

Abstract
A multiunit floating drug delivery system of rosiglitazone maleate has been developed by encapsulating the drug into Eudragit® RS100 through nonaqueous emulsification/solvent evaporation method. The in vitro performances of microspheres were evaluated by yield (%), particle size analysis, drug entrapment efficiency, in vitro floating behavior, surface topography, drug–polymer compatibility, crystallinity of the drug in the microspheres, and drug release studies. In vitro release was optimized by a {3, 3} simplex lattice mixture design to achieve predetermined target release. The in vivo performance of the optimized formulation was evaluated in streptozotocin-induced diabetic rats. The results showed that floating microspheres could be successfully prepared with good yields (69–75%), high entrapment (78-97%), narrow size distribution, and desired target release with the help of statistical design of experiments from very small number of formulations. In vivo evaluation in albino rats suggested that floating microspheres of rosiglitazone could be a promising approach for better glycemic control.
Key words: in vivo, microsphere, mixture design, optimization, rosiglitazone maleate
INTRODUCTION
Oral route remains so far the most convenient route of administration mainly because of its ease of administration, patient compliance, and flexibility of formulation. The overall performance of oral controlled-release drug delivery system is limited by several factors such as physiological variability such as gastrointestinal (GI) transit in addition to gastric retention time, variation in the physiological pH throughout gastrointestinal tract (GIT), and narrow absorption window of the drug molecule. If the absorption window of a drug molecule is very narrow, it is desired to retain the delivery system at the site of absorption for a longer period of time in order to obtain controlled release of the drug. There are several approaches to retain the drug delivery system at the GIT such as floatation, sedimentation, expansion, mucoadhesion, and modified shape system (1–7). In literature, both single- and multiple-unit gastroretentive systems have been reported (8–10). Multiple-unit floating polymeric drug delivery systems such as floating microspheres offer advantages of retaining the dosage form in the upper part of GIT for prolonged period and thereby releasing the drug in a controlled manner. Such floating devices show more reproducible release profiles over fortuitous (all-or-nothing emptying) nature or dose-dumping phenomenon associated with single-unit system (11). They decrease intersubject variability in absorption and minimize the possibility of dose dumping by uniform distribution within the gastric content and provide longer duration of action (12).
Rosiglitazone maleate (RZM; Fig. 1) is an antidiabetic drug for type II diabetes that improves insulin sensitivity in muscle and adipose tissues through activation of peroxisome proliferator-activated γ receptor (PPARγ) that are involved in transcription of insulin-responsive genes responsible for glucose production, transport, and utilization (13–15). The drug shows linear pharmacokinetics over a dose of 0.2–20 mg with biological half-life of 3–4 h with oral bioavailability of 99.8% (16). The drug is highly soluble in simulated gastrointestinal fluid (SGF). But the solubility gradually decreases with increment of pH. Above pH 7, the solubility of the drug is very low. Therefore, the rate and extent of absorption viz. bioavailability of the drug is mainly controlled by its dissolution rate. Following rosiglitazone monotherapy for 8 to 12 weeks, the dose should be increased to 8 mg/day in case of insufficient glycemic control, which results in higher incidents of dose-dependent side effects such as gastrointestinal disturbances, headache, altered blood lipids, edema, and hypoglycemia (17). Further, clinically significant adverse effects such as edema, anemia, and weight gain are frequently reported with conventional dosage forms of the drug (18,19). Clinical studies showed that 4-mg twice-per-day regimen compared to 8 mg once a day provides statistically greater improvement in glycemic control (20).
Fig. 1.

Chemical structure of rosiglitazone maleate
The objective of the present study was to develop a controlled-release oral drug delivery system of rosiglitazone maleate, which would control blood glucose level for prolonged period to achieve better glycemic control over immediate-release dosage formulation. To achieve controlled delivery of RZM, floating microspheres were prepared by emulsion solvent evaporation method and drug release was optimized as per target release using a simplex lattice mixture design. Finally, in vivo experiments were carried out to show the efficacy of the dosage form to achieve prolonged glycemic control. In the literature, very few reports of RZM formulations such as carbopol-based mucoadhesive tablet (21) and intragastric floating sustained-release tablet (22) based on hydrodynamically balanced system are available. The efficacy of carbopol-based mucoadhesive dosage is restricted by its nonspecific mucoadhesion and mucin turnover in GIT (23). Single-unit floating dosage form like tablet is associated with all-or-nothing emptying nature or dose-dumping phenomenon. However, no attempt has been reported yet to develop floating microspheres of rosiglitazone maleate for its controlled-release delivery utilizing statistical optimization technique to stomach and upper part of GIT from where the drug is predominantly absorbed.
MATERIALS AND METHODS
Materials
RZM was obtained as gift sample from M/S Torrent Pharmaceuticals Ltd. (Ahmedabad, India). Eudragit®RS100 (RS100) granules and tributyl citrate (TBC) were kindly provided by Degussa India Pvt. Ltd. (Mumbai, India) and East India Pharmaceuticals Works Ltd. (Kolkata, India), respectively. Heavy liquid paraffin (HLP) and petroleum ether (40–60°) were purchased from Merck (India). All other chemicals were of analytical grade and used without further purification.
Methods
Preparation of Microspheres
Microspheres were prepared by emulsification (oil-in-oil type)-solvent evaporation method with minor modification reported by Bogataj et al., using acetone/liquid paraffin solvent system (24). Ammonio methacrylate copolymer (Eudragit® RS100) was used as coating polymer of RZM. Required amount of RS100 with TBC (20% w/w of RS100) was dissolved in acetone to prepare 6.66%, 10%, and 13.32% (w/v) polymeric solution using a cyclomixer (CM-100, Remi, India) in a stoppered glass tube. Appropriate quantity of RZM (passed through # 120 ASTM) was added to this polymeric solution and a smooth suspension was prepared. The suspension was then poured into 30 mL of HLP kept at 30 ± 1°C while stirring at 800 rpm by a PMDC stirrer (RQ-121/D, Remi, India) fitted with stirring shaft (6 × 250 mm) with pitched-blade-type impeller. The stirring was continued for 3 h, until acetone evaporated completely. The ratio of drug and polymer was changed to obtain spherical microspheres of three different formulations F1, F2, and F3 with drug polymer ratio 1:1, 1:2, and 2:1 (w/w), respectively. After evaporation, microspheres formed were collected by filtration and washed three times with petroleum ether and dried under vacuum at room temperature overnight. All microsphere formulations were prepared in triplicate. Microspheres dried at room temperature were then weighed and the yield of the preparation was calculated using the following formula:
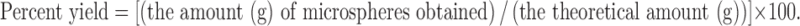 |
1 |
Quantitative Analysis of Rosiglitazone Maleate
High-performance liquid chromatography (HPLC) analyses were performed according to our previously reported method (25) with a Jasco HPLC system (JASCO, Japan) equipped with a Jasco-PU-980 pump and a Jasco-UV-975 UV detector (set at the wavelength of 260 nm) and Clarity Lite® software. For analysis, a reversed-phase Fine Pak SIL C8 steel column (250 × 4.6 mm, JASCO, Japan; average particle size 5 µm) was eluted with acetonitrile/methanol/acetate buffer with pH 4.0 (30:20:50 v/v/v) in isocratic mode. The flow rate of 1.5 mL/min was maintained. The injection volume was 20 μL. The retention time of RZM was found to be 5.81 min.
