

Abstract
The purpose of this research was to generate, characterize, and investigate the in vivo efficacy of budesonide (BUD) microparticles prepared by spray-drying technology with a potential application as carriers for pulmonary administration with sustained-release profile and improved respirable fraction. Microspheres and porous particles of chitosan (drug/chitosan, 1:2) were prepared by spray drying using optimized process parameters and were characterized for different physicochemical parameters. Mass median aerodynamic diameter and geometric standard deviation for conventional, microspheres, and porous particles formulations were 2.75, 4.60, and 4.30 µm and 2.56, 1.75, and 2.54, respectively. Pharmacokinetic study was performed in rats by intratracheal administration of either placebo or developed dry powder inhalation (DPI) formulation. Pharmacokinetic parameters were calculated (Ka, Ke, Tmax, Cmax, AUC, and Vd) and these results indicated that developed formulations extended half life compared to conventional formulation with onefold to fourfold improved local and systemic bioavailability. Estimates of relative bioavailability suggested that developed formulations have excellent lung deposition characteristics with extended T1/2 from 9.4 to 14 h compared to conventional formulation. Anti-inflammatory activity of BUD and developed formulations was compared and found to be similar. Cytotoxicity was determined in A549 alveolar epithelial cell line and found to be not toxic. In vivo pulmonary deposition of developed conventional formulation was studied using gamma scintigraphy and results indicated potential in vitro–in vivo correlation in performance of conventional BUD DPI formulation. From the DPI formulation prepared with porous particles, the concentration of BUD increased fourfold in the lungs, indicating pulmonary targeting potential of developed formulations.
Key words: budesonide, dry powder inhaler, gamma scintigraphy, microparticles, spray drying
INTRODUCTION
Inhaled corticosteroids (ICSs) are effective in controlling inflammation and improving lung function and asthma symptoms (1) and are recommended as first-line therapy for asthma patients. There is considerable evidence that treatment with anti-inflammatory ICSs reduces morbidity and mortality in asthma (2). ICSs appear to have a place in the management of severe chronic obstructive pulmonary disease (COPD), perhaps by decreasing and reducing the frequency of exacerbations and improve quality of life in patients with COPD (3). Budesonide (BUD) has a high ratio of topical anti-inflammatory to systemic activity and is one of the most extensively used inhaled glucocorticoids (4). BUD decreases airway hyper-responsiveness and reduces the number of inflammatory cells and mediators present in airways of patients with asthma. There are many advantages for developing sustained-release formulations for pulmonary drug delivery, which include reduced dosing frequency, improved patient compliance, and reduction of side effects (5–7).
Microparticles (microspheres and porous particles) combine several advantages. Positive features of potential use of microparticles as drug carrier systems include the possibility of controlled drug release and drug targeting, providing protection of incorporated active compounds against degradation, lack of toxicity as its solid matrix is composed of physiological and well-tolerated polymers, e.g., chitosan and gelatin, and they allow hydrophilic and/or hydrophobic drugs to be incorporated (5,8). The drug solubility, chemical and physical structure of polymers, and their polymorphic state determine the loading capacity of drug in microparticles. Several sustained-release systems that include liposome and other biodegradable microspheres have been investigated as potential pulmonary carriers (9). Particle size and potential toxicity of excipients are critical factors that limit the use of microparticles for pulmonary administration. The deepest penetration of particles into airways and their deposition in peripheral regions are achieved when the particle size ranges between 1 and 5 µm (7,10–19). Excipients must be physiologically acceptable, biodegradable, and nonimmunogenic and might not induce inflammatory and alloreactive responses (18).
In the present paper, BUD has been chosen as drug model, a potent corticosteroid with high glucocorticoid receptor affinity and prolonged tissue retention, inhibits inflammatory symptoms such as edema and vascular hyperpermeability (3). Spray-dried powders that exhibit sustained drug release properties were generated through inclusion of drug release modifiers such as chitosan (5). Chitosan, a polysaccharide derived from deacetylation of naturally occurring polymer chitin, is a promising excipient that can be employed in a wide range of applications, including sustained-release preparations (20). Given that chitosan not only acts as a drug release modifier but also has mucoadhesive properties, it would appear to be a useful excipient while preparing sustained-release formulations for pulmonary drug delivery (21).
Tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) are potent inflammatory cytokines, which have been implicated in a variety of pulmonary diseases including idiopathic pulmonary fibrosis, asthma, and COPD (22–24). Most cells in the body express receptors for TNF-α, which can elicit a number of different responses depending on many factors including the dose, route of administration, and duration of TNF-α. Release of TNF-α is triggered primarily by inflammatory stimuli. In the lungs, TNF-α production arises from numerous cell types including epithelium, endothelium, activated macrophages, and monocytes and probably also smooth muscle. TNF-α is released in allergic responses from both mast cells and macrophages via IgE-dependent mechanisms, elevated levels of TNF-α are frequently observed in bronchoalveolar fluid of asthmatic subjects undergoing allergen challenge. Besides mast cells and macrophages, many other cell types that appear to play a contributory role in the pathogenesis of asthma are also a significant source of TNF-α, including eosinophils, epithelial cells, and neutrophils. In addition, T cells from asthmatic airways constitutively produce large amounts of TNF-α both at the protein and mRNA levels. IL-6 is known as a proinflammatory cytokine involved in immune response, inflammation, and hematopoiesis. IL-6 is a multifunctional cytokine acting in various cells. It induces the differentiation of β cells to antibody-producing plasma cells, T cell growth and differentiation, the differentiation of myeloid leukemic cell lines into macrophages, megakaryocyte maturation, and the neuronal differentiation of PC12 cells development (25,26). BUD is reported to exhibit anti-inflammatory activity via the inhibition of inflammatory mediators TNF-α and IL-6. In order to prove that the spray-drying process, polymers, and excipients did not alter the therapeutic activity of drug, BUD and developed formulations were tested against TNF-α and IL-6 in A549 alveolar epithelial cells (27).
