

Abstract
The present study demonstrates feasibility of preparation of nanoparticles using a novel polymer, polyethylene sebacate (PES), and its application in the design of drug-loaded nanocarriers. Silymarin was selected as a model hydrophobic drug for the present study. Two methods of preparation, viz., nanoprecipitation and emulsion solvent diffusion, were evaluated for preparation of nanoparticles. Effect of surfactants polyvinyl alcohol (PVA), lutrol F 68, and Tween 80 on the preparation of blank and silymarin-loaded PES nanoparticles was evaluated. Nanoprecipitation resulted in the formation of nanoparticles with all the surfactants (<450 nm). Increase in surfactant concentration resulted in decrease in entrapment efficiency and particle size except with PVA. The type and concentration of surfactant was critical to achieve low size and adequate drug entrapment. While increase in concentration of PES resulted in larger nanoparticles, inclusion of acetone in the organic phase resulted in particles of smaller size. In case of emulsion solvent diffusion, nanoparticles were obtained only with lutrol F 68 as surfactant and high surfactant concentration. The study revealed nanoprecipitation as a more versatile method for preparation of PES nanoparticles. Scanning electron microscopy studies revealed spherical shape of nanoparticles. Freeze-dried nanoparticles exhibited ease of redispersion, with a marginal increase in size. Differential scanning calorimetry and X-ray diffraction analysis revealed amorphous nature of the drug. The study demonstrates successful design of PES nanoparticles as drug carriers.
Key words: biodegradable polymer, emulsion solvent diffusion, nanoparticles, nanoprecipitation, silymarin
INTRODUCTION
Synthetic biodegradable polymers most investigated for drug delivery systems include lactide/glycolide polymers, polylactide, polyglycolide, copolymer of lactide, and glycolide referred to as poly (lactide-co-glycolide), polyanhydrides, polycaprolactones, polyorthoesters, and polyphosphazones (1,2).
Families of polyesters of diols and diacids with even number of carbon atoms are well known as being biodegradable and nontoxic to living animals (3). These polyesters are degraded by the mechanism of fat degradation in humans or animals and hence can be used for wide pharmaceutical applications. We have recently reported synthesis and characterization of polyethylene sebacate (PES) from ethylene glycol and sebacic acid by conventional condensation and trans-esterification reaction. Enzymatic degradation studies with lipase revealed PES as biodegradable, while toxicity studies as per Organization for Economic Cooperation and Development guidelines confirmed PES as nontoxic (4). PES is also found to be nongenotoxic and nonmutagenic (5).
Biodegradable polymeric nanoparticles have been widely investigated for enhancing bioavailability of poorly soluble drugs, controlled release of drugs, protection of drugs especially peptides and protein from degradation, targeted delivery of drugs, as carriers for intracellular drug delivery, and for coating stents (1,6–8). Development of PES as nanoparticles would open up application of PES for a whole range of drug delivery applications. The present study therefore explores the feasibility of design of PES nanoparticles for application as drug carrier.
Two methods were evaluated for preparation of PES nanoparticles, nanoprecipitation, and emulsion solvent diffusion. Nanoprecipitation is a straightforward, rapid, and easy to perform one-step procedure for preparation of nanoparticles using water-miscible solvents. Nanoparticle formation is spontaneous with nanoparticles of small size and unimodal distribution (9–13). Additionally, the method requires no or low surfactant concentration (10). Emulsion solvent diffusion utilizes partially water-miscible solvents. Initially, an oil-in-water emulsion is formed in the presence of stabilizer or surfactants, and subsequent addition of water will allow diffusion of solvent, causing precipitation of polymer as nanoparticles (8,14–17). Though it is a two-step procedure, it is reported to be a versatile and efficient method allowing precise size control and higher entrapment of lipophilic drugs (10,16).
PES being a new polymer, comparison of methods of preparation will enable design of PES nanoparticles with optimum physicochemical characteristics. The data generated in the course of the study would form the basis for further nanoparticle preparation using PES as polymer for a range of drugs. The objective of the present study includes evaluation of the feasibility of preparation of PES nanoparticles and their application in the preparation of drug-loaded carriers. Silymarin was selected as a model hydrophobic drug for the present study. Low aqueous solubility and poor bioavailability of silymarin (18,19) make it a suitable candidate for design into nanoparticulate drug delivery systems.