Solubility Measurement
RZM in an amount of excess of its solubility was added to 20 mL of simulated gastric fluid without pepsin (pH 1.2 and 2.0) or different buffered media with pH 4.0, 6.0, 6.8, 7.2, and 8.0 in a 50-mL stoppered conical flask in order to understand the solubility behavior of RZM over the usual pH range encountered in the GIT. Conical flasks were maintained at 37 ± 0.5°C and shaken in a thermostatically controlled water bath for 24 h. Aliquots of 1 mL were taken from the dissolution medium at suitable time intervals, filtered through 0.22-μm membrane filter, and then analyzed by HPLC. The result is expressed as milligram of drug dissolved per milliliter of dissolution media. Similarly, the solubility of free rosiglitazone base was also determined.
Drug Loading and Entrapment Efficiency
For the determination of drug loading and entrapment efficiency, microspheres containing RZM equivalent to 10 mg of the drug based on theoretical drug content were crushed in a glass mortar. The content was carefully transferred to a 100-mL volumetric flask with 50 mL of acetate buffer (pH 4.0). The glass mortar was washed with the same buffer and added to the volumetric flask to make up the volume of 100 mL. The acetate buffer (pH 4.0) containing crushed microsphere was stirred in thermostatically controlled water bath for 45 min in order to extract the drug efficiently in acetate buffer. Then, it was filtered through 0.22-μm membrane filter. A 5-mL volume of this solution was properly diluted with mobile phase and then analyzed by HPLC (25). Drug concentration was determined with the help of calibration curve. The range of drug concentration in calibration curve was 5–100 μg/mL. The results were expressed as percentage of RZM entrapment efficiency determined using the following formula:
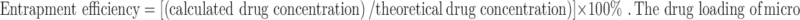 |
2 |
Particle Size Analysis
Different sizes of microspheres and their distribution in each batch were measured by sieving in mechanical shaker using a nest of standard sieves (ASTM) and the shaking period of 15 min. The particle size distribution was determined for all formulations and mean particle size of microspheres was calculated by using the following formula (26)
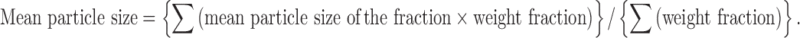 |
3 |
Surface Topography (SEM)
The surface morphology of the microspheres was examined by scanning electron microscopy (SEM; JSM-5200, Jeol, Japan) operated at 15 kV on samples gold-sputtered for 120 s at 10 mA, under argon low pressure. The morphology of the microspheres was analyzed by direct observation.
In Vitro Buoyancy
An in vitro floating ability of the microspheres was carried out in simulated gastric fluid USP containing 1% Tween® 80 as dispersing medium. Microspheres were spread over the surface of 900-mL dispersing medium taken in a USP XXIV dissolution apparatus (type II) at 37 ± 0.5°C. The medium was agitated with a paddle rotating at 50 rpm for 12 h. The floating and settled portions of the microspheres were collected, dried, and weighed separately. The buoyancy percentage was calculated as the ratio of mass of microspheres that remained floating and the total mass of microspheres.
Solid-State Characterization: X-ray Powder Diffraction
X-ray powder diffraction was carried out with a Miniflex (Rigaku, Japan) powder diffractometer. A Cu Kα source operation (30 kV, 15 mA) was employed. The diffraction patterns were recorded over 2θ angular range of 5–40° with scan speed of 2°/min at room temperature.
Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) experiments were carried out to characterize the physical state of RZM in microspheres as well as to find out the presence of any interaction among drug and the excipients. Five to ten milligrams of rosiglitazone, Eudragit® RS100, physical mixture of rosiglitazone, and Eudragit® RS100 and microspheres were put separately in aluminum pan and hermetically sealed. The heating rate was 10°C/min; nitrogen served as purged gas and the system was cooled down by liquid nitrogen. The differential thermal analyzer (Pyris Diamond TG/DTA, Perkin Elmer Instruments, USA) was used for this purpose.
Fourier Transform Infrared Spectroscopy
The Fourier transform infrared (FT-IR) spectra of samples were obtained using FT-IR spectrophotometer (Shimadzu, Japan). About 2–3 mg of samples was mixed with dried potassium bromide of equal weight and compressed to form a KBr disk. The samples were scanned from 400 to 4,000 cm−1.
In Vitro Dissolution Study
In vitro dissolution study was carried out in a paddle-type six-station dissolution test apparatus (TDT-06P, Electrolab, India) with stirring speed of 50 rpm using simulated gastric fluid as dissolution medium under sink conditions. Accurately weighed microspheres equivalent to 30 mg of RZM was added to dissolution medium kept at 37 ± 0.5°C. Periodically, solution withdrawn from the dissolution medium was filtered with a 0.45-μm hydrophilic filter disk and analyzed by HPLC. Same volume of dissolution medium was replaced back after each sampling in order to maintain sink condition. The kinetic data obtained from the release rates were also evaluated by fitting into different kinetic models. After the dissolution study, microspheres were filtered, dried, and observed under the SEM to examine any changes in surface topography.
Curve Fitting
Release data were fitted to different mathematical models to reveal the release mechanism from the microspheres. Zero-order (27) first order and Higuchi release models (28) were used for this purpose.
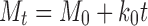 |
4 |
Mt = amount of drug released at time t; M0 = concentration of drug in the solution at t = 0; k0 = zero-order release rate constant.
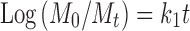 |
5 |
Mt = amount of drug released at time t; M0 = concentration of drug in the solution at t = 0; k1 = first-order release rate constant.