BUD has a low molecular weight of 430.53 Da and an oral bioavailability of 6–11% with a half life of 2–3 h (21). BUD forms esters in all tissues; pharmacokinetic modeling has shown that ester formation occurs primarily in the large airways and lungs, sustaining local anti-inflammatory activity. High doses of ICSs are recommended in the treatment of moderate to severe persistent asthma. It is well accepted that ICSs have fewer systemic side effects than oral or parenteral glucocorticosteroids (28), but there is still some concern about the long-term safety of high doses of ICSs (29). Long-term systemic side effects of high doses of ICSs include thinning of the skin and easy bruising. The aim of inhaled administration of corticosteroids in respiratory disease is to achieve high local concentrations of active drug in the lungs while limiting systemic exposure. A dose–response relationship has been demonstrated for BUD (30,31) and an increase in the dose and frequency of BUD administration has been shown to be beneficial in quickly reducing inflammation and bronchoconstriction in patients with unstable asthma (28). Thus, inhaled corticosteroids should preferentially combine a high fraction of the dose that reaches the airways with a low swallowed fraction (32,33). Respirable fraction of currently available dry powder inhalation (DPI) formulations is not more than 30% (34–36), which means that only 30% of the total dose reaches the site of action, thus increasing frequency and dose of drug administration. This also increases the systemic side effects mentioned above. Conventional DPI formulations of this drug are available in the market but not as controlled-release formulations. This necessitates the development of novel formulations. Pulmonary targeting can be achieved by prolongation of pulmonary residence time either by reducing dissolution rate of drug particle (drug lipophilicity or crystal structure), reducing release from drug delivery system (liposomes or microparticles), or initiation of biological interaction resulting in prolonged pulmonary residence time (ester formation or capturing in membrane structures). Thus, development of useful controlled-release formulations for use in the respiratory tract presents additional challenges because, apart from controlling drug release in the lung environment, drug particles need to avoid removal by the lung clearance mechanisms for the period of drug delivery.
No report is available comparing the effect of various carriers on respirable fraction of drug and particle engineering technology in order to generate sustained-release microparticles using chitosan as polymer with improved deposition profiles of BUD. As a part of our continuing efforts to develop novel drug delivery systems (37–41) and based on literature reports, the objectives of our current work were (1) to develop and characterize conventional formulations (using various grades of inhalable lactose) and novel spray-dried microparticles viz. pulmosols (prepared with various sugars), microspheres, and porous particles (generated using natural polymers viz. gelatin and chitosan) in order to achieve sustained-release profile and to improve respirable fraction of formulation to the lungs; (2) assessment of acute pulmonary toxicity of microparticles in vivo by analyzing bronchoalveolar lavage fluid (BALF) after intratracheal instillation of microparticles in rats; (3) to perform in vitro anti-inflammatory activity and cytotoxicity of BUD and developed formulations to prove tolerance of novel formulations in living systems; and (4) in vivo pulmonary deposition pattern of developed conventional formulation in human volunteers by gamma scintigraphy.
MATERIALS AND METHODS
Materials
BUD was obtained from Lupin Ltd., Mumbai; chitosan was procured from S.D. Fine Chemicals, Mumbai; and different grades of inhalable lactose were obtained as gift sample from DMV Int., The Netherlands. Alveolar epithelial cancer cell line A549 was obtained from NCCS Pune. Cell culture reagent, DMEM/F12 and MTT (3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide, triazolyl blue) were obtained from Sigma Chemicals, Maharashtra, India. Flasks and cell culture plates were purchased from Nunc, Roskilde, Denmark.
Assay for Degree of Deacetylation of Chitosan
It has been proposed to define chitosan and chitin as soluble or insoluble in 0.1 M acetic acid, respectively, or by degree of deacetylation. Greater than 20% of deacetylation is the proposed definition of chitosan (42). Chitosan is made by alkaline N-deacetylation of chitin. The term chitosan does not refer to a uniquely defined compound; it merely refers to a family of copolymers with various fractions of acetylated units. It consists of two types of monomers: chitin monomers and chitosan monomers. Chitin is a linear polysaccharide consisting of (1-4)-linked 2-acetamido-2-deoxy-β-d-glucopyranose. Chitosan is a linear polysaccharide consisting of (1-4)-linked 2-amino-2-deoxy-β-d-glucopyranose. Degree of deacetylation of chitosan affects overall charge density, an increasing presence of ammonium groups resulting in decrease in the cross-linking density related to hydrogen bonding and hydrophobic interactions (20,43). An increase in degree of deacetylation also results in increased swelling due to an increase in number of ionic sites and their counterions. Degree of deacetylation dictates the reactivity, solubility, and viscosity of chitosan solutions. It was calculated using the infrared (IR) frequency-based equation proposed by Baxter et al. (43; Eq. 1):
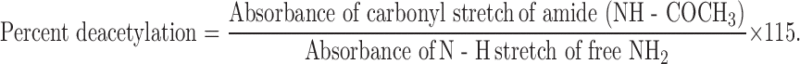 |
1 |
Results of the IR spectra of chitosan showed the characteristic peaks for N–H stretch (νmax 3,346 cm−1) for free amine and C=O stretch (νmax 1,633 cm−1) for amide carbonyl. From the Eq. 1, percent deacetylation was calculated as 45% which is within the range definition for chitosan.
Optimization of Spray-Drying Process Parameters
Microparticles were prepared at laboratory scale by spray drying using the Labultima Mini Spray Dryer (Mumbai, India). Spray drying is a one-step process that converts liquid feed (solution, coarse suspension, colloidal dispersion) to a dried particulate form. Principal advantages of spray drying with respect to pulmonary drug delivery are ability to manipulate and control particle size and size distribution, particle shape, and density in addition to macroscopic powder properties such as bulk density, flowability, and dispersibility (44). Various process parameters were optimized by 32 factorial design. Surface response curves were plotted using Stat-Ease Design-Expert v.7 software (Fig. 1). Effect of aspirator rate (varied from 40% to 60%), spraying air flow pressure (2–4 bar), and inlet temperature (varied from 100°C to 130°C) on moisture content of product, yield, and entrapment efficiency of drug were studied. For all these experiments, concentration of chitosan solution was 0.5% w/v (45,46). Following were the conditions of spray drying: inlet temperature, 130°C; outlet temperature, 80°C; aspirator rate, 240 mWc (60%); solution feed rate, 2 ml/min; spraying air flow pressure, 2 bar. These process parameters were selected from batch CH3 where the concentration of chitosan solution was 0.5% w/v.
Fig. 1.