MATERIALS AND METHODS
Materials
Silymarin was provided by Vav Life Sciences India; polyethylene sebacate (molecular weight 9,625) was synthesized in our laboratory (4,5). Polyvinyl alcohol (PVA) and polyoxyethylene-polyoxypropylene block-co-polymer (lutrol F 68), were obtained from Colorcon Asia Pvt Ltd and BASF India, respectively. Polyoxyethylene sorbitan monooleate (Tween 80), tetrahydrofuran (THF), and methyl ethyl ketone (MEK) were purchased from S.D. Fine Chemicals, India. Distilled water was used throughout the experiments. All the other chemicals and reagents were either spectroscopic or analytical grade.
Nanoparticle Preparation
Nanoprecipitation
Briefly, PES (125 mg) was dissolved in THF (10 mL) and added dropwise to the aqueous phase (25 mL) comprising surfactant in water under continuous stirring. The surfactants selected for the study included PVA, lutrol F 68, and Tween 80. The resulting dispersion was stirred for 4 h to allow complete evaporation of organic solvent. In case of drug-loaded nanoparticles, silymarin (25 mg) was dissolved in THF.
Emulsion Solvent Diffusion
PES (125 mg) was dissolved in MEK (10 mL). This organic phase was added under stirring to aqueous phase (10 mL) containing surfactant. The surfactants screened for the study included PVA, lutrol F 68, and Tween 80. The resulting oil-in-water emulsion was stirred for a period of 5 min. Furthermore, 40 mL of water was introduced so as to allow diffusion of MEK into the external aqueous phase leading to the formation of nanoparticles. The dispersion was stirred for 4 h to allow complete evaporation of the organic solvent. In case of drug-loaded nanoparticles, silymarin (25 mg) was dissolved in MEK.
Entrapment Efficiency
Nanoparticles were separated from dispersion by centrifugation (Eltek 4100 D Research Centrifuge) at 15,000 rpm for 30 min. The supernatant obtained after centrifugation was suitably diluted and analyzed for free silymarin by UV spectrophotometry (Shimadzu, Japan) at 288 nm. The percent entrapment efficiency was calculated as follows 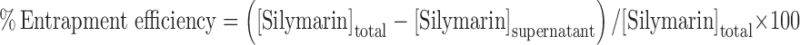 .
.
Equilibrium Solubility of Silymarin in Surfactant Solution
Excess of silymarin was added to the surfactant solutions (0.1%, 0.5%, 4% w/v) with intermittent sonication and vortexing. After 24 h, solutions were centrifuged and the supernatant analyzed for silymarin by UV spectrophotometry (Shimadzu, Japan) at 288 nm.
Particle Size Analysis
Particle size was determined by photon correlation spectroscopy using N4 plus submicron particle size analyzer (Beckman Coulter) at 25°C. All the measurements were taken by scattering the light at 90°. The nanoparticle dispersion was sonicated using probe sonicator (Vibronic India). The particle size was determined by diluting the nanoparticle dispersion with water to obtain final counts per second (Intensity), 5 × 104 to 1 × 106. The water used for dilution was filtered through 0.22 μm membrane filter. Freeze-dried nanoparticles were dispersed with sonication prior to particle size determination.
Viscosity
Viscosities of the aqueous phases were determined by capillary tube viscometer (Oswald viscometer) at 25°C using water as the reference (viscosity of water = 0.008 centipoise). Each measurement was made in triplicate.
Surface Tension
Surface tension of the aqueous phases was determined by Stalagmometer at 25°C using water as the reference (surface tension of water = 72 dyne/cm). Each measurement was made in triplicate.
In Vitro Drug Release
The release of silymarin from the nanoparticles prepared by nanoprecipitation was determined by dialysis bag method using USP dissolution apparatus I, in phosphate buffer pH 7.4(900 mL), at 50 rpm and 37 ± 0.5°C. Drug-loaded nanoparticles corresponding to 10 mg of silymarin were filled in a dialysis bag (HIMEDIA®, Molecular weight cutoff 12,000–14,000 Da) which was placed in the basket of the USP dissolution apparatus I. An aliquot of 5 ml was withdrawn at suitable time intervals and replaced with the same amount of medium. The samples were analyzed for silymarin by UV spectrophotometer at 326.5 nm.
Scanning Electron Microscopy
A drop of nanoparticle dispersion was deposited on an aluminum grid and dried in vacuum. The samples were analyzed using the scanning electron microscope equipped with a field emission, a JEOL JSM 6380 after sputtering the sample with platinum using a coater JEOL JSM 1600.
Freeze-Drying
Nanoparticle pellet obtained after centrifugation was dispersed in solutions containing different concentrations of lutrol F 68 as stabilizer. Trehalose 5% w/v was used as cryoprotectant. The samples were lyophilized (REVA-03, Ref-Vac Consultancy, Vadodara, India) for 36 h at a temperature of 25°C and vacuum of 0.23 mbar.