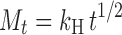 |
6 |
Mt = amount of drug released at time √t; kH = Higuchi release rate constant.
All curve fitting, simulation, and plotting were performed using commercially available Microsoft® Excel Solver (Microsoft Corporation, USA).
Simplex Lattice Mixture Design and Statistical Optimization of In Vitro Release
The in vitro drug release of rosiglitazone from the floating microspheres was optimized to achieve a target release profile by blending three microsphere formulations of F1, F2, and F3 according to a {3,3} simplex lattice mixture design with five additional runs for replication. The content of all parent formulations was varied from 0% to 100%. The target release profiles at different time intervals (first hour, second hour, fourth hour, sixth hour, and eighth hour) were set based on pharmacokinetic data available in the literature (16,29). Drug release at different time intervals was optimized through desirability function approach (30,31).
For controlled release of the drug, the total dose of the drug required was calculated based on conventional dose of 4 mg. The total dose was calculated by the following equation (32).
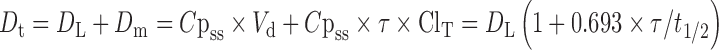 |
7 |
where Dt = total dose of drug; DL = immediate-release dose; Dm = maintenance dose, Cpss = steady-state plasma concentration; Vd = volume of distribution; τ = total time period (h) during which the controlled release is desired (12 h); and t1/2 = half-life of the drug (3.5 h). For rosiglitazone maleate, Dt = 4[1 + (0.693 × 12) / 3.5] = 13.504 mg rosiglitazone which is present in 17.8895 mg of rosiglitazone maleate. The loading dose (DL) should be released in the first hour and the maintenance dose will be available in the subsequent hours. Based on this assumption, the target release at first hour, second hour, fourth hour, sixth hour, and eighth hour was set as 33.7%, 39.23%, 52%, 61.23%, and 72.38%, respectively.
Statistical Analysis
The experimental results were expressed as mean ± SD. The differences were considered statistically significant at p < 0.05. The effect of independent variables on the different responses was statistically evaluated by commercially available software package Design Expert® version 6.0.10 (Stat-Ease, Inc., Minneapolis, MN, USA). The simplex lattice mixture design was evaluated by quadratic model, which bears the Eq. 8
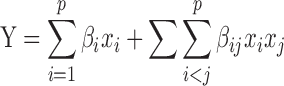 |
8 |
where Y is the response variable; βi represents expected responses due to pure blending of components and βij represents response due to synergistic or antagonistic blending.
In Vivo Evaluation
In vivo evaluation studies of the optimized formulation and pure drug were carried out on normal healthy male albino rats selected with average body weight of about 200–250 g. They were housed individually in polypropylene cages, maintained under standard conditions (12-h light and 12-h dark cycle; 25 ± 30°C; 35–60% humidity); the animals were fed with standard rat pellet diet and water ad libitum. The study was approved by the Institutional Animal Ethical Committee of Jadavpur University, Kolkata, India. Noninsulin-dependent diabetes mellitus (NIDDM) was induced in overnight fasted animals by a single intraperitoneal injection of 60 mg/kg of streptozotocin (Sigma Aldrich, Germany), 15 min after the intraperitoneal administration of 120 mg/kg nicotinamide (Ranbaxy Chemicals Ltd., Mumbai, India). Hyperglycemia was confirmed by the elevated glucose levels in plasma, determined at 72 h and then on day 7 after the injection. Each animal with a blood glucose concentration level above 250 mg/dL (13.8 mM) was considered to be diabetic and used in the experiments. Only the rats found with permanent NIDDM were used for in vivo evaluation studies.
Animals were divided into four groups of six rats each such as group 1: normal control rats administered with drinking water; group 2: diabetic control rats administered with drinking water; group 3: diabetic rats administered with pure rosiglitazone (4 mg/kg body weight); and group 4: diabetic rats administered with optimized formulation of microspheres equivalent to the dose of the drug, 4-mg/kg body weight using intragastric tube. For the control (groups 1 and 2), the fasting was done overnight and water ad libitum was allowed. For group 3 and group 4, pure drug and microspheres were administered orally in the morning following overnight fasting. No food and liquid except water (ad libitum) were given to the animals during the experiment. After collection of zero-hour blood sample, optimized formulation of microspheres was administered orally through intragastric tube. Blood samples (0.1 mL) were withdrawn from the tail vein of the rats up to 12 h at 1-h interval. Plasma glucose levels were determined using OneTouch® Horizon™, Blood Glucose monitoring system, LifeScan, Inc., Milpitas, USA.
RESULTS AND DISCUSSION
RZM belongs to class 1 drugs of Biopharmaceutical Classification System (33). The drug is highly soluble in aqueous solution at pH 1.2. So a good release retardant is necessary to control the release of RZM from the floating microspheres at low pH. Eudragit® RS100 copolymer is insoluble in water and digestive juices. Because of its low permeability, the active ingredient is released slowly by diffusion. It shows pH-independent release profile, which means that drug release takes place independently of individual variation. For this reason, this copolymer has been used to prolong RZM release from the microsphere formulation. The polymer also has been used by other researchers to prolong the release of several drugs from floating dosage form (34–38).
Solubility of Rosiglitazone in Different pH of GIT
From solubility–pH profile (Fig. 2), it was clear that solubility of RZM was highly pH dependent. The maximum solubility was found at pH 1.2 and thereafter solubility gradually decreased up to pH 4.0. At pH 6.0 and higher, pH solubility had been reduced drastically. Such solubility behavior could be explained by the following equation (39):
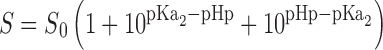 |
9 |
where S denotes molar solubility of RZM at a particular pH; S0 = molar solubility of free base rosiglitazone (0.002889 mol/L); pHp = pH above which RZM starts to precipitate as rosiglitazone base; pKa1 and pKa2 are 6.1 and 6.8, respectively. The aqueous solubility of RZM at pH 1.2 is 0.035861 mol/L. Solving Eq. 9 yielded pHp = 5.76 and 7.13. So, the solubility of RZM started to fall drastically after pH 6.0 (>5.76) and hence the drug precipitated as rosiglitazone base.
Fig. 2.