Factorial design for optimization of process parameters
Preparation of Microparticles
Conventional DPI Formulations
BUD DPI formulations were developed on laboratory scale (100 g) using various grades of novel inhalable lactose like pharmatose, lactohale, inhalac, and mannitol in various combinations (fine lactose/coarse lactose, 60:40 and fine lactose/coarse lactose, 70:30). Earlier, Heng et al. (47) used the sieving process for the development of DPI formulations of salbutamol sulfate; therefore, we used this process to develop BUD DPI formulations on laboratory scale. BUD was initially mixed with fine lactose using mesh #100 and this premix was blended with coarse lactose to homogeneity in geometric proportions using mesh #80. In all these 16 developed batches (Table I), 25 mg of formulation was equivalent to 200 µg of BUD. Effect of particle size of excipients (fine or coarse) on fine particle fraction (FPF) of drug was assessed using twin stage impinger (TSI) study (48).
Table I.
Development and Evaluation of BUD DPI Formulations
| Formulation type | Batch code | Excipients/drug/polymer ratio | Excipients | Percent drug content/percent drug loading (% ± SD) | %FPF (% ± SD) | EI | %CI | Hausner ratio |
|---|---|---|---|---|---|---|---|---|
| Conventional fine lactose/coarse lactose | L1 | 60:40 | A + B | 105.7 ± 0.98 | 31.8 ± 0.22 | 46.3 | 28.6 | 1.4 |
| L2 | C + B | 109.6 ± 0.27 | 34.5 ± 0.34 | 49.2 | 25.0 | 1.3 | ||
| L3 | D + B | 115.7 ± 1.27 | 26.1 ± 0.09 | 44.8 | 42.9 | 1.8 | ||
| L4 | F + B | 113.0 ± 1.02 | 22.2 ± 0.12 | 43.9 | 28.6 | 1.4 | ||
| L5 | A + E | 101.1 ± 2.03 | 12.8 ± 0.08 | 31.8 | 22.2 | 1.3 | ||
| L6 | C + E | 103.4 ± 0.88 | 27.2 ± 0.83 | 36.5 | 20.0 | 1.3 | ||
| L7 | D + E | 107.0 ± 1.92 | 24.6 ± 0.03 | 43.1 | 18.2 | 1.2 | ||
| L8 | F + E | 103.5 ± 1.32 | 31.7 ± 0.01 | 51.8 | 37.5 | 1.6 | ||
| Conventional fine lactose/coarse lactose | K1 | 70:30 | A + B | 109.3 ± 2.09 | 31.3 ± 0.03 | 49.6 | 22.2 | 1.3 |
| K2 | C + B | 112.6 ± 2.21 | 30.9 ± 0.35 | 43.6 | 18.2 | 1.2 | ||
| K3 | D + B | 109.9 ± 0.02 | 30.8 ± 0.45 | 48.2 | 25.0 | 1.3 | ||
| K4 | F + B | 106.0 ± 0.21 | 31.9 ± 0.08 | 48.8 | 33.3 | 1.5 | ||
| K5 | A + E | 101.7 ± 1.03 | 18.4 ± 0.65 | 37.8 | 25.0 | 1.3 | ||
| K6 | C + E | 104.7 ± 1.45 | 33.3 ± 0.09 | 45.6 | 22.2 | 1.3 | ||
| K7 | D + E | 99.1 ± 0.93 | 31.5 ± 0.83 | 48.5 | 25.0 | 1.3 | ||
| K8 | F + E | 108.6 ± 2.01 | 30.4 ± 0.98 | 48.3 | 27.3 | 1.4 | ||
| Pulmosols drug/mannitol | PS1 | 1:50 | 10% w/w | 31.3 ± 0.34 | – | – | – | – |
| PS2 | 1:200 | 20% w/w | 10.0 ± 0.38 | – | – | – | – | |
| PS3 | 1:100 | 20% w/w | 49.6 ± 0.65 | – | – | – | – | |
| PS4 | 1:50 | 20% w/w | 87.7 ± 1.03 | 29.3 ± 0.01 | 48.6 | 12.5 | 1.1 | |
| Microspheres drug/gelatin | G1 | 1:1 | 1% w/w | 56.9 ± 1.01 | – | – | – | – |
| G2 | 1:2 | 88.6 ± 1.34 | 18.2 ± 0.09 | 38.3 | 30.0 | 1.4 | ||
| G3 | 1:5 | 38.8 ± 1.09 | – | – | – | – | ||
| G4 | 1:7 | 24.1 ± 0.09 | – | – | – | – | ||
| Microspheres drug/gelatin/HPβ-CD | G5 | 1:0.5:1.5 | 92.5 ± 0.99 | – | – | – | – | |
| Microspheres drug/chitosan | CH1 | 1:2 | 1% w/w | 7.3 ± 0.82 | – | – | – | – |
| CH2 | 1:5 | 106.7 ± 0.45 | – | – | – | – | ||
| CH3 | 1:2 | 0.5% w/w | 86.0 ± 0.98 | 35.7 ± 0.23 | 54.5 | 30.8 | 1.4 | |
| CH4 | 1:4 | 90.6 ± 1.08 | – | – | – | – | ||
| CH5 | 1:7 | 101.4 ± 1.45 | – | – | – | – | ||
| Microspheres drug/chitosan/HPβ-CD | CH6 | 1:1:1 | 0.5% w/w | 101.4 ± 1.98 | – | – | – | – |
| CH7 | 1:0.5:1.5 | 102.9 ± 2.04 | – | – | – | – | ||
| Microspheres drug/gelatin/chitosan | GC1 | 1:2:2 | 1% w/w | 83.0 ± 1.04 | – | – | – | – |
| GC2 | 1:2:5 | 109.8 ± 1.38 | – | – | – | – | ||
| GC3 | 1:1:1 | 55.7 ± 1.34 | – | – | – | – | ||
| Microspheres drug/compritol 888 ATO | CM1 | 1:1 | 1% w/w | 44.7 ± 1.00 | – | – | – | – |
| CM2 | 1:2 | 49.3 ± 0.92 | – | – | – | – | ||
| CM3 | 1:5 | 91.1 ± 0.66 | – | – | – | – | ||
| Porous particles drug/gelatin | P1 | 1:2 | 1% w/w | 98.4 ± 0.46 | 12.2 ± 0.02 | 31.8 | 25.0 | 1.3 |
| Porous particles drug/chitosan | PC1 | 1:2 | 0.5% w/w | 95.9 ± 0.78 | 46.8 ± 0.09 | 64.2 | 26.7 | 1.4 |
According to the BP procedure, ten capsules were used for each deposition experiment
A pharmatose 125 M, B pharmatose 150 M, C lactohale 300, D lactohale 200, E lactohale 100, F inhalac, FPF fine particle fraction, EI effective index, CI Carr's index
Pulmosol Formulations
Based on the report on spray-dried composites of the drug bendroflumethiazide with polyethylene glycol by Corrigan et al. (49), we attempted development composites of BUD with different sugars. Different sugars like lactose, sucrose, mannitol, fructose, and dextrose and carriers like albumin and PEG 4000 were used in formulation development of pulmosols. Initially, blank batches were spray dried to determine the optimum concentration of sugar as well as to determine powder flow properties. Mannitol was chosen as carrier in formulations as it yielded free flowing powder with maximum drug loading. Different ratios of drug/mannitol (formulations PS1–PS4), as shown in Table I, were spray dried at optimized process conditions.
