3e illustrated that as the surfactant concentration in the continuous phase was increased, the drug release rate from the microspheres tended to be slower in alkaline dissolution medium. A statistically significant difference (p < 0.05) existed in their mean t50% values (Table I). All formulations were capable of retarding drug release below 27.23% for 2 h in pH 1.2 HCl solution. The drug diffusion from the microspheres was non-Fickian, however, became Fickian with the decrease in concentration of Tween 80 from 1.0% to 0.3% (Table I).
Effect of External Aqueous Phase Volume
Increase in volume of the external aqueous phase from 60 to 140 ml resulted in a decrease in yield of microspheres (Table I). The decrease in yield may be attributed to a higher rate of drug leaching from the internal phase of the primary emulsion to the external phase of the secondary emulsion during preparation of the microspheres. With large volumes, a decrease in mixing efficiency may be anticipated due to reduction in agitation and finally, the particle size of the microspheres was increased (Table I). A reduction in mixing efficiency probably produced an increase in the size of emulsion droplets which could form large microspheres (22). No effect of external phase volume was evident on the gross morphology of the microspheres (Fig. 2e). An increase in volume of the external aqueous phase decreased the drug entrapment efficiency of the microspheres (Table I). The emulsion droplets probably more freely moved in the medium and the drug was extracted at a higher rate by the external phase of secondary emulsion and lowered the drug entrapment efficiency at higher external phase volume. Schlicher et al. (28) reported a similar decrease in drug encapsulation efficiency of the microspheres. The differences between release profiles of drug-loaded microspheres in acidic and alkaline dissolution media with increasing external phase volume has been presented in Fig. 3f. Faster drug release profile was observed in alkaline dissolution medium for the microspheres prepared using 60 ml of external water phase, followed by 100 and 140 ml. At lower phase volume, the t50% value was decreased (Table I). The variation in external phase volume introduced a statistically significant difference in their mean t50% values. However, the drug release patterns were more or less similar in acidic dissolution medium. In comparison to alkaline medium for a period of 2 h, the drug release was less in acidic medium and varied between 19.06% and 25.10%. In order to simulate the gastrointestinal environment, the microspheres prepared using 60 ml of external aqueous phase were transferred into alkaline medium (PBS, pH 7.4) following dissolution in pH 1.2 HCl solution for 2 h (Fig. 3f). The change of the medium from acidic to alkaline was associated with a burst release of drug (releasing about 20% drug within half an hour). Such pH-dependent release behavior could be explained by their pH-dependent swelling properties. As indicated in Fig. 4, the swelling ratio of the microspheres was comparatively higher in alkaline medium, and higher drug release could be expected in alkaline dissolution medium. Moreover, when the blank microspheres were transferred into alkaline medium, there was a sudden increase in swelling ratio from 2.11 to 4.86 within half an hour. Hence, the increased swelling of the microspheres may be accounted for a burst drug release of 20% following change of the dissolution medium. The release behavior from microspheres conformed best to Korsmeyer–Peppas semiempirical model and the drug release was Fickian type irrespective of external phase volume (Table I). Fickian drug transport is usually predominated by a matrix-controlled diffusional process, where tortuosity and porosity of the diffusional path controlled the drug release. Surface analysis of the drug-loaded microspheres by scanning electron microscopy before and after dissolution study revealed a large number of pores and supported micropore diffusion controlled drug release mechanism (Fig. 2f–h).
Fig. 4.

Swelling study of the microspheres prepared using 60 ml external aqueous phase volume. Triangle pH 7.4 PBS solution; square pH 1.2 HCl solution (2 h) followed by pH 7.4 PBS solution (3 h)
The influence of a number of processing variables has been critically reviewed and it was found that as high as 80.03% fluconazole entrapment efficiency could be achieved only with an external aqueous phase to internal aqueous phase volume ratio of 5:1. Of all the formulation variables studied, the smallest particle size and a slower uniform drug release were obtained under above processing condition. The values of Carr’s index (11.13%) and Hausner ratio (1.13) were also within limits to indicate good flow property of these microspheres. The microspheres prepared under this condition were considered best and hence, further clarifications were done with this formulation. The effect of aging on drug dissolution was studied following storage of these microspheres in closed amber glass container in a desiccator for 3 months at room temperature. For comparison, t50% value (h) was calculated and found to be 4.87 ± 0.15 and 5.27 ± 0.42, respectively before and after aging of the microspheres in pH 7.4 PBS solution (Fig. 5). On the other hand, the percentage drug release in 2 h was considered as a parameter for comparison of the dissolution rate in pH 1.2 HCl solution and was found to be 26.42 ± 0.08 and 24.43 ± 1.55, respectively, for the freshly prepared and aged microspheres (Fig. 5). Therefore, it was evident that the drug release pattern was slightly slower in both dissolution media; however, there were no statistically significant difference in drug release before and after aging (p > 0.05). Also, the drug release mechanism in pH 7.4 PBS solution was negligibly affected by aging because the calculated n values before (0.2847) and after (0.2772) aging were very close to each other.