Solubility–pH profile of rosiglitazone maleate
Therefore, to achieve the controlled release of the drug as well as to avoid drug dissolution problem at higher pH, retention of drug molecule at low pH, i.e., stomach or upper part of GI tract and retardation of drug release with the help of suitable delivery system might be the suitable strategy. In this respect, floating microparticulate delivery system might be a good choice. Hence, floating microspheres were prepared to achieve the controlled release of rosiglitazone maleate using Eudragit® RS100 polymer as release retardant material.
Microsphere Formation and Morphology
RZM-loaded microspheres were prepared by oil-in-oil emulsion solvent evaporation method. Briefly, a smooth suspension of RZM in Eudragit® RS100–TBC–acetone mixture taking drug–polymer in the ratio of 1:1, 1:2, or 2:1 was added to heavy liquid paraffin as immiscible continuous phase. Since no stabilizer was used, high stirring rate (800 rpm) could produce smaller droplets of acetone, which was utilized to produce stable emulsion for sufficient period of time required for the evaporation of acetone. During this period, acetone will diffuse from the dispersed droplets into continuous phase and evaporate. As the acetone evaporates, there will be precipitation of the polymer over the insoluble particle of RZM and microspheres form. However, diffusion of liquid paraffin into polymer-rich acetone also causes the precipitation of Eudragit® RS100.
Preformulation study revealed that the drug would precipitate and form needle-shaped crystal below the concentration of 25.60 mg RZM per milliliter of acetone and hence needle-shaped polymeric particles would form instead of spherical particles. To avoid this problem, high concentration of drug (66.6 mg/mL of acetone) was used to prepare the microspheres. The optimized concentration of TBC was found as 20% w/w of polymer, which improved the mechanical properties of the polymeric film and produced free-flowing microspheres. The surface structure of the microspheres produced by the o/o emulsion solvent evaporation method was found spherical as observed by scanning electron microscope (Fig. 3a, b). The surface of the microspheres was smooth and revealed the presence of few pores in drug-loaded microspheres. The cross section of microspheres showed formation of hollow core (Fig. 3c), which, in part, might be responsible for their floating behavior. The hollow core formation inside the microspheres probably caused the reduction of their bulk density.
Fig. 3.

Scanning electron micrograph of microspheres: shape and surface a, b of the microspheres; c cross section of the microspheres showing hollow core
After the dissolution study, microspheres were filtered, dried, and observed under the SEM. SEM revealed that the microspheres retained their size intact. They were spherical in nature with smooth surface. There is no significant change in their surface topography.
Yield and Drug Entrapment Efficiency
List of formulations and their yields and drug loading as well as entrapment efficiencies are shown in Table I. It was observed that the yields of preparations and RZM entrapment efficiencies were high for all microspheres. It was further observed that there was a correlation between the initial drug loading and drug entrapment efficiency. With the increase of initial drug loading, entrapment efficiency also increases. Such correlation was also evidenced by Zaghloul et al. (40).
Table I.
Yields and Drug Encapsulation Efficiencies of Different Formulations
| Formulation | F1 | F2 | F3 |
|---|---|---|---|
| Yield (%)a | 75.23 ± 6.81 | 75.23 ± 6.81 | 72.67 ± 5.64 |
| Preparation drug loading (%) | 33.33 | 50 | 66.66 |
| Experimental drug loading (%)a | 26.03 | 40.62 | 64.85 |
| DEE (%)a | 78.11 ± 7.54 | 81.23 ± 4.93 | 97.29 ± 9.59 |
| Mean diametera (μm) | 813.12 ± 29.94 | 737.09 ± 43.84 | 596.48 ± 44.21 |
| Buoyancy (%)a | 96.34 ± 1.03 | 65.32 ± 3.13 | 80.49 ± 1.36 |
DEE drug entrapment efficiency
aMean ± SD, n = 3
Particle Size Analysis
Microspheres were prepared by using a gradually increasing concentration of RS100 with the fixed concentration of RZM in order to assess the effect of polymer concentration on the size of the microspheres. The mean particle size of the microspheres significantly increased with increase in the polymer concentration and found in the size ranges of 596.48 to 813.12 μm. The average particle size of the formulations was shown in Table I. All formulations showed narrow size distribution, which was evident from size distribution curve (figures not shown).
In Vitro Buoyancy
The aim of preparing floating microspheres is to extend the gastric residence time of a drug. In order to assess their floating properties, microspheres were placed in simulated gastric fluid containing surfactant 1% Tween® 80 to simulate gastric conditions. Tween® 80 was added in order to mimic the wetting effect of the natural surface-active agents in the GIT. The microspheres floated for a prolonged time over the surface of the dissolution medium. Buoyancy percentage of the microspheres was in the range of 65.32% to 96.34% at the end of 12 h (Table I).
Drug–Excipient Interaction Study: FT-IR Spectroscopy
Potential chemical interaction between drug and polymer may change the therapeutic efficacy of the drug. To investigate the possibility of chemical interaction between drug and polymer FT-IR spectra of pure RZM, pure polymer and drug-loaded formulations were analyzed over the range 400–4,000 cm−1 (Fig. 4) The IR spectrum of pure RZM showed strong absorption bands attributable to carbonyl stretching at 1,703 cm−1 and C=N stretching of pyridine moiety at 1,512 cm−1 and pure polymer showed characteristic absorption band at 1,734 cm−1 due to the carbonyl stretching of the ester moiety of RS100. FT-IR spectra of the RZM-loaded formulations displayed all the characteristic bands of both drug and polymer, without any significant spectral shift. This suggested that there was no potential chemical interaction between the components of the microspheres.
Fig. 4.

FT-IR spectra of pure rosiglitazone maleate (a), Eudragit® RS100 (b), and drug-loaded formulation F1 (c), F2 (d), and F3 (e)
Solid-State Characterization: X-ray Power Diffraction
The transformation from crystalline to amorphous form of the drug during the preparation of microspheres may lead to change in its dissolution pattern. To assess whether RZM was incorporated in the microspheres in its crystalline or amorphous form, powder X-ray diffraction studies were carried out. The degree of crystallinity of pure drug and its formulations showed that there was little loss in crystallinity of the drug upon microencapsulation. But total drug was not changed to amorphous form, which was confirmed by d and 2θ values of pure and microencapsulated drug from X-ray diffractometry (Table II). The peak intensity, however, decreased due to lesser fraction of pure drug in the microspheres (Fig. 5).