Microspheres Formulation
BUD and natural polymers viz. chitosan and gelatin were spray dried at optimized process parameters. Corrigan et al. reported chitosan cospray-dried multiparticulates of salbutamol using varying concentrations of 0.5–2% chitosan and this group observed that 0.5% w/v was the optimum concentration of polymer based on entrapment efficiency and release profile from developed composites (48). Hence, gelatin (1 wt.%/ml solution in water), chitosan (0.5 wt.%/ml solution in 0.1 M HCl) were spray dried and the drug/polymer ratio was optimized based on the percent drug entrapment and release profile (Table I). The final concentration of the solution to be spray dried was adjusted to 1 and 0.5 wt.%/ml for gelatin and chitosan, respectively. BUD and polymer were dissolved in equal parts of methanol and water. The polymeric phase was mixed using Ultra-Turrax at 13,000 rpm to which the methanolic phase was slowly added and the solution was stirred to homogeneity. This solution was spray dried to get microspheres.
Porous Particle Formulations
BUD and natural polymers viz. chitosan and gelatin were spray dried in water/methanol (1:1) as 1.0% and 0.5% w/v, respectively. Solution containing BUD, polymer, and a blowing agent was atomized into the drying chamber and brought in contact with a hot air stream. Blowing agent which is trapped in droplets decomposes at higher temperatures creating a void in the center of the particle (50,51). Since air stream temperature is greater than that of the droplet, the droplet temperature increases until the evaporation temperature of the solvent is reached. Solvent at the surface (blowing agent) begins to evaporate causing solvent below the surface of droplet to diffuse to the surface. The droplet, as it passed through the spray chamber forms a hollow particle (52). Hence, porous particles were generated by adding chloroform (5% v/v) as a blowing agent. The drug/polymer ratio was optimized based on the percent drug entrapment and release profile. The effect of HPβ-cyclodextrin (HPβ-CD) on entrapment efficiency and drug release of microspheres and porous particles was also assessed as it is reported in literature that HPβ-CD improves entrapment efficiency along with release profile (53). Microspheres and porous particles obtained by spray drying were formulated with inhalable lactoses.
In Vitro Assessment of Developed Aerosol Formulations
In vitro deposition of dry powders for inhalation was determined using a twin impinger [Copley Instruments (Nottingham) Ltd.]. The 25-mg formulation was weighed and loaded into size 3 hydroxypropyl methyl cellulose (HPMC) stick-free capsule (Associated Capsules Pvt. Ltd., India), which was individually installed in a Rotahaler® device. Rotahaler® was attached to the impinger which contained 7 and 30 ml of collecting solvent [acetonitrile/buffer (disodium hydrogen orthophosphate, pH 3.0) 650:350] in stages 1 and 2, respectively. Capsule contents were released by twisting the Rotahaler® and the system was vacuumed to produce air streams of 60 l/min for 5 s. Liquid in stages 1 and 2 was collected, diluted to 100 mL, and measured by ultraviolet (UV) spectrophotometry at 244 nm. Each deposition experiment involved aerosolization of ten capsules. Respirable fraction was calculated as the amount deposited in the lower stage as a percentage of the emitted dose (ED; amount emitted into the upper and lower stages excluding the amount remaining in the device). All formulations were analyzed in triplicates. Statistical analysis was carried out using Sigma Stat-2.0, Jandel Scientific Software.
Formulations were also subjected to Anderson cascade impactor (ACI) [Copley Instruments (Nottingham) Ltd.] studies to determine the mass median aerodynamic diameter (MMAD) and geometric standard deviation (GSD; 54). ACI utilizes eight jet stages enabling the classification of aerosols from 9 µm and above to 0.4 µm (at 60 L/min) and allows airborne particulates to impact upon stainless steel impaction surfaces. A final filter collects all particles smaller than 0.4 µm. Rotahaler® device was filled with a no. 3 HPMC stick-free capsule (Associated Capsules Pvt. Ltd., India) loaded with 25 mg of powder (200 µg BUD). Test was conducted at a flow rate of 60 L/min for 4 s. Three fine-particle determinations were performed on each test formulation and analyzed by UV spectrophotometry. Starting at the filter, a cumulative mass deposition (undersize in percentage) vs cut-off diameter of the respective stages was derived. Calculation by interpolation of mass of active ingredient with an aerodynamic diameter of <5 µm gave the FPF. It is considered to be directly proportional to the amount of drug able to reach the pulmonary tract in vivo: consequently, the higher the percentage of FPF, the deeper the estimated lung deposition will be. Data was statistically analyzed using Sigma Stat-2.0, Jandel Scientific Software.
Drug content of BUD formulations recovered from twin impinger apparatus was determined by UV spectrophotometry. Absorbance was measured at λmax of 244 nm for BUD analysis. Concentration was determined by reference to a calibration curve prepared from dilutions of stock solutions of BUD.