Fig. 5.

Effect of aging on the dissolution behaviors in different dissolution media for the microspheres prepared with an external to internal phase volume ratio of 5:1. Release study in pH 1.2 HCl solution: closed triangle freshly prepared microspheres; closed square aged microspheres. Release study in pH 7.4 PBS solution: open triangle freshly prepared microspheres; open square aged microspheres
The physical state of drug in these microspheres was examined by X-RD and DSC analysis. The important crystallographic characteristics of fluconazole were observed at scattering angles of 15.770°, 16.400°, 17.030°, 21.470°, 24.260°, and 25.880° with their associated d values of 5.6147, 5.4004, 5.2020, 4.1352, 3.6656, and 3.4397, respectively (Fig. 6a) (34). The respective signal intensities were in the order of 522, 248, 1,547, 399, 537, and 950 cps. Conversely, the physical mixture of drug/excipients showed very weak signals owing to some drug crystals at scattering angles of 16.670°, 20.180°, and 20.630° with their respective intensities of 43, 56, and 27 cps (Fig. 6b). Even the fluconazole-loaded microspheres showed very weak signals at scattering angles of 21.530° and 21.980° with their respective intensities of 62 and 68 cps (Fig. 6c). Therefore, it was realized that the degree of crystallinity of fluconazole reduced in the physical mixture as well as in the microspheres. DSC thermogram of pure fluconazole showed a clear endothermic peak associated with crystal melting at a temperature of 140.1°C (Fig. 7a). However, the DSC thermogram of fluconazole-loaded ethyl cellulose microspheres showed a wide weak melting endotherm at temperature between 150°C and 200°C apart from a broad signal around 45–60°C (Fig. 7b). The amorphous blank polymer did not show any fusion peak or phase transition, however, showed a broad signal around 45–60°C due to a partial loss of residual humidity (Fig. 7c). The thermal behavior indicated that the polymers probably inhibited the melting of drug crystals close to its reported melting point (140.1°C) (35). The thermal behavior coupled with the X-ray crystallographic data suggested that the drug was able to disperse almost homogenously in the microspheres. As the drug is slightly water soluble, a slight excess drug was not able to form a homogeneous solid solution with the polymer at a polymer/drug weight ratio of 1:0.08. The distribution of drug substance in different sieve fractions (sieve nos. 44, 60, and 85) of polymer particles was evaluated by calculating drug entrapment efficiencies of these fractions. It was observed that the entrapment efficiencies were in between 77.12% and 82.05% suggesting identical distribution of drug in different sieve fractions. Another point of interest was the influence of physical interaction between drug and excipients on drug dissolution behavior. It was observed that the dissolubility of free drug was affected by pH of the dissolution medium. While the drug dissolubility in acidic medium was 14.12%, the same was found 8.81% in alkaline dissolution medium in 0.5 h (Fig. 8). This is expected because the drug is weakly basic in nature. Within a same timeframe, the drug dissolubility from the physical mixture was 18.73% in acidic medium. It was clear that the dissolubility of drug from the physical mixture increased slightly in the acidic dissolution medium. Because sodium alginate and ethyl cellulose remained insoluble in the acidic dissolution medium, the improved drug dissolubility could be attributed to the relative decrease in size of the drug crystals (36). On the other hand, the drug dissolubility from the physical mixture was higher (33.91%) in alkaline medium than in acidic dissolution medium (Fig. 8). These anomalies could be explained as follows: Firstly, the size of the drug crystals decreased relatively in the physical mixture, and secondly, the hydrocolloid sodium alginate became ionized/hydrated in alkaline dissolution medium and probably facilitated faster dissolution of the slightly water soluble drug from the mixture by forming a water soluble film around the drug particle. Hence, it could be suggested that the physical interaction between drug and polymers affected the drug dissolution behaviors depending upon the pH of the dissolution medium.