Table II.
d and 2θ Values of Pure and Microencapsulated Drug (from X-ray Diffractometry)
| Peak no. | Pure RZM | Formulation F1 | Formulation F2 | Formulation F3 | ||||
|---|---|---|---|---|---|---|---|---|
| d | 2θ | d | 2θ | d | 2θ | d | 2θ | |
| 1 | 5.646 | 15.68 | 5.625 | 15.74 | 5.679 | 15.59 | 5.646 | 15.68 |
| 2 | 3.929 | 22.61 | 3.924 | 22.64 | 3.949 | 22.49 | 3.934 | 22.58 |
| 3 | 3.507 | 25.37 | 3.491 | 25.49 | 3.511 | 25.34 | 3.507 | 25.37 |
| 4 | 2.988 | 29.87 | 2.991 | 29.84 | 2.994 | 29.81 | 2.988 | 29.87 |
Fig. 5.

X-ray diffractograms of rosiglitazone maleate (a), Eudragit® RS100 (b), physical mixture of drug and polymer (c), and drug-loaded formulation F1 (d), F2 (e), and F3 (f)
Differential Scanning Calorimetry
The thermal properties of the drug and the mixture of drug and excipients are of important interest since this can help to ascertain the crystalline or amorphous status of the entrapped drug in the excipients, to assess the interaction among different components of the formulations during the fabrication process. Figure 6 shows the DSC thermograms corresponding to rosiglitazone, Eudragit® RS100, and rosiglitazone-loaded microspheres. The Eudragit® RS100 thermogram displayed a broad endothermic peak at about 50°C, corresponding to the glass polymer transition (Tg), from a more fragile state (glassy state) to a rubbery one (41). The DSC curve of rosiglitazone showed a single endothermic peak at 126.82°C (−103.10 J/g) corresponding to its melting point (MP 122–123°C) being started at 123.63°C and ended at 129.76°C. The physical mixture of drug–polymer showed also similar endothermic peak at 126.65°C (−55.39 J/g), which started at 122.23°C and ended with 129.20°C (figure not shown). In drug-loaded microspheres, the sharp endothermic peak of the drug was shifted to a lower temperature (119.05°C), where a less defined and broader peak was found. Such behavior suggests a partial loss of drug crystallinity when drug was loaded into the microspheres (42), which were also supported by X-ray diffractograms that showed less intensity of peaks corresponding to rosiglitazone from formulations. Since there was no major shift in the Tg (∼50°C) of the polymer in drug-loaded formulation, it can be concluded that there was no significant interaction occurring between the drug and the polymer.
Fig. 6.

A: thermogram of rosiglitazone maleate (endothermic peak at 126.82°C); B: thermogram of Eudragit® RS100 (T g ∼50°C); C: thermogram of drug-loaded microspheres (broader endothermic peak at 119.05°C)
Dissolution Study and In Vitro Drug Release Kinetics
The equilibrium solubility of RZM was found to be 16.98 ± 1.43 mg/mL in simulated gastric fluid (pH 1.2), indicating the sink condition limit for the dissolution studies. The in vitro drug release from the microspheres was studied in simulated gastric fluid USP (SGF) at pH 1.2 for 12 h. Figure 7 shows the release profiles of RZM from microspheres with same size range (weight fraction retained by #30) and different drug loadings (F1–F3). It was observed that an initial burst effect in the first 2 h of drug release occurred in case of F2 and F3 in SGF, which may be explained by the presence of uncovered drug crystals on the surface of the microsphere. After 2 h, drug released slowly. In case of F1, similar burst release from the microspheres was not observed as compared to F2 and F3 probably due to high polymer concentration (66.66% w/w), which was sufficiently high to encapsulate the drug particles. With increasing polymer load, the cumulative drug release slowed down as expected. The increased density of the polymeric matrix at higher polymer concentration results in an increased diffusional path length, which may reduce the cumulative release of drug from the polymer matrix. Moreover, when the polymer concentration was low, smaller particles with large surface area were produced and hence drug release was faster during exposure to dissolution media.
Fig. 7.

Release profiles of rosiglitazone maleate microspheres with different drug loading (F1, F2, and F3 with initial drug loading of 33.33%, 50%, and 66.66%, respectively) in simulated gastric fluid (pH 1.2)
The data obtained for in vitro release were fitted into equations for the zero-order, first-order, and Higuchi release models. It was observed that the regression coefficient was highest for the Higuchi model. So, the release pattern of RZM from the microspheres followed Higuchi release kinetics. Since the microspheres are actually monolithic or matrix systems, the release of the drug from the microspheres should be expected to follow diffusion-controlled model in accordance with Higuchi.
The results of kinetic evaluation SGF were presented in Table III based on different models. In case of SGF, appropriate kinetic model was chosen without considering the first 2 h of release profiles due to burst effect (43). From the tabulated results, it was clear that the release kinetics of all formulations followed Higuchi model. The results indicated that various drug–polymer ratios did not change the release kinetics. But there is significant difference in the release rate constants between F1, F2, and F3 formulations. It was further observed that higher drug–polymer ratio has an influence on the amount of drug crystals deposited on the surface of the microsphere. Therefore, the microspheres with high drug–polymer ratio showed higher burst effect in F2 and F3 in SGF.
Table III.
Correlation Coefficients of Different Mathematical Models and Release Rate Constant (k) for Rosiglitazone Maleate Microspheres in Simulated Gastric Fluid
| Formulation | Correlation coefficient (R 2) | k a (mg h−0.5) | ||
|---|---|---|---|---|
| Zero order | First order | Higuchi equation | ||
| F1 | 0.7551 | 0.7927 | 0.9070 | 18.588 |
| F2 | 0.8716 | 0.8847 | 0.9265 | 2.228b |
| F3 | 0.9258 | 0.9386 | 0.9637 | 1.456b |
a k was calculated based on the Higuchi model
bRelease rate constant was calculated excluding first 2 h of burst release
Statistical Optimization of In Vitro Drug Release
The microspheres prepared with varying polymer–drug ratios showed that drug release is proportional to the drug loading. Hence, the next step was to optimize the release profile to achieve a desired controlled-release profile. Classical mixture design has been adopted to optimize the drug release at different time periods (44–47). A {3, 3} simplex lattice mixture design with five additional runs was adopted to obtain ten different blends of microspheres and their release profile was obtained (Table IV). The weight fraction of microspheres with a definite drug–polymer ratio in a blend was taken as independent variable and the percent cumulative drug release at first hour, second hour, fourth hour, sixth hour, and eighth hour were taken as the dependent variables to optimize the in vitro release for achieving target release. Polynomial models were generated for each response variables via multiple least-square regression analysis. The models were validated through analysis of variance (ANOVA) as well as other diagnostic statistics (Table V).