Drug Content, Content Uniformity, and In Vitro Release Studies
Drug content for conventional formulations, drug entrapment of developed novel formulations, and content uniformity was determined by the method as described above. In vitro release profile of microparticles was performed using the Flow-Through Cell Apparatus-USP IV at 37°C with a flow rate of 16 mL/min (in 100 mL of phosphate buffer, pH 7.0). Five-milliliter samples were taken, filtered through a 0.4-µm filter, and replaced with 5 mL of fresh medium at 37°C. Samples were analyzed by UV spectroscopy at 244 nm. Mechanism of drug release was determined using various kinetic models using the PCP Disso V3 software.
Fourier Transform Infrared Spectroscopy
IR spectra were recorded from 4,000 to 500 cm−1 with a Fourier transform infrared spectrometer (Nicolet, USA) to confirm drug entrapment in the polymer. Samples were prepared by processing compressed KBr disks (55).
Characterization of Particle Shape by Scanning Electron Microscopy
Morphology of particles was evaluated by scanning electron microscopy using JSM-840A/WDS/EDS Sys-Jeol, Japan. Powders were scattered onto a thin film of a two-component epoxy resin and coated with a gold layer (52). SEM micrographs are shown in Fig. 2.
Fig. 2.

SEM micrographs: a pulmosols, b chitosan microspheres, c chitosan porous particles
Characterization of Microparticles by Differential Scanning Calorimetry and Crystalline State by X-ray Powder Diffraction
Thermal behavior of microparticles was investigated using a Perkin-Elmer DSC-7 differential scanning calorimeter/TAC-7 thermal analysis controller with an Intracooler-2 cooling system (Perkin-Elmer Instruments, USA) to prove drug–excipients compatibility in the formulations (Fig. 3). Samples of about 3 mg were placed in 50-µL perforated aluminum pans and sealed. Heat runs for each sample were set from 50°C to 300°C using nitrogen as the blanket gas. The apparatus was indium–cyclohexane calibrated.
Fig. 3.

DSC spectra of developed formulations: a BUD porous particles, b BUD microspheres, c BUD pure, d chitosan pure, e BUD conventional formulation
X-ray powder diffraction (XRPD) is another powerful and widely used tool for crystalline state evaluation. Diffraction patterns of BUD, excipients, and microparticles were determined using a Siemens Diffractometer D5000 (Siemens, Germany) with a Cu line as the source of radiation (WL1Z1.5406 A, WL2Z1.54439 A). XRPD patterns are shown in Fig. 4.
Fig. 4.

XRPD patterns of a BUD, b microspheres, c porous particles, d chitosan
Evaluation of Other Physical Characteristics
Effective index (EI) is the geometric mean of total ED and FPF, represented by Eq. 2 (36):
 |
2 |
where DF is the device fraction (amount of drug retained in the DPI device).
Bulk and tapped densities were measured using a tap density tester (Thermonik, Campbell Electronics). Apparent volume occupied by a mass of powder of about 10 mg, carefully placed into a 5-mL graduated cylinder, was determined before and after packing (tapped more than 500 times in order to obtain the closest packed densities). Bulk and tapped density values allow the determination of Carr's compressibility index by Eq. 3 (46):
 |
3 |
The Hausner ratio is a measure of flowability of the drug and is calculated using Eq. 4. A low Hausner ratio means that the drug has a high flowability (46):
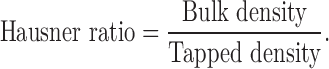 |
4 |
Percent porosity (ε) is one of the methods used to determine the compressibility of powder that is the degree of volume reduction due to an applied pressure, is the measurement of porosity changes during compaction, and is calculated using Eq. 5 (56):
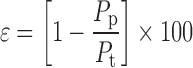 |
5 |
where Pp and Pt are bulk density and tapped density, respectively.
Moisture content was determined by the Karl Fischer method of analysis.
HPLC Method Development for Analysis of BUD and its Metabolite(s) in Plasma
The method used to quantify BUD in plasma samples employed a Phenomenex C18 analytical column (5 µ, 250 × 4.6 mm) connected to a high-performance liquid chromatography (HPLC; model SPD-M2OA 230 V) system (Shimadzu Corporation, Japan) consisting of a LC-8A pump, SPD-M2OA PDA detector, C3M-20A flow cell, a degasser unit, and a 20-µL injection loop. The LC Solution software was used for data collection. The mobile phase consisted of methanol/water (80:20 v/v). The flow rate was 1.5 mL/min. The wavelength of detection was 244 nm (λmax for BUD). The injection volume was 20 µL. The method was validated for linearity, precision, repeatability, sensitivity, and selectivity (57). Plasma/BALF/tissue samples were analyzed using the developed HPLC method. HPLC and UV spectra are shown in Fig. 5.
Fig. 5.

HPLC and UV chromatograms: a BUD microspheres formulation, b metabolite of BUD in plasma, c metabolite of BUD in BALF, d metabolite of BUD in tissue
In Vivo Studies After Intratracheal Administration of Developed Formulations
On the basis of results of FPF and particle size analysis, formulations L2, CH3, and PC1 were selected for in vivo studies (batch L2 and PC1 were blended with inhalable lactose based on the entrapment efficiency such that 25 mg of formulation ≈ 200 µg of BUD). Animals used in these experiments were male/female Wistar rats (n = 60), weighing 250 to 350 g. The Animal Ethics Committee of Bombay Veterinary College, Parel, Mumbai approved the experimental protocol.