Fig. 6.

X-ray diffraction pattern of a pure fluconazole, b physical mixture (w/w) of fluconazole and excipients (0.08:1), and c fluconazole-loaded microspheres
Fig. 7.

DSC thermograms of a pure fluconazole, b fluconazole-loaded microspheres, and c blank microspheres
Fig. 8.

Influence of physical interaction between drug and excipients on drug dissolution behaviors in different dissolution medium. Dissolubility in pH 1.2 HCl solution: closed triangle free drug; closed square physical mixture. Dissolubility in pH 7.4 PBS solution: open triangle free drug; open square physical mixture
The compatibility of fluconazole in this formulation was evaluated qualitatively through FTIR analysis. In the FTIR spectrum of pure fluconazole (Fig. 9a), the frequencies associated with C–O stretching vibration consistent with a tertiary alcohol, aromatic C–F stretching vibrations, aromatic C=C stretching vibrations, and aromatic C–H stretching vibrations were identified at 1,140.06, 1,275.92, 1,619.38, and 3,117.11 cm−1, respectively (34). Similar vibrational peaks of fluconazole were detected in the spectrum of fluconazole-loaded microspheres with minor differences in frequencies (Fig. 9b). This suggested that the drug was apparently stable in the microspheres. To evaluate the drug stability quantitatively in this formulation, the sample stored at 40°C/75%RH for 3 months was analyzed for drug entrapment efficiency. The drug entrapment efficiency of this formulation decreased from 80.03 ± 1.63(%) to 78.24 ± 1.86 (%) after 3 months of storage in this condition. Although the entrapment efficiency slightly decreased, the difference was statistically insignificant (p > 0.05). The characteristics λmax of pure fluconazole also appeared at 261 nm on the ultraviolet spectra of fluconazole, extracted from these microspheres in pH 7.4 PBS solution.
Fig. 9.

FTIR spectra of a pure fluconazole, b fluconazole-loaded ethyl cellulose microspheres, and c blank ethyl cellulose microspheres
Finally, this formulation was evaluated for its in vitro antifungal activity. For the assessment of a possible medical application, it is important to confirm the nature of the compound released from the microspheres as fluconazole. The release of functionally active fluconazole from the optimized formulation was confirmed by its effectiveness to inhibit the fungal colony of C. albicans. This was tested by spread plate method, in order to establish the minimum inhibitory concentration (MIC) at which the extracted fluconazole could prevent the fungal growth. The reported MIC50 value of fluconazole against C. albicans was 0.5 μg/ml and MIC ranges from 0.125 to >64 μg/ml (37). Although the MIC50 value obtained with the fluconazole-loaded microspheres was comparatively higher (5.69 μg/ml), the lowest concentration (1.29 μg/ml) at which a definite decrease in colony count was observed was in good agreement with the reported MIC range and even this concentration could be achieved within half an hour following dissolution of the microspheres. A good correlation (r2 = 0.9502) existed between the concentration of fluconazole released (as a function of time) and the percentage inhibition (Fig. 10). This suggested that the optimized formulation possessed good in vitro antifungal activity and may have a therapeutic potential in the treatment of systemic fungal infections in humans.
Fig. 10.