Differential Scanning Calorimetry
Powdered samples were accurately weighed (5 mg) in aluminum pans, sealed, and subjected to differential scanning calorimetry (DSC) under nitrogen flow using a Perkin Elmer Pyris 6 DSC thermal analysis instrument. Thermograms were recorded by heating samples from 35°C to 250°C at a heating rate of 10°C min−1 with empty aluminum pan as the reference.
X-Ray Diffraction
X-Ray diffraction (XRD) spectra of the formulations were recorded at room temperature using Philips Pro Expert diffractometer, with nickel filtered Cu Kα radiation operated at a voltage of 3 kV, 5 mA current, 4°/min scanning speed, and 5°–40° (2θ) range.
Statistical Analysis
All data in tables and the figures are expressed as mean ± standard deviation and mean ± standard error, respectively. Statistical analysis was performed using the one-way analysis of variance (ANOVA) with Tukey–Kramer honestly significant difference and Student’s t tests. p < 0.05 was the criterion for statistical significance.
RESULTS AND DISCUSSION
Nanoprecipitation
Nanoprecipitation technique developed and patented by Fessi et al. (9) involves addition of polymer in water-miscible organic solvent into large amount of nonsolvent, usually water. Diffusion of solvent into water facilitates formation of small nanoparticles and higher yield of transformed nanoparticles. Among the various solvents reported, THF and acetone are commonly employed solvents for nanoprecipitation (13). In our study, THF was selected as the organic phase as both silymarin and PES exhibited good solubility in THF.
Preparation of Blank Nanoparticles
Feasibility of preparation of nanoparticles of PES was evaluated using three surfactants, PVA, lutrol F 68, and Tween 80 at three different concentrations 0.1%, 0.5%, and 4% w/v of the aqueous phase. The organic/aqueous phase ratio was maintained at 0.4, and PES concentration was maintained at 12.5 mg/mL.
Reduction in surface tension due to surfactants allows formation of smaller droplet and thus small mean size of nanoparticles. Further addition of surfactants increases the viscosity of the aqueous phase promoting hydrodynamic stabilization by preventing coalescence and aggregation of droplets formed. This effect is more pronounced above the critical micelle concentration (CMC) of the surfactant. Above CMC, excess surfactant present in the bulk solution is available for droplet coverage resulting in better hydrodynamic stability and thereby lower particle size.
A sharp decrease in particle size from greater than 1,000 nm to around 300 nm (Fig. 1) was observed as PVA concentration increased from 0.1% to 0.5%. PVA exerts its stabilizing effect by adsorbing at the droplet interface, thus reducing surface tension and promoting mechanical and steric stabilization. Further increase in PVA concentration to 4% led to increase in particle size (p < 0.05) and was related to increase in viscosity. Increase in viscosity of external aqueous phase hinders effective diffusion of organic phase leading to larger droplet formed, thus increasing mean size. Similar results at higher PVA concentrations have been reported (12,14,20,21).
Fig. 1.

Effect of surfactant concentration and viscosity of aqueous phase on particle size of blank PES nanoparticles prepared using nanoprecipitation (mean ± SE, n = 3)
Lutrol F 68 and Tween 80 as surfactants enabled nanosize at all the concentrations evaluated. This is explained by the low CMC value of lutrol F 68 (0.1%) (22) and Tween 80 (0.0013%) (23). In general, at the same concentration, mean size of nanoparticles was in the order of PVA > lutrol F 68 > Tween 80. PES nanoparticles with size ranging from ∼200 to 330 nm were obtained by suitably manipulating surfactant concentration and type. The above data confirmed the feasibility of preparation of PES nanoparticles by nanoprecipitation.
Preparation of Silymarin-Loaded Nanoparticles
Effect of Surfactant Concentration and Type
Similar effect as observed with blank nanoparticles was seen with silymarin-loaded nanoparticles. Figure 2 illustrates the effect of surface tension of the aqueous phase on mean particle size of silymarin-loaded nanoparticles prepared by nanoprecipitation.
Fig. 2.

Effect of surface tension of the aqueous phase on mean size of nanoparticles prepared by nanoprecipitation technique (mean ± SE, n = 3)
A good correlation between particle size and surface tension was observed with lutrol F 68 (r2 = 0.911) and Tween 80 (r2 = 0.744). However, with PVA, such correlation was observed only up to 2% (r2 = 0.739). At higher PVA concentration, significant increase (p < 0.05) in particle size was observed as seen with blank nanoparticles. Mean size of nanoparticles was found to be in the order of PVA > lutrol F 68 > Tween 80, which was similar to the data obtained for blank nanoparticles.