Intratracheal Administration of Developed Formulations
All animals were weighed prior to anesthesia. Rats were anesthetized by intraperitoneal injection of ketamine (50 mg/kg) and xylazine (5 mg/kg). Intratracheal instillation was performed using vein flow which was inserted into the distal part of the trachea. Control rats (n = 4) were instilled with placebo formulation, whereas treated rats were instilled with developed formulations (0.4–0.5 mg of formulation ≈ 3–4 µg BUD). Here, we assumed 40% of deposition fraction of formulation which was same during in vitro studies. To have appropriate comparison of intratracheal dose with the other routes for bioavailability, we calculated the dose based on the surface area of the animal since it is a more accurate method than dose calculation based on weight (58,59). In order to favor lower airway penetration of the instilled solution, all animals were placed in dorsal recumbence during recovery of anesthesia (9). At different time intervals, blood was collected (0.5, 1, 2, 3, and 4 h) in a centrifuge tube containing ethylenediaminetetraacetic acid (EDTA) and stored at refrigeration conditions (60,61). Blood samples were centrifuged and plasma was collected. BUD metabolite was precipitated out (white precipitate) from plasma using dichloromethane (DCM), supernatant was decanted, and this precipitate was dissolved in methanol. Methanol was evaporated under vacuum and residue was injected into the HPLC system after diluting with the mobile phase. The volume of plasma/DCM/methanol used for extraction was 1:1:1 (62–65). Animals were then euthanized, respiratory tract (trachea along with lungs) were removed very carefully, and washed with 3 mL of sterile saline (0.9% w/w). Several researchers used 0.9% sterile solution to collect BALF (9). BUD forms active metabolite 6β-hydroxy budesonide in biological fluids. The formation of this metabolite was noticed from the HPLC analysis of BALF sample (structure 2 in the “Results and Discussion” section). Hydroxylated metabolite of BUD being more polar in nature than BUD enhanced its solubility in normal saline (60,61,66,67). The collected BALF was centrifuged to remove cell debris and supernatant was collected for the extraction of metabolite from BALF as mentioned above. Cytological analysis of BALF was performed (9,68). Lungs were separated from the trachea and processed for extraction of metabolite. Before subjecting to extraction procedure, lungs were weighed and were homogenized with the aid of water (2 mL). This homogenized tissue material was centrifuged; supernatant was collected and subjected to extraction procedure as mentioned for plasma (69). All these samples were diluted with mobile phase and then injected into the HPLC system for estimation of BUD metabolite. Time–concentration profiles after intratracheal administration of formulations was plotted (Fig. 6). Pharmacokinetic parameters viz. Ka, Ke, Tmax, Cmax, AUC (using trapezoidal rule), and Vd were calculated (see Table II) from the time–concentration profiles (70–72). Histopathological study of the trachea and lungs (at the end of 4 h) was performed for all formulations to observe morphological changes in the respiratory tract.
Fig. 6.

Time vs concentration profile of formulations: a conventional, b microspheres, c porous particles
Table II.
Pharmacokinetic Parameters in Rats After Intratracheal Administration of Developed BUD Formulations
| Pharmacokinetic parameters | Intratracheal administration (n = 4) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Conventional formulation (batch L2) | Microspheres formulation (batch CH3) | Porous particles formulation (batch PC1) | |||||||
| Plasma | Lungs | BALF | Plasma | Lungs | BALF | Plasma | Lungs | BALF | |
| Ka (h−1) | 0.31 | 0.07 | 0.08 | 0.29 | 0.33 | 0.33 | 2.26 | 0.66 | 1.29 |
| T 1/2(Ka) (h) | 2.23 | 9.64 | 8.18 | 2.37 | 2.10 | 2.09 | 0.31 | 1.04 | 0.54 |
| Ke (h−1) | 0.07 | 0.06 | 0.06 | 0.08 | 0.04 | 0.06 | 0.050 | 0.13 | 0.03 |
| T 1/2(Ke) (h) | 9.04 | 12.52 | 11.22 | 9.42 | 15.61 | 11.28 | 14.00 | 5.45 | 22.77 |
| T max (h) | 1.0 | 0.5 | 2.0 | 2.0 | 0.5 | 2.0 | 3.0 | 1.0 | 1.0 |
| C max (ng/ml) | 3.95 | 1.54 | 0.97 | 4.19 | 3.04 | 0.50 | 10.49 | 5.43 | 1.15 |
| AUC0–4 h (ng h/ml) | 6.12 | 3.24 | 2.01 | 8.78 | 3.32 | 1.34 | 24.15 | 11.29 | 4.11 |
| Vd (L) | 0.81 | 2.08 | 3.31 | 0.76 | 1.05 | 4.19 | 0.31 | 0.59 | 0.88 |
| Relative bioavailability (F) | – | 0.65 (1.2-fold compared to conventional formulation) | 1.92 (3.5-fold compared to conventional formulation) | ||||||
Ka absorption rate constant, T 1/2 half life, Ke elimination rate constant, T max time required to reach maximum concentration, C max concentration maximum, AUC area under the curve, Vd apparent volume of distribution
Acute Toxicity Studies After Intratracheal Administration
Acute toxicity study of batch PC1 (porous particles DPI formulation) was conducted as it was the best formulation among all developed formulations. Toxicity studies were conducted in Wistar rats to prove safety of developed DPI formulation after single exposure by intratracheal administration as per OECD guidelines (9). Animals were sacrificed after 14 days. Histopathological examination of the trachea, lungs, heart, brain, adrenals, esophagus, stomach, duodenum, small intestine, large intestine, and kidney was performed (Fig. 7). Hematological analysis was conducted to check any abnormalities with respect to white blood cells (WBC), red blood cells (RBC), platelet count, and hemoglobin percentage.
Fig. 7.

Histopathology of lungs (first column) and trachea (second column): a control, b conventional formulation, c microspheres formulation, d porous particle formulation
In Vitro Anti-Inflammatory Activity and Cytotoxicity of Developed Formulations
Novel formulations of BUD viz. microspheres and porous particles were generated using a particle engineering technology that is spray-drying technology. The process of generation of microparticles involves the use of organic solvents and polymers. Microparticles were generated by spraying the feed into the drying chamber which was maintained at 130°C. Drug is exposed to higher temperature hence to prove that the spray-drying process, polymers, and excipients did not alter the therapeutic activity of the drug, BUD, and developed formulations tested against TNF-α and IL-6.
Anti-Inflammatory Activity Against TNF-α and IL-6
Screening in LPS Stimulated THP-1 Cells
Proinflammatory cytokine production by lipopolysaccharide (LPS) in THP-1 cells was measured. Briefly, THP-1 cells were cultured in RPMI 1640 culture medium (Gibco BRL, Pasley, UK) containing 100 U/ml penicillin and 100 mg/ml streptomycin, (100X solution, Sigma Chemical Co., St Louis, MO, USA) containing 10% fetal bovine serum (JRH). The cells were differentiated with phorbol myristate acetate (Sigma). Following cell plating, the test compounds or vehicle (0.5% dimethyl sulfoxide [DMSO]) were added to each well and the plate was incubated for 30 min at 37°C. Finally, LPS (Escherchia coli 0127:B8; Sigma Chemical Co., St. Louis, MO, USA) was added at a final concentration of 1 µg/ml. Plates were incubated at 37°C for 24 h, 5% CO2. Supernatants were harvested and assayed for TNF-α and IL-6 by enzyme-linked immunosorbent assay (ELISA) as described by the manufacturer (BD Biosciences). Cells were simultaneously evaluated for cytotoxicity using CCK-8 from Dojindo Laboratories. Percent inhibition of cytokine release compared to the control was calculated (Table III; 73).