Table IV.
Blended Formulations as Per Simplex Lattice Mixture Design
| Blended Formulations | Parent formulations of rosiglitazone | Responses modelleda | ||||||
|---|---|---|---|---|---|---|---|---|
| Code | F1 | F2 | F3 | CR1 | CR2 | CR4 | CR6 | CR8 |
| R1 | 1.000 | 0.000 | 0.000 | 17.82 | 33.70 | 51.53 | 58.47 | 61.69 |
| R2 | 0.500 | 0.500 | 0.000 | 37.91 | 53.22 | 63.46 | 67.57 | 69.65 |
| R3 | 0.500 | 0.000 | 0.500 | 48.34 | 56.91 | 66.58 | 70.55 | 72.42 |
| R4 | 0.000 | 1.000 | 0.000 | 58.00 | 72.73 | 75.39 | 76.66 | 77.60 |
| R5 | 0.000 | 0.500 | 0.500 | 68.43 | 76.43 | 78.51 | 79.65 | 80.37 |
| R6 | 0.000 | 0.000 | 1.000 | 78.86 | 80.12 | 81.63 | 82.63 | 83.14 |
| R7 | 0.667 | 0.167 | 0.167 | 34.69 | 47.94 | 60.52 | 65.53 | 67.92 |
| R8 | 0.167 | 0.667 | 0.167 | 54.78 | 67.46 | 72.45 | 74.62 | 75.87 |
| R9 | 0.167 | 0.167 | 0.667 | 65.21 | 71.15 | 75.57 | 77.61 | 78.64 |
| R10 | 0.333 | 0.333 | 0.333 | 51.56 | 62.18 | 69.52 | 72.59 | 74.14 |
aCRx = % cumulative drug release at xth hour
Table V.
Summary of ANOVA of Different Responses and Diagnostic Statistics
| Response parameter | Model equation | SS | df | MS | F value | p > F | R 2adj | R 2pred | PRESS |
|---|---|---|---|---|---|---|---|---|---|
| CR1 | Eq. 10 | 3.10869 | 4 | 0.77717 | 3422.77 | <0.0001 | 0.999 | 0.9989 | 3.57E−03 |
| CR2 | Eq. 11 | 0.000448 | 5 | 8.97E−05 | 1741.707 | <0.0001 | 0.9984 | 0.9981 | 8.33E−07 |
| CR4 | Eq. 12 | 57.0352 | 4 | 14.2588 | 287.7427 | <0.0001 | 0.9879 | 0.9866 | 0.77 |
| CR6 | Eq. 13 | 49.94231 | 4 | 12.48558 | 347.7429 | <0.0001 | 0.9900 | 0.9890 | 0.55 |
| CR8 | Eq. 14 | 46.62577 | 4 | 11.65644 | 382.7688 | <0.0001 | 0.9909 | 0.99 | 0.47 |
The model equations indicate a complex nonlinear relationship between the independent variables and the response variables and the responses.
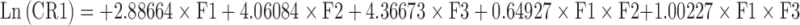 |
10 |
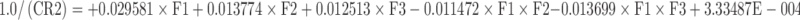 |
11 |
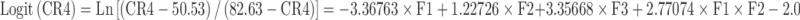 |
12 |
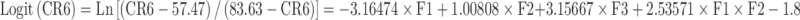 |
13 |
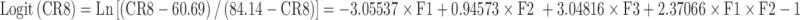 |
14 |
Then, the numerical optimization (Table VI) of the model equations (Eqs. 10–14) was done with target set as CR1 = 33.7%, CR2 = 39.23%, CR4 = 52%, CR6 = 61.23%, and CR8 = 72.38%. The predicted blend of microspheres was obtained as F1/F2/F3 = 0.712:0:0.288 with high desirability statistics. By blending the parent formulations in the said ratios, dissolution study was then carried out. The residual value between release of predicted optimum blends and that of experimental blends was found minimal. The release from experimental blend has also good agreement with target release (Table VII).
Table VI.
Numerical Optimization: Constraints
| Parameter | Goal | Lower limit | Upper limit |
|---|---|---|---|
| F1 | In range | 0 | 1 |
| F2 | In range | 0 | 1 |
| F3 | In range | 0 | 1 |
| Ln(CR1) | Target = 3.5175 | 2.88032 | 4.36767 |
| 1.0/(CR2) | Target = 0.02549 | 0.0124813 | 0.0296736 |
| Logit(CR4) | Target = −3.03672 | −3.4372078 | 3.4372078 |
| Logit(CR6) | Target = −1.78464 | −3.2252554 | 3.2252554 |
| Logit(CR8) | Target = −0.00597 | −3.11129 | 3.11129 |
Table VII.
Predicted and Optimized Microsphere Blends and Release Parameters
| Microspheres blends (F1/F2/F3) | Response parameters | Desirability statistics | |||||
|---|---|---|---|---|---|---|---|
| CR1 | CR2 | CR4 | CR6 | CR8 | |||
| Predicted optimum blends | 0.712:0:0.288 | 33.73 | 45.75 | 56.72 | 62.88 | 65.72 | 0.780 |
| Experimental blends | 0.712:0:0.288 | 35.40 | 47.07 | 60.20 | 65.43 | 67.87 | |
| Residual | − | 1.67 | 1.32 | 3.48 | 2.55 | 2.15 | |
| Target release | − | 33.70 | 39.23 | 52.00 | 61.23 | 72.38 | |
In Vivo Evaluation
In vivo evaluation of the microspheres were carried out in healthy male albino rat by measuring blood glucose level after oral administration with optimized formulation of microspheres equivalent to the dose of the drug, 4-mg/kg body weight in comparison with administration of pure drug at same dose. The antihyperglycemic effect of formulation and pure drug in diabetic rats were assessed at different time intervals (Fig. 8). When rosiglitazone solution (standard) was given orally, the blood glucose level started to decrease from the second hour. After the fourth hour, blood glucose level reached to almost normal level but after the fifth hour blood glucose level started to increase again. On the contrary, the optimized formulation of RZM blood glucose level started to decrease from the third hour and this decrease continued up to the ninth hour until blood glucose reached to normal level. This was maintained up to the 12th hour and blood glucose was found to be 4.86 mM. The lowering of blood glucose level was slower, as expected, in case of RZM microspheres than pure RZM due to its higher dissolution rate in pure form in gastric fluid of the rats.