Silymarin-loaded nanoparticles exhibited larger mean size compared to blank nanoparticles with PVA and Tween 80 (p < 0.05). The solute loading in the organic phase influences the nanoparticle size. The higher the solute concentration, the larger the size of nanoparticles formed (24).
Figure 3 shows effect of solubility of silymarin in the aqueous phase on entrapment efficiency of nanoparticles. It is apparent that entrapment efficiency of silymarin in nanoparticles was found to decrease significantly (p < 0.05) with increase in surfactant concentration and was in the order of PVA > lutrol F 68 > Tween 80. Higher aqueous solubility of silymarin at higher surfactant concentration favored increased partition into the nonsolvent phase thereby resulting in decrease in entrapment efficiency. Selection of surfactant type and concentration is therefore extremely crucial to strike a balance between low size and adequate drug loading.
Fig. 3.

Effect of silymarin solubility in the aqueous phase on entrapment efficiency of nanoparticles prepared by nanoprecipitation and emulsion solvent diffusion (mean ± SE, n = 3)
Effect of Polymer Concentration
The effect of polymer concentration was studied using lutrol F 68 as the surfactant at a concentration of 0.5%. As the polymer concentration in the organic phase increased, both particle size and entrapment efficiency increased (Fig. 4). Significant increase in entrapment efficiency (p < 0.001) of silymarin at higher polymer concentration can be related to availability of higher amount of polymer for entrapment. On the other hand, increase in particle size (p < 0.001) can be explained due to increased polymer–polymer interaction coupled with increase in viscosity of aqueous phase which prevents effective diffusion of solvent into aqueous phase thereby increasing the particle size (24).
Fig. 4.

Effect of polymer concentration on entrapment efficiency and particle size of silymarin nanoparticles prepared by nanoprecipitation method (mean ± SE, n = 3). Organic phase: THF + ACT (5 mL + 5 mL)/THF (10 mL)
Effect of Organic/Aqueous Phase Ratio
Figure 5 shows the effect of organic/aqueous phase ratio on particle size and entrapment efficiency of silymarin nanoparticles. A significant decrease in entrapment efficiency as well as particle size was observed when organic/aqueous phase ratio increased from 0.2 to 0.4 (p < 0.001). Further increase in ratio significantly affected neither entrapment efficiency nor the size (p > 0.05). Increased concentration of organic phase while promoting rapid diffusion of solvent into aqueous phase to obtain small size nanoparticles also favors partition of the drug in the aqueous phase to decrease the entrapment efficiency.
Fig. 5.

Effect of organic/aqueous phase ratio on entrapment efficiency and particle size of silymarin nanoparticles prepared by nanoprecipitation method (mean ± SE, n = 3; ***p < 0.001, ns nonsignificant p > 0.05 ANOVA followed by Tukey test)
Effect of Solvent Type
Solvents of high polarity like acetone are reported to form small size nanoparticles (13,25) by promoting rapid diffusion into the aqueous phase. Furthermore, the lower the dielectric constant of the solvent, the larger the size of nanoparticles (11). Since PES is not soluble in acetone, THF/acetone (1:1) was evaluated. Particle size decreased significantly (p < 0.05) when THF and acetone were used in combination as solvent compared to THF alone at all the polymer concentrations (Fig. 4). Rapid diffusion of the more polar solvent acetone into the nonsolvent phase favors the formation of smaller nanoparticles.
The dielectric constant of THF is 7.5, and that of acetone is 21 (13). Dielectric constant of THF and acetone mixture was determined theoretically from the following formula (26).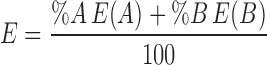 Where
Where
E = dielectric constant of medium
E(A) = dielectric constant of pure solvent A; E(B) = dielectric constant of pure solvent B
% A = percent content of A in mixture; % B = percent content of B in mixture
The dielectric constant of THF/acetone (1:1) was found to be 14.5, which explained the formation of smaller nanoparticles.
On the other hand, significant decrease (p < 0.05) in entrapment efficiency was observed with THF and acetone (1:1; Fig. 4). Increased diffusivity of solvent due to addition of acetone may cause leaching of drug into aqueous phase, thus decreasing the entrapment. This illustrates that, though modulating polarity and dielectric constant of the solvent phase can be used effectively to control the particle size, it has to be used with caution to ensure adequate drug loading. The batch prepared using THF/acetone ratio of 1:1 with PES concentration of 7.5 mg/ml and lutrol F 68 (0.5%) was further characterized by scanning electron microscopy (SEM) and subjected to freeze drying.