In vitro antifungal activity of fluconazole-loaded microspheres prepared using 60 ml of external aqueous phase
CONCLUSION
This study suggested that the use of aqueous sodium alginate solution as an internal phase in conventional double emulsion solvent evaporation technique may be an alternative approach for the successful incorporation of slightly water soluble drugs in ethyl cellulose microspheres. This investigation has also provided an understanding of the effects of some processing parameters on particle size and release characteristics of the microspheres. Proper control of the processing variables allowed the entrapment of as high as 80% fluconazole in the microspheres and enabled the preparation of smooth, spherical, free-flowing microspheres having a size range between 228 and 592 μm. The microspheres were able to provide drug release for an extended period of time (8 h or more) in pH 7.4 PBS solution. The drug release in pH 1.2 HCl solution could also be controlled by selection of the appropriate conditions. The systematic investigation of various process variables helped to conclude that a higher level of fluconazole entrapment could be attained in the ethyl cellulose microspheres, when prepared with an external aqueous phase to internal aqueous phase volume ratio of 5:1. The drug release from these microspheres was predominantly controlled by Fickian-diffusion mechanism and the mechanism did not differ significantly even after aging. X-RD and DSC analysis indicated that most of the entrapped drug existed in amorphous form and distributed homogeneously in these microspheres. The drug distribution in different sieve fractions of polymer particles was more or less identical. However, the physical interaction of drug with excipients increased the drug dissolubility greatly in alkaline dissolution medium. Both qualitative and quantitative analysis demonstrated that the drug was stable in these polymeric microspheres. The microspheres with high entrapment levels of fluconazole (80.03%) showed a good in vitro antifungal activity and hence could be effective in the oral treatment of systemic fungal infections. Therefore, it was inferred that proper control of the processing variables involved in this modified technology could allow effective incorporation of slightly water soluble drugs into ethyl cellulose microspheres. In addition, this technology had a potential to replace the more simple process technologies for the effective incorporation of drugs of this class into ethyl cellulose microspheres.
Acknowledgments
The authors are grateful to the authority of Gupta College of Technological Sciences, Asansol, India and Jadavpur University, Department of Pharmaceutical Technology, Kolkata, India for providing necessary facilities for this work.
References
- 1.Follonier N, Doelkar E. Biopharmaceutical comparison of oral multiple unit and single unit sustained release dosage forms. STP Pharm Sci. 1992;2:141–58. [Google Scholar]
- 2.Davis SS, Hardy JG, Taylor MJ, Whalley DR, Wilson CG. A comparative study of the gastrointestinal transit of a pellet and tablet formulation. Int J Pharm. 1984;21:167–77. doi: 10.1016/0378-5173(84)90091-7. [DOI] [Google Scholar]
- 3.Hincall AA, Calis S. Microsphere preparation by solvent evaporation method. In: Wise DL, editor. Handbook of pharmaceutical controlled release technology. Cambridge, MA: Taylor and Francis; 2000. pp. 329–43. [Google Scholar]
- 4.Sanadrap P, Moes AJ. Influence of manufacturing parameters on the size characteristics and release properties of nifedipine from poly (dl-lactide-co-glycolide) microspheres. Int J Pharm. 1993;98:157–64. doi: 10.1016/0378-5173(93)90052-H. [DOI] [Google Scholar]
- 5.Arshady R. Microspheres and microcapsules, a survey of manufacturing techniques. III. Solvent evaporation. Polym Eng Sci. 1990;30:915–24. doi: 10.1002/pen.760301506. [DOI] [Google Scholar]
- 6.Ogawa Y, Yamamoto M, Okada H, Yashiki T, Shimamoto T. A new technique to efficiently entrap leuprolide acetate into microcapsules of poly lactic acid of copolylactic/glycolic acid. Chem Pharm Bull. 1988;36:1095–103. doi: 10.1248/cpb.36.1095. [DOI] [PubMed] [Google Scholar]
- 7.Parikh RH, Parikh JR, Dubey RR, Soni HN, Kapadia KN. Poly (D, L-lactide-co-glycolide) microspheres containing 5-fluorouracil: optimization of process parameters. AAPS PharmSciTech. 2003;4:14–21. doi: 10.1208/pt040213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sipos P, Csóka I, Srčič S, Pintye-Hódi K, Erõs I. Influence of preparation conditions on the properties of Eudragit microspheres produced by a double emulsion method. Drug Dev Res. 2005;64:41–54. doi: 10.1002/ddr.10425. [DOI] [Google Scholar]
- 9.Alex R, Bodmeier R. Encapsulation of water soluble drugs by a modified solvent evaporation method. I. Effect of process and formulation variables on drug entrapment. J Microencapsul. 1990;3:347–55. doi: 10.3109/02652049009021845. [DOI] [PubMed] [Google Scholar]
- 10.Nagareyan N, Uchida T, Matsuyama K. Preparation and characterization of enteric microspheres containing bovine insulin by a w/o/w emulsion solvent evaporation method. Chem Pharm Bull. 1998;46:1613–7. doi: 10.1248/cpb.46.1613. [DOI] [PubMed] [Google Scholar]
- 11.Gibaly-El I, Safwat SM, Ahmed MO. Microencapsulation of ketoprofen using w/o/w complex emulsion technique. J Microencapsul. 1996;13:67–87. doi: 10.3109/02652049609006804. [DOI] [PubMed] [Google Scholar]
- 12.J.S. Young, K.I. Chan, and K. Yong-Hee. Preparation method for biodegradable polymeric microspheres using solvent extraction and preparation method for microspheres for treating local inflammation using the same. US Patent no. 6149944 (2000).