Fig. 8.

Comparison of in vivo plasma glucose level in streptozotocin-induced diabetic albino rat following oral administration of pure drug (group 3) and rosiglitazone microspheres (group 4) with plasma glucose level of normal rat (group 1) and streptozotocin-induced diabetic rat without drug (group 2)
CONCLUSION
The present study showed that floating microspheres could be a promising approach for control of blood glucose level in hyperglycemic condition for prolonged period. The method yielded microspheres with good yield and high entrapment efficiency. There was no chemical interaction among the drug and the excipients found. The use of simple lattice mixture design helped to develop a statically validated product to achieve the target release profiles. In vivo results suggested that such drug delivery system of rosiglitazone could be a better alternative to the physicians over the existing immediate-release formulations.
Acknowledgements
Financial support from the University Grants Commission (India) is gratefully acknowledged. Authors are grateful to M/S Torrent Pharmaceuticals Ltd. (Ahmedabad, India) for providing rosiglitazone maleate and Degussa India Pvt. Ltd. (Mumbai, India) for generous supply of Eudragit® RS100. Authors are also thankful to Dr. Dipankar Datta, Scientist, Indian Association for the Cultivation of Science (Kolkata, India) for carrying out the FT-IR study.
References
- 1.Choudhury PK, Kar M, Chauhan CS. Cellulose acetate microspheres as floating depot systems to increase gastric retention of antidiabetic drug: formulation, characterization and in vitro–in vivo evaluation. Drug Dev Ind Pharm. 2008;34:349–54. doi: 10.1080/03639040701542531. [DOI] [PubMed] [Google Scholar]
- 2.Deshpande AA, Shah NH, Rhodes CT, Malick W. Development of a novel controlled-release system for gastric retention. Pharm Res. 1997;10:14815–9. doi: 10.1023/a:1012171010492. [DOI] [PubMed] [Google Scholar]
- 3.Lee JH, Park TG, Choi HK. Development of oral drug delivery system using floating microspheres. J Microencapsul. 1999;16:715–29. doi: 10.1080/026520499288663. [DOI] [PubMed] [Google Scholar]
- 4.Rednick B, Tucker SJ. Sustained release bolus for animal husbandry. United States Patent US 3507952; 1970, Apr 21.
- 5.Urquhart J, Theeuwes F. Drug delivery system comprising a reservoir containing a plurality of tiny pills. United States Patent US 4434153; 1984, Feb 28.
- 6.Mamajek RC, Moyer ES. Drug-dispensing device and method. United States Patent US 4207890; 1980, June 17.
- 7.Fix JA, Cargill R, Engle K. Controlled gastric emptying III: gastric residence time of a non-disintegrating geometric shape in human volunteers. Pharm Res. 1993;10:1087–9. doi: 10.1023/A:1018939512213. [DOI] [PubMed] [Google Scholar]
- 8.Fujimori J, Machida Y, Nagai T. Preparation of magnetically-responsive tablet and confirmation of its gastric residence in beagle dogs. STP Pharm Sci. 1994;4:425–30. [Google Scholar]
- 9.Nur AO, Zhang JS. Captopril floating and/bioadhesive tablets: design and release kinetics. Drug Dev Ind Pharm. 2000;26:965–9. doi: 10.1081/DDC-100101323. [DOI] [PubMed] [Google Scholar]
- 10.Streubel A, Siepmann J, Bodmeier R. Floating matrix tablets based on low density foam powder: effect of formulation and processing parameters on drug release. Eur J Pharm Sci. 2003;18:37–45. doi: 10.1016/S0928-0987(02)00223-3. [DOI] [PubMed] [Google Scholar]
- 11.Acikgoz M, Kas HS, Hascelik Z, Milli U, Hincal AA. Chitosan microspheres of diclofenac sodium, II: in vitro and in vivo evaluation. Pharmazie. 1995;50:275–7. [PubMed] [Google Scholar]
- 12.Iannuccelli V, Coppi G, Bernabei MT, Cameroni R. Air compartment multiple-unit system for prolonged gastric residence. Part I. Formulation study. Int J Pharm. 1998;174:47–54. doi: 10.1016/S0378-5173(98)00229-4. [DOI] [Google Scholar]
- 13.Wagstaff AJ, Goa KL. Rosiglitazone—a review of its use in the management of type 2 diabetes mellitus. Drugs. 2002;62:1805–37. doi: 10.2165/00003495-200262120-00007. [DOI] [PubMed] [Google Scholar]
- 14.Oakes ND, Kennedy CJ, Jenkins AB, Laybutt DR, Chisholm DJ, Kraegen EW. A new antidiabetic agent, BRL 49653, reduces lipid availability and improves insulin action and glucoregulation in the rat. Diabetes. 1994;43:1203–10. doi: 10.2337/diabetes.43.10.1203. [DOI] [PubMed] [Google Scholar]
- 15.Smith SA. Peroxisomal proliferator activated receptors (PPARs): molecular targets for hypolipidaemic agents and insulin sensitisers. Pharmacol Rev Com. 1996;8:57–64. [Google Scholar]
- 16.Cox PJ, Ryan DA, Hollis FJ, Harris AM, Miller AK, Vousden M, Cowley H. Absorption, disposition, and metabolism of rosiglitazone, a potent thiazolidinedione insulin sensitizer, in humans. Drug Metab Dispos. 2000;28:772–80. [PubMed] [Google Scholar]
- 17.Diamant M, Heine RJ. Thiazolidinediones in type-II diabetes mellitus: current clinical evidence. Drugs. 2003;63:1373–406. doi: 10.2165/00003495-200363130-00004. [DOI] [PubMed] [Google Scholar]
- 18.Idris I, Gray S, Donnelly R. Rosiglitazone and pulmonary oedema: an acute dose-dependent effect on human endothelial cell permeability. Diabetologia. 2003;46:288–90. doi: 10.1007/s00125-002-1008-1. [DOI] [PubMed] [Google Scholar]
- 19.Werner AL, Travaglini MT. A review of rosiglitazone in type 2 diabetes mellitus. Pharmacotherapy. 2001;21:1082–99. doi: 10.1592/phco.21.13.1082.34615. [DOI] [PubMed] [Google Scholar]
- 20.Phillips LS, Grunberger G, Miller E, Patwardhan R, Rappaport EB, Salzman A. Once- and twice-daily dosing with rosiglitazone improves glycemic control in patients with type 2 diabetes. Diabetes Care. 2001;24:308–15. doi: 10.2337/diacare.24.2.308. [DOI] [PubMed] [Google Scholar]