Emulsion–Solvent Diffusion
Emulsion–solvent diffusion technique differs from nanoprecipitation in that it employs a partially water-miscible solvent. It is widely adapted for preparation of polymeric nanoparticles (14), lipidic nanospheres (16), solid lipid nanoparticle (17), nanocapsules (10,15,20), and drug nanosuspensions (27). For the present study, methyl ethyl ketone was selected as the solvent because of high solubility of silymarin and PES. Table I summarizes effect of surfactant type and concentration on particle size of PES nanoparticles prepared by emulsion–solvent diffusion technique.
Table I.
Effect of Surfactant Type and Concentration on Particle Size of Nanoparticles Prepared by Emulsion–Solvent Diffusion (Mean ± SD, n = 3)
| Particle Size (nm) | ||||||
|---|---|---|---|---|---|---|
| Concentration (% w/v) | PVA | Lutrol F 68 | Tween 80 | |||
| Blank | Silymarin-loaded | Blank | Silymarin-loaded | Blank | Silymarin-loaded | |
| 0.1 | >1,000 | >1,000 | >1,000 | >1,000 | >1,000 | >1,000 |
| 0.5 | >1,000 | >1,000 | >1,000 | >1,000 | >1,000 | >1,000 |
| 1 | – | 613 ± 84.3 | – | >1,000 | – | >1,000 |
| 2 | – | 283.6 ± 2.2 | – | 534.9 ± 6.9 | – | >1,000 |
| 4 | >1,000 | >1,000 | 336.5 ± 3.8 | 273.5 ± 23.4 | >1,000 | 852.8 ± 24.6 |
Nanoparticles (<500 nm) were obtained at 2% w/v PVA and 4% w/v lutrol F 68 and PVA, respectively. At higher PVA concentrations, size increased and is attributed to viscosity of PVA. Tween 80 did not show nanoparticle formation. Similar observations with Tween 80 have been reported by D. Quintanar-Guerrero et al. (14), who studied poly (d,l-lactic acid) nanoparticles using emulsion–solvent diffusion technique. The final size of nanoparticles prepared by emulsion–solvent diffusion technique is largely governed by the characteristics of the primary emulsion. The more stable the primary emulsion, the smaller is the size obtained (15). The difference in the particle size observed with the different surfactants is related to ability of the surfactant to form a stable primary emulsion.
When compared to nanoprecipitation, emulsion–solvent diffusion technique required higher concentrations of the surfactant to achieve nanosize. In fact with nanoprecipitation, nanosize was obtained even at surfactant concentrations of 0.1%. Literature reports available on emulsion–solvent diffusion technique have achieved nanosize at surfactant concentrations usually >1% (10,14,15,20,24). Entrapment efficiency was found to be in the order lutrol F 68 > PVA > Tween 80. Nanoprecipitation appears to be a more versatile method for preparation of PES nanoparticles as smaller size and adequate entrapment efficiency is achieved at lower surfactant concentration.
In Vitro Drug Release
In vitro drug release profile of silymarin from nanoparticles is depicted in Fig. 6. The drug release was characterized by employing four basic models: zero-order kinetic model—cumulative percent drug release vs. time (r2 = 0.9909), first-order kinetic model—log cumulative of percent drug retained vs. time (r2 = 0.1598), Higuchi kinetic model—percent drug release vs. square root of time (r2 = 0.9227), Korsmeyer–Peppas model—log cumulative percent drug release vs. log time (r2 = 0.9526). In vitro drug release studies revealed zero-order release of silymarin from nanoparticles with 50% drug release in about 3 h (t 50 = 2.86 h).
Fig.6.

In vitro release profile of silymarin-loaded nanoparticles (mean ± SE, n = 3)
Scanning Electron Microscopy
Figure 7 shows SEM image of nanoparticles. The particles revealed spherical morphology and a size which is well correlated to photon correlation spectroscopy measurements.
Fig. 7.