- 13.Takada S, Yamagata Y, Masaki M, Taira K, Kurokawa T. Sustained release of human growth hormone from ms prepared by a solvent evaporation technique. J Control Rel. 2003;88:229–42. doi: 10.1016/S0168-3659(02)00494-7. [DOI] [PubMed] [Google Scholar]
- 14.Yeh MK, Chen JL, Chiang CH. In vivo and in vitro characteristics for insulin-loaded PLA microparticles prepared by w/o/w solvent evaporation method with electrolytes in the continuous phase. J Microencapsul. 2004;21:719–28. doi: 10.1080/02652040400008481. [DOI] [PubMed] [Google Scholar]
- 15.Gibaud S, Gaia A, Astier A. Slow-release melarsoprol microparticles. Int J Pharm. 2002;243:161–6. doi: 10.1016/S0378-5173(02)00278-8. [DOI] [PubMed] [Google Scholar]
- 16.Mandal TK, Shekleton M, Onyebueke E, Washington L, Penson T. Effect of formulation and processing factors on the characteristics of biodegradable microcapsules of zidovudine. J Microencapsul. 1996;13:545–57. doi: 10.3109/02652049609026040. [DOI] [PubMed] [Google Scholar]
- 17.Uetrecht J, Walmsley SL. Antimicrobial and antifungal agents that act on cell membrane. In: Kalant H, Roschlau WHE, editors. Principles of medical pharmacology. New York: Oxford University Press; 1998. p. 677. [Google Scholar]
- 18.Parrott EL. Milling. In: Lachman L, Lieberman HA, Kanig JL, editors. The theory and practice of industrial pharmacy. Washington Square, Philadelphia: Lea & Febiger; 1991. p. 28. [Google Scholar]
- 19.Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35. doi: 10.1016/0378-5173(83)90064-9. [DOI] [PubMed] [Google Scholar]
- 20.Ritger PI, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Control Rel. 1987;5:37–42. doi: 10.1016/0168-3659(87)90035-6. [DOI] [PubMed] [Google Scholar]
- 21.Jeffery H, Davis SS, O’Hagan DT. The preparation and characterization of poly (lactide-co-glycolide) microparticles. I. Oil-in-water emulsion solvent evaporation. Int J Pharm. 1991;77:169–75. doi: 10.1016/0378-5173(91)90314-E. [DOI] [Google Scholar]
- 22.Benoit MA, Baras B, Gillard J. Preparation and characterization of protein loaded poly (ε-caprolactone) microspheres for oral vaccine delivery. Int J Pharm. 1999;184:73–84. doi: 10.1016/S0378-5173(99)00109-X. [DOI] [PubMed] [Google Scholar]
- 23.Rafati H, Coombes AGA, Adler J, Holland J, Davis SS. Protein-loaded poly (dl-lactide-co-glycolide) microparticles for oral administration: formulation, structural and release characteristic. J Control Rel. 1997;43:89–102. doi: 10.1016/S0168-3659(96)01475-7. [DOI] [Google Scholar]
- 24.Sah HK, Toddywala R, Chien YW. The influence of biodegradable microcapsule formulations on the controlled release of a protein. J Control Rel. 1994;30:201–11. doi: 10.1016/0168-3659(94)90026-4. [DOI] [Google Scholar]
- 25.Melzer E, Kreuter J, Daniels R. Ethylcellulose: a new type of emulsion stabilizer. Eur J Pharm Biopharm. 2003;56:23–7. doi: 10.1016/S0939-6411(03)00025-0. [DOI] [PubMed] [Google Scholar]
- 26.Schügers C, Larucelle N, Wihant N, Grandfils CH. Effect of emulsion stability on the morphology and porosity of semicrystalline poly-1-lactide microparticles prepared by w/o/w double emulsion evaporation. J Control Rel. 1994;32:161–76. doi: 10.1016/0168-3659(94)90055-8. [DOI] [Google Scholar]