Table III.
Results of Anti-Inflammatory Screening of BUD and Developed Formulations
| Sample | LPS-induced cytokine production in THP-1 cells (percent inhibition) | hPBMC (IC50)a | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TNF-α | IL-6 | Toxicity | TNF-α | IL-6 | ||||||||||
| 0.1 | 1 | 10 | 30 | 0.1 | 1 | 10 | 30 | 0.1 | 1 | 10 | 30 | |||
| BUD | 0 | 0 | 3 | 25 | 69 | 68 | 87 | 90 | 0 | 0 | 0 | 0 | 0.03 | 0.1 |
| Batch L2 | 0 | 0 | 2 | 26 | 62 | 61 | 75 | 84 | 0 | 0 | 0 | 0 | 0.035 | 0.09 |
| Batch CH3 | 0 | 0 | 3 | 24 | 64 | 66 | 78 | 89 | 0 | 0 | 0 | 0 | 0.033 | 0.11 |
| Batch PC1 | 0 | 0 | 5 | 28 | 67 | 6 | 82 | 88 | 0 | 0 | 0 | 0 | 0.036 | 0.09 |
Batch L2 conventional formulation, Batch CH3 microspheres formulation, Batch PC1 porous particles formulation
aIC50 values are expressed in micrometers
hPBMC Assay
TNF-α Assay
Human peripheral blood mononuclear cells assay (hPBMCs; 74,75): TNF-α is a key cytokine produced primarily by monocytes and macrophages which is involved in host immune response (76). Hence, the 50% inhibitory concentration (IC50) value for TNF-α and IL-6 inhibition was determined in human monocytes. Peripheral blood mononuclear cells (PBMC) were harvested from human blood and suspended in RPMI 1640 culture medium (Gibco BRL, Pasley, UK) containing 10% FCS, 100 U/ml penicillin (Sigma Chemical Co., St Louis, MO, USA), and 100 mg/ml streptomycin (Sigma Chemical Co., St Louis, MO, USA) at 1 × 106 cells per milliliter. Cells were added to three wells of a 96-well culture plate. Following cell plating, hPBMCs were pretreated with various concentrations of test compound or vehicle (0.5% DMSO) for 30 min and stimulated with 1 µg/ml LPS (E. coli 011:B4; Sigma Chemical Co., St. Louis, MO, USA) and the incubation was continued for 5 h at 37°C, 5% CO2. In each experiment, the viability, as determined by trypan blue exclusion, was uniformly >98%. Supernatants were harvested and assayed for TNF-α by ELISA as described by the manufacturer (OptiEIA ELISA sets, BD BioSciences). The IC50 values were calculated by a nonlinear regression method (Table III).
IL-6 Assay
hPBMCs were harvested from human blood and suspended in RPMI 1640 culture medium containing 10% FCS, 100 U/ml penicillin, and 100 mg/ml streptomycin at 10 × 106 cells per milliliter. In accordance with a previous study (75), in preliminary experiments, we established that treatment of hPBMCs with 20 ng/ml recombinant human IL-6 (R&D Systems; Minneapolis, MN, USA) for 10 min at 37°C robustly induces phosphorylation of STAT3. Accordingly, to investigate the effect of developed formulations on IL-6 signaling pathway, hPBMCs were pretreated with varying concentrations of test samples (0.1, 1, 10, and 30 µM) for 1 h and subsequently stimulated with 20 ng/ml IL-6. Following 10 min stimulation, hPBMCs were extensively washed with ice-cold PBS (without Ca2+ and Mg2+). Cells were then lysed with cell lysis reagent solution (Sigma Aldrich, St. Louis, MO, USA) supplemented with a protease inhibitor cocktail (Roche, Indianapolis, IN, USA). Lysates were used immediately or were aliquoted and stored at −20°C. Effect on IL-6 bioactivity by the sample is represented as percent inhibition compared with the control. Data was statistically compared using Bartlett statistics and normality test (Table III).
MTT Cytotoxicity Assay
Cell Culture
Alveolar epithelial cancer cell line A549 was obtained from NCCS Pune. Cell cultures were grown to confluence in DMEM/F12 media supplemented with 10% FCS, 100 IU/ml penicillin, 50 µg/ml streptomycin, and 10 mM glutamine in humidified atmosphere under 5% CO2 at 37°C. Trypsin–EDTA solution (containing 2.5 g trypsin and 0.2 g EDTA per liter of Hank's balanced salt solution) was used for subculturing and cell isolation.
Method
Cells were harvested from confluence cultures on the fourth day after subculturing. Cells (5 × 103) were seeded and grown in 96-well tissue culture plates in a final volume of 150 µL in humidified atmosphere for 48 h (37°C and 5% CO2). Developed formulations, namely, conventional, microspheres, and porous particles formulation of BUD along with pure BUD were evaluated for cytotoxic effect. The placebo formulation was also studied for comparison using similar dilutions. Each formulation was tested for varying concentrations over the range of 10 nM–500 µM of drug concentration. Cells were treated with varying concentrations of drug in each formulation for 24 h; each concentration was studied in replicates of three wells. The whole experiment was repeated twice to get statistically valid results. After incubation, 10 µL of MTT labeling agent (5 mg/ml in PBS) was added to each well. The microplate was incubated for 4 h in humidified atmosphere. After incubation, 100 µL of solubilizing solution (10% SDS in 0.01 M HCl) was added to each well. The plate was incubated overnight. The OD570 nm was measured with a reference wavelength at 630 nm using an ELISA reader (77–79).
In Vivo Pulmonary Deposition Pattern of Developed Conventional Formulation in Human Volunteers by Gamma Scintigraphy
Pulmonary scintigraphy is a noninvasive method for visualizing deposition pattern and quantifying the amount of drug deposited and concept of proof of bioavailability (13, 80–82). The study was carried out at Radiation Medicine Centre (BARC), Tata Hospital, Mumbai, based on protocol approved by the Ethical Committee of the Department of Clinical Pharmacology, TNMC and BYL Nair Charitable Hospital, Mumbai, 400 008, India (IEC/20/04). The scintigraphy study was conducted for developed conventional formulation as radiolabeling of pure drug was easy compared with microspheres and porous particles.