SEM image of nanoparticles prepared by nanoprecipitation
Freeze Drying
Physical and chemical stability of nanoparticles can be improved by freeze drying. In recent years, a large number of studies have been reported on optimization of freeze drying of nanoparticles (28–32). However, increase in particle size is often observed after freeze drying (28,30,33). Selection of proper additives which can control the increase in size during freeze drying is absolutely critical. Among the various sugars and excipients evaluated for freeze drying, trehalose appears to be the most effective and frequently used cryoprotectant (28,34). Hence, trehalose 5% w/v was selected as cryoprotectant for the present study. Nanoparticle dispersion obtained upon suspending the pellet isolated after centrifugation revealed increase in size, suggesting tendency of nanoparticles to agglomerate. Hence, the dispersions were stabilized using lutrol F 68. The particle size obtained before and after freeze drying is depicted in Table II. Use of trehalose alone did not restrain increase in particle size upon freeze drying. However, addition of lutrol F 68 to nanoparticle dispersion prior to freeze drying enabled control of this growth in size. This effect was related to concentration of lutrol F 68. There are conflicting reports in the literature regarding stabilization effect of lutrol F 68 during freeze drying of nanoparticles. While few studies report inability of lutrol F 68 to maintain integrity of nanoparticles in absence of cryoprotectant (30), others report the cryoprotective effect of lutrol F 68 and attribute it to dehydration of surfactant present in the bulk solution which forces surfactant to the particle surface, providing cryoprotective effect (35). Hence, we used lutrol F 68 in combination with trehalose. Indeed, only marginal increase in size was observed at 2% w/v concentration of lutrol F 68. The ratio of the size after freeze drying (268 ± 1.5 nm) to the size before freeze drying (284.5 ± 7.7 nm) was found to be 1.061, which is well below the suggested limit ∼1 ± 0.3 (30). Presence of lutrol F 68 prevented nanoparticles from aggregating as well as promoted their ready dispersion when freeze-dried nanoparticles were suspended in water.
Table II.
Optimization of Freeze Drying (Mean ± SD, n = 3)
| Batch | Lutrol F 68% w/v | Trehalose % w/v | Particle Size (nm) | |
|---|---|---|---|---|
| Before Freeze drying (Size in dispersion) | After Freeze drying | |||
| 1 | – | 5 | 332.8 ± 3.8 | 715 ± 28.7 |
| 2 | 1 | 5 | 252.4 ± 9.7 | 426.4 ± 15.9 |
| 3 | 2 | 5 | 268 ± 1.5 | 284.5 ± 7.7 |
DSC and XRD Analysis
DSC and XRD analysis was carried out on freeze-dried samples. The DSC and XRD pattern of silymarin nanoparticles and other excipients are reported in Fig. 8 and Fig. 9, respectively. Silymarin did not reveal sharp endotherm suggesting poorly crystalline nature of drug. On the other hand, PES, lutrol F 68, and trehalose revealed sharp melting endotherms. DSC thermogram of silymarin-loaded nanoparticles showed an endotherm corresponding to lutrol F 68. The XRD spectra of silymarin nanoparticles without trehalose and lutrol F 68 corresponded to that of silymarin. It is now apparent that the peaks seen in XRD spectra of silymarin nanoparticles with trehalose and lutrol F 68 are due to the excipients which are crystalline in nature.
Fig. 8.

Comparative DSC thermograms of a trehalose, b lutrol F 68, c PES, d silymarin, e silymarin nanoparticles without trehalose and lutrol F 68, f silymarin nanoparticles with trehalose and lutrol F 68
Fig. 9.

Comparative XRD spectra of a trehalose, b lutrol F 68, c PES, d silymarin, e silymarin nanoparticles without trehalose and lutrol F 68, f silymarin nanoparticles with trehalose and lutrol F 68
CONCLUSION
The study presents successful design of PES nanoparticles and their application as a drug carrier. Nanoprecipitation was found to be a better approach for the preparation of PES nanoparticles. PES nanoparticles were readily freeze dried with minimum increase in particle size, using relatively low concentration (5%) of cryoprotectant. The study confirmed the feasibility of PES for the design of nanoparticulate drug delivery system.
Acknowledgments
We would like to thank Department of Chemical Engineering, Institute of Chemical Technology for carrying out SEM studies and Dr. B.N. Thorat Department of Chemical Engineering, Institute of Chemical Technology, Mumbai, India for allowing use of freeze dryer. Swati A. Guhagarkar would like to acknowledge Lady Tata Memorial Trust, Mumbai India, for the Fellowship.
References
- 1.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinsk WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70:1–20. doi: 10.1016/S0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 2.Bala I, Hariharan S, Kumar MNVR. PLGA nanoparticles in drug delivery. The state of art Pharm Res. 2004;21:387–422. doi: 10.1615/critrevtherdrugcarriersyst.v21.i5.20. [DOI] [PubMed] [Google Scholar]
- 3.Edlund U, Albertsson A-C. Polyesters based on diacid monomers. Adv Drug Deliv Rev. 2003;55:585–609. doi: 10.1016/S0169-409X(03)00036-X. [DOI] [PubMed] [Google Scholar]
- 4.Malshe VC, Devarajan PV, Shastri SR. Novel biodegradable aliphatic polyesters and pharmaceutical compositions and applications thereof. US Patent application No. 20060286138 (2006).