- 27.Jiao YY, Ubrich N, Hoffart V, Marchand-Arvier M, Vigneron C, Hoffman M, Maincent P. Preparation and characterization of heparin-loaded polymeric microparticles. Drug Dev Ind Pharm. 2002;28:1033–41. doi: 10.1081/DDC-120014740. [DOI] [PubMed] [Google Scholar]
- 28.Schlicher JAM, Postma NS, Zuidema J, Talsma H, Hennik WE. Preparation and characterization of poly (D, L-lactic-co-glycolic acid) microspheres containing desferrixamine. Int J Pharm. 1997;153:235–45. doi: 10.1016/S0378-5173(97)00116-6. [DOI] [Google Scholar]
- 29.Jeffery H, Davis SS, O’Hagan DT. Preparation and characterization of poly (lactide-co-glycolide) microparticles: II: entrapment of a model protein using a (water in oil) in water emulsion solvent evaporation technique. Pharm Res. 1993;10:362–8. doi: 10.1023/A:1018980020506. [DOI] [PubMed] [Google Scholar]
- 30.Leo E, Pecquet S, Rojas J, Couvruer P, Fattal E. Changing the pH of the external aqueous phase may modulate protein entrapment and delivery from poly (lactide-co-glycolide) microspheres prepared by w/o/w solvent evaporation method. J Microencapsul. 1998;15:421–30. doi: 10.3109/02652049809006869. [DOI] [PubMed] [Google Scholar]
- 31.Yeh MK, Tung SM, Lu DW, Chiang CH. Formulation factors for preparing ocular biodegradable delivery system of 5-fluorouracil microparticles. J Microencapsul. 2001;18:507–19. doi: 10.1080/02652040010018100. [DOI] [PubMed] [Google Scholar]
- 32.Crotts G, Gwan Park T. Protein delivery from poly (lactic-co-glycolic acid) biodegradable microspheres: release kinetics and stability issues. J Microencapsul. 1998;15:699–713. doi: 10.3109/02652049809008253. [DOI] [PubMed] [Google Scholar]
- 33.Blanco-Prieto MJ, Delie F, Fattal E, Tartar A, Puisieux F, Gulik A, Couvreur P. Characterization of V3 BRU peptide-loaded small PLGA microspheres prepared by a (w1/o/w2) emulsion solvent evaporation method. Int J Pharm. 1994;111:137–45. doi: 10.1016/0378-5173(94)00104-9. [DOI] [Google Scholar]
- 34.Dash AK, Elmquist WF. Fluconazole. In: Brittain H, editor. Analytical profiles of drug substances and excipients. San Diego: Academic; 2001. pp. 67–113. [Google Scholar]
- 35.Pignatello R, Spadaro D, Vandelli MA, Forni F, Puglisi G. Characterization of the mechanism of interaction in ibuprofen–eudragit RL100® coevaporates. Drug Dev Ind Pharm. 2004;30:277–88. doi: 10.1081/DDC-120030421. [DOI] [PubMed] [Google Scholar]
- 36.Kim K-H, Kim K-S, Lim J-S, Shin J-S, Chung K-H. Effect of dissolution properties of p-aminosalicylic acid with chitin and chitosan mixtures. Polymer (Korea) 1988;12:56–62. [Google Scholar]
- 37.Carrillo-Munoz AJ, Quindos G, Tur C, Ruesga MT, Miranda Y, delValle O, Cossum PA, Wallace TL. In-vitro antifungal activity of liposomal nystatin in comparison with nystatin, amphotericin B cholesteryl sulphate, liposomal amphotericin B, amphotericin B lipid complex, amphotericin B desoxycholate, fluconazole and itraconazole. J Antimicrob Chemother. 1999;44:397–401. doi: 10.1093/jac/44.3.397. [DOI] [PubMed] [Google Scholar]