- 21.Krishna SS, Ray S, Thakur RS. Formulation and evaluation of mucoadhesive dosage form containing rosiglitazone maleate. Pak J Pharm Sci. 2006;19:208–13. [PubMed] [Google Scholar]
- 22.Feng H, Wang Z, Chen D. Studies on rosiglitazone maleate intragastric floating sustained release tablet. J China Pharm Univ. 2002;33:196–9. [Google Scholar]
- 23.Ponchel G, Irache JM. Specific and non-specific bioadhesive particulate system for oral delivery to the gastrointestinal tract. Adv Drug Del Rev. 1998;34:191–219. doi: 10.1016/S0169-409X(98)00040-4. [DOI] [PubMed] [Google Scholar]
- 24.Bogataj M, Mrhar A, Kristl A, Kozjek F. Eudragit E microspheres containing bacampicillin: preparation by solvent removal methods. J Microencapsul. 1991;8:401–6. doi: 10.3109/02652049109069567. [DOI] [PubMed] [Google Scholar]
- 25.Kamila MM, Mondal N, Ghosh LK, Gupta BK. Drug dissolution studies and determination of rosiglitazone maleate in tablets and polymeric microspheres by a rapid, validated RP-HPLC method. J Liq Chromatogr Rel Technol. 2008;31:2503–16. doi: 10.1080/10826070802319800. [DOI] [Google Scholar]
- 26.Badri VN, Thomas PA, Pandit JK, Kulkarni MG, Mashelkar RA. Preparation of non-porous microspheres with high entrapment efficiency of proteins by a (water-in-oil)-in-oil emulsion technique. J Control Release. 1999;58:9–20. doi: 10.1016/S0168-3659(98)00140-0. [DOI] [PubMed] [Google Scholar]
- 27.Lee PI. Novel zero order drug delivery via immobilized nonuniformed drug distribution in glassy hydrogels. J Pharm Sci. 1984;73:1344–7. doi: 10.1002/jps.2600731004. [DOI] [PubMed] [Google Scholar]
- 28.Higuchi T. Mechanism of sustained-action medication: theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52:1145–9. doi: 10.1002/jps.2600521210. [DOI] [PubMed] [Google Scholar]
- 29.Goodman LS, Hardman JG, Limbird LE, Gillman AG. Goodman and Gillman’s the pharmacological basis of therapeutics. 10. New York: McGraw-Hill; 2001. [Google Scholar]
- 30.Armstrong NA, James KC. Pharmaceutical Experimental Design and Interpretation. London: Taylor & Francis; 1996. [Google Scholar]
- 31.Lewis GA, Mathieu D, Phan-Tan-Luu R. Pharmaceutical experimental design. New York: Marcel Dekker; 1999. [Google Scholar]
- 32.Bentley AO, Rawlins EA. Bentley’s text book of pharmaceutics. 8. London: Bailliere Tindall; 1977. [Google Scholar]
- 33.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 34.Ma N, Xu L, Wang Q, Zhang X, Zhang W, Li Y, et al. Development and evaluation of new sustained-release floating microspheres. Int J Pharm. 2008;358:82–90. doi: 10.1016/j.ijpharm.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 35.Yin L, Cheng K, Wang G, Zhang L, Zang H, Zhou J. Effect of film coating on cephalexin gastric-floating tablets. J Chin Pharm Univ. 2008;39:37–41. [Google Scholar]
- 36.Lunio R, Sawicki W. Influence of acrylic esters and methacrylic esters on flotation of pellets and release rate of verapamil hydrochloride. Acta Pol Pharm. 2006;63:69–74. [PubMed] [Google Scholar]
- 37.Fukuda M, Peppas NA, McGinity JW. Floating hot-melt extruded tablets for gastroretentive controlled drug release system. J Control Rel. 2006;115:121–9. doi: 10.1016/j.jconrel.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 38.Streubel A, Siepmann J, Bodmeier R. Multiple unit gastroretentive drug delivery systems: a new preparation method for low density microparticles. J Microencapsul. 2003;20:329–47. doi: 10.1080/0265204021000058384. [DOI] [PubMed] [Google Scholar]
- 39.Avdeef A. Absorption and drug development: solubility, permeability, and charge state. Hoboken: Wiley; 2003. pp. 91–3. [Google Scholar]
- 40.Zaghloul AA, Mustafa F, Siddiqu A, Khan M. Biodegradable microparticulates of beta-estradiol: preparation and in vitro characterization. Drug Dev Ind Pharm. 2005;31:803–11. doi: 10.1080/03639040500217624. [DOI] [PubMed] [Google Scholar]
- 41.Lehmann KOR. Chemistry and application properties of polymethacrylate coating systems. In: McGinity JW, editor. Aqueous polymeric coatings for pharmaceutical dosage forms. New York: Marcel Dekker; 1997. pp. 101–76. [Google Scholar]
- 42.Pignatello R, Amico D, Chiechio S, Spadaro C, Puglisi G, Giunchedi P. Preparation and analgesic activity of Eudragit® RS100 microparticles containing diflunisal. Drug Deliv. 2001;8:35–45. doi: 10.1080/107175401300002748. [DOI] [PubMed] [Google Scholar]
- 43.Bolourtchian N, Karimi K, Aboofazeli R. Preparation and characterization of ibuprofen microspheres. J Microencapsul. 2005;22:529–38. doi: 10.1080/02652040500161941. [DOI] [PubMed] [Google Scholar]
- 44.Bhattacharyya S, Ray S, Gupta BK, Ghosh LK. Design, evaluation and statistical optimisation of a controlled release multiparticulate acyclovir delivery system. Latin Am J Pharm. 2007;26:852–8. [Google Scholar]
- 45.Fernández CM, Marín YL, Fernández EG, Pérez SI, Jiménez CB. Optimization of a meprobamate fast released tablet formulation using mixture design. Latin Am J Pharm. 2008;27:62–7. [Google Scholar]
- 46.Plaizier-Vercammen J, Dauwe D, Brioen P. Possibility of the use of Eudragit® RS as a sustained release matrix agent for the incorporation of water soluble active compounds at high percentages. STP Pharma Sci. 1997;7:491–7. [Google Scholar]
- 47.Tillotson J, Sakr A. Application of multiple-response optimization to fluoride release from an extended-release matrix tablet. Pharm Ind. 2004;66:601–6. [Google Scholar]