- 5.More AB, Chilgunde SN, Patil PS, Kamble JC, Malshe VC, Vanage GR, et al. Polyethylene sebacate: genotoxicity, mutagenicity evaluation and application in periodontal drug delivery system, J Pharm Sci. 2009; doi:10.1002/jps.21796 [DOI] [PubMed]
- 6.Jain R. The manufacturing techniques of various drug loaded biodegradable poly (lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21:2475–90. doi: 10.1016/S0142-9612(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 7.Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55:329–47. doi: 10.1016/S0169-409X(02)00228-4. [DOI] [PubMed] [Google Scholar]
- 8.Couvreur P, Vauthier C. Nanotechnology: intelligent design to treat complex disease. Pharm Res. 2006;23:1417–50. doi: 10.1007/s11095-006-0284-8. [DOI] [PubMed] [Google Scholar]
- 9.Fessi H, Puisieux F, Devissaguet J-P, Ammoury N, Benita S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int J Pharm. 1989;55:R1–4. doi: 10.1016/0378-5173(89)90281-0. [DOI] [Google Scholar]
- 10.Moinard-Checot D, Chevalier Y, Briançon S, Fessi H, Guinebretière S. Nanoparticles for drug delivery: review of the formulation and process difficulties illustrated by the emulsion–diffusion process. J Nanosci Nanotech. 2006;6:2664–81. doi: 10.1166/jnn.2006.479. [DOI] [PubMed] [Google Scholar]
- 11.Bilati U, Allémann E, Doelker E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur J Pharm Sci. 2005;24:67–75. doi: 10.1016/j.ejps.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Lamprecht A, Ubrich N, Yamamoto H, Schäfera U, Takeuchi H, Lehr CM, Maincent P, Kawashima Y. Design of rolipram-loaded nanoparticles: comparison of two preparation methods. J Control Release. 2001;71:297–306. doi: 10.1016/S0168-3659(01)00230-9. [DOI] [PubMed] [Google Scholar]
- 13.Legrand P, Lesieur S, Bochot A, Gref R, Raatjes W, Barratt G, Vauthier C. Influence of polymer behaviour in organic solution on the production of polylactide nanoparticles by nanoprecipitation. Int J Pharm. 2007;344:33–43. doi: 10.1016/j.ijpharm.2007.05.054. [DOI] [PubMed] [Google Scholar]
- 14.Quintanar-Guerrero D, Fessi H, Allémann E, Doelker E. Influence of stabilizing agents and preparative variables on the formation of poly (d,l-lactic acid) nanoparticles by an emulsification–diffusion technique. Int J Pharm. 1996;143:133–41. doi: 10.1016/S0378-5173(96)04697-2. [DOI] [Google Scholar]
- 15.Quintanar-Guerrero D, Allémann E, Doelker E, Fessi H. Preparation and characterization of nanocapsules from preformed polymers by a new process based on emulsification–diffusion technique. Pharm Res. 1998;15:1056–62. doi: 10.1023/A:1011934328471. [DOI] [PubMed] [Google Scholar]
- 16.Quintanar-Guerrero D, Tamayo-Esquivel D, Ganem-Quintanar A, Allémann E, Doelker E. Adaptation and optimization of the emulsification–diffusion technique to prepare lipidic nanospheres. Eur J Pharm Sci. 2005;26:211–8. doi: 10.1016/j.ejps.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Trotta M, Debernardi F, Caputo O. Preparation of solid lipid nanoparticles by a solvent emulsification–diffusion technique. Int J Pharm. 2009;257:153–60. doi: 10.1016/S0378-5173(03)00135-2. [DOI] [PubMed] [Google Scholar]
- 18.El-Samaligy MS, Afifi NN, Mahmoud EA. Increasing bioavailability of silymarin using a buccal liposomal delivery system: preparation and experimental design investigation. Int J Pharm. 2006;308:140–8. doi: 10.1016/j.ijpharm.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 19.Wu W, Wang Y, Que L. Enhanced bioavailability of silymarin by self-microemulsifying drug delivery system. Eur J Pharm Biopharm. 2006;63:288–94. doi: 10.1016/j.ejpb.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 20.Moinard-Chécot D, Chevalier Y, Briançon S, Beney L, Fessi H. Mechanism of nanocapsules formation by the emulsion–diffusion process. J Colloid Interface Sci. 2008;317:458–68. doi: 10.1016/j.jcis.2007.09.081. [DOI] [PubMed] [Google Scholar]
- 21.Murakami H, Kawashima Y, Niwa T, Hino T, Takeuchi H, Kobayashi M. Influence of the degrees of hydrolyzation and polymerization of poly(vinylalcohol) on the preparation and properties of poly(dl-lactide-co-glycolide) nanoparticle. Int J Pharm. 1997;149:43–9. doi: 10.1016/S0378-5173(96)04854-5. [DOI] [Google Scholar]
- 22.Youan BC, Hussain A, Nguyen NT. Evaluation of sucrose esters as alternative surfactants in microencapsulation of proteins by the solvent evaporation method. AAPS PharmSciTech. 2003;5:1–9. doi: 10.1208/ps050222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Türk M, Lietzow R. Stabilized nanoparticles of phytosterol by rapid expansion from supercritical solution into aqueous solution. AAPS PharmSciTech. 2004;4:1–10. doi: 10.1208/pt050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galindo-Rodriguez S, Allémann E, Fessi H, Doelker E. Physicochemical parameters associated with nanoparticle formation in the salting-out, emulsification–diffusion, and nanoprecipitation methods. Pharm Res. 2004;8:1428–39. doi: 10.1023/B:PHAM.0000036917.75634.be. [DOI] [PubMed] [Google Scholar]
- 25.Thioune O, Fessi H, Devissaguet JP, Puisieux F. Preparation of pseudolatex by nanoprecipitation: influence of the solvent nature on intrinsic viscosity and interaction constant. Int J Pharm. 1997;146:233–8. doi: 10.1016/S0378-5173(96)04830-2. [DOI] [Google Scholar]
- 26.Moreno A, Bolanos-Garcia VM, Soriano-Garcia M. The influence of dielectric constant upon protein crystallization by dynamic light scattering investigations. J Biomol Tech. 1998;9:18–20. [Google Scholar]
- 27.Trotta M, Gallarate M, Pattarino F, Morel S. Emulsions containing partially water-miscible solvents for the preparation of drug nanosuspensions. J Control Release. 2001;76:119–28. doi: 10.1016/S0168-3659(01)00432-1. [DOI] [PubMed] [Google Scholar]
- 28.Abdelwahed W, Degobert G, Stainmesse S, Fessi H. Freeze-drying of nanoparticles: formulation, process and storage considerations. Adv Drug Deliv Rev. 2006;58:1688–713. doi: 10.1016/j.addr.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 29.Shahgaldian P, Gualbert J, Aýssa K, Coleman AW. A study of the freeze-drying conditions of calixarene based solid lipid nanoparticles. Eur J Pharm Biopharm. 2003;55:181–4. doi: 10.1016/S0939-6411(02)00196-0. [DOI] [PubMed] [Google Scholar]
- 30.Saez A, Guzman M, Molpeceres J, Aberturas MR. Freeze-drying of polycaprolactone and poly(-lactic-glycolic) nanoparticles induce minor particle size changes affecting the oral pharmacokinetics of loaded drugs. Eur J Pharm Biopharm. 2000;50:379–87. doi: 10.1016/S0939-6411(00)00125-9. [DOI] [PubMed] [Google Scholar]
- 31.Hinrichs WLJ, Mancenido FA, Sanders NN, Braeckmans K, De Smedt SC, Demeester J, Frijlink HW. The choice of a suitable oligosaccharide to prevent aggregation of PEGylated nanoparticles during freeze thawing and freeze drying. Int J Pharm. 2006;311:237–44. doi: 10.1016/j.ijpharm.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 32.Schaffazicka SR, Pohlmannb AR, Dalla-Costaa T, Guterresa SS. Freeze-drying polymeric colloidal suspensions: nanocapsules, nanospheres and nanodispersion: a comparative study. Eur J Pharm Biopharm. 2003;56:501–5. doi: 10.1016/S0939-6411(03)00139-5. [DOI] [PubMed] [Google Scholar]
- 33.Schwarz C, Mehnert W. Freeze-drying of drug-free and drug-loaded solid lipid nanoparticles (SLN) Int J Pharm. 1997;157:171–9. doi: 10.1016/S0378-5173(97)00222-6. [DOI] [PubMed] [Google Scholar]
- 34.De Jaeghere F, Allémann E, Feijen J, Kissel T, Doelker E, Gurny R. Freeze-drying and lyopreservation of diblock and triblock poly (lactic acid)-poly (ethylene oxide) (PLA-PEO) copolymer nanoparticles. Pharm Dev Tech. 2000;5:473–83. doi: 10.1081/PDT-100102031. [DOI] [PubMed] [Google Scholar]
- 35.Quintanar-Guerrero D, Ganem- Quintanar A, Allémann E, Fessi H, Doelker E. Influence of the stabilizer coating layer on the purification and freeze-drying of poly (d,l-lactic acid) nanoparticles prepared by an emulsion–diffusion technique. J Microencapsul. 1998;15:107–19. doi: 10.3109/02652049809006840. [DOI] [PubMed] [Google Scholar]