BUD was radiolabeled with 99mTc by physical adsorption of radioactivity then blended with inhalable lactose and filled in size 3 capsule. Dose administered to each volunteer was one capsule (200 µg of BUD labeled with ≅5 mCi of technetium). The following method was used for radiolabeling of BUD: (1) Selection of solvent for radiolabeling—Initially, drug was suspended in saline, water, methyl ethyl ketone (MEK), and chloroform to see the effect of these solvents on the particle size of the drug. It was observed that, in water and saline, particle morphology remained unchanged and, therefore, it was decided to use water and saline as suitable solvents for radiolabeling of the drug. When chloroform was used, particle morphology was found to be changed and particles became needle-shaped, while in case of MEK, particle size was reduced. Therefore, chloroform and MEK were not chosen for radiolabeling of BUD. (2) Scintigraphic measurement after DPI inhalation—The radioactivity was obtained from RMC and was measured by dosimeter. BUD was added to saline containing 99mTc for physical adsorption and then evaporated to dryness using magnetic stirrer along with hot plate. This radiolabeled BUD was then mixed with inhalable lactose by sieving. During in vitro deposition studies, 1 mCi per dose of 99mTc was used and filled in capsules. These capsules were analyzed for drug content and percent respirable fraction using TSI. In order to avoid radioactivity contamination, FPF was determined and not MMAD. Radiolabeled formulation was prepared at RMC and was kept in a lead pot. The volunteer inhaled one radiolabeled capsule containing 200 µg of BUD labeled with 5 mCi of activity. All radiological information was acquired with the gamma camera Infinia. Subjects were asked to inhale in front of the gamma camera with midpoint of chest at the center of the field of view. During the study, dynamic images were obtained for 10–15 min with a split of 30 s time frame so that the movement of labeled compound could be followed. Gamma scintigraphic images obtained with DPI for one representative volunteer are depicted in Fig. 8a, b. The region of interest (ROI) was drawn from the figure using the computer software attached to the camera. Counts were corrected for attenuation correction factor, lung clearance, and time for elimination of radioactivity from the lungs. The amount of drug deposited into the lungs was calculated based upon the counts of radioactivity in the formulation (83,84). Analysis of the images yielded data for the lungs, oropharynx, trachea, and inhalation device in terms of radioactive counts, termed as “primary counts” (85). As gamma rays pass through the body to the gamma camera, they are absorbed and scattered by different body tissues through which they pass. Therefore, in order to quantify aerosol deposition accurately, it is necessary to further correct “primary counts” for attenuation by utilizing dimensionless correction factors, termed as tissue attenuation correction factors (ACFs) (86); correction factor for radioactive decay, lung clearance, and absorption and these counts are termed as “secondary counts.” Counts corrected for tissue attenuation = “primary counts” × ACFs. The ACF is obtained to correct the scattering of gamma rays by that particular tissue and calculated on the basis of the thickness of that tissue. The correction factor for the mouth and oropharynx is 1.88; for the lungs is 2, for the trachea is 1, and for the stomach is 3.35 (87).
Fig. 8.

Scintigraphic image obtained with DPI: a for one representative volunteer, b scintigraphic scan after administration of BUD from DPI
RESULTS AND DISCUSSION
Optimization of Spray-Drying Process Parameters
Effect of aspirator rate, spraying air flow pressure, and inlet temperature on the moisture content of product, percent yield, and percent drug entrapment was studied (46) by plotting surface response curves (Fig. 1) and interpreted in terms of percent contribution of each factor and from equations obtained from the Stat-Ease Design-Expert v.7 software. Effect of aspirator rate and spraying air flow pressure on percent yield was graphically shown in Fig. 1a and contribution of aspirator rate, spraying air flow pressure, and both parameters on yield of product was found to be 98.3197%, 1.0541%, and 0.6261%, which was calculated from Eq. 6. This proved that aspirator rate was a major parameter affecting yield of product:
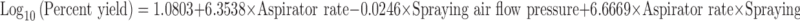 |
6 |
Effect of aspirator rate and spraying air flow pressure on percent drug entrapment was graphically shown in Fig. 1b and contribution of aspirator rate, spraying air flow pressure, and both parameters on drug entrapment was found to be 51.4420%, 1.6225%, and 46.9354%, which was calculated from Eq. 7. This proved that aspirator rate alone and aspirator rate and spraying air flow pressure together were major parameters affecting drug entrapment:
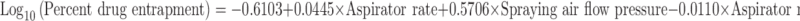 |
7 |
Effect of aspirator rate and inlet temperature on percent moisture content was graphically shown in Fig. 1c and contribution of aspirator rate, inlet temperature, and both parameters on moisture content was found to be 7.3979%, 15.5549%, and 77.0471%, which was calculated from Eq. 8. This proved that aspirator rate and inlet temperature together were major parameters affecting moisture content:
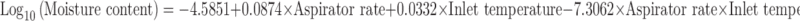 |
8 |
Preparation of Microparticles
All 16 conventional batches (Table I) developed using inhalable lactose in combination of fine lactose/coarse lactose, 60:40 and 70:30, were evaluated for various parameters, but the optimized batch was selected based on the %FPF. Batch L2 (lactohale 300 M/pharmatose 150 M, 60:40) was selected as it gave the maximum FPF (34.50%) compared to other batches. For optimization of pulmosols, initially blank batches of lactose, sucrose, mannitol, fructose, dextrose, albumin, and PEG 4000 were prepared. Except mannitol, all other carriers viz. lactose, sucrose, fructose, dextrose, albumin, and PEG 4000 yielded very sticky product, which could not be removed from the cyclone, so mannitol was chosen for drug loading as it gave free flowing powder. Drug/carrier ratios of 1:50, 1:100, and 1:200 were tried out at concentrations of 10% and 20% w/w solution (Table I). Batch PS4 was selected to determine the %FPF (10 mg of formulation ≈ 200 µg of BUD) because drug/carrier ratio was minimum (1: 50) with optimum drug loading of about 88.0% w/w compared with other batches of pulmosols.
Microspheres of gelatin and chitosan, alone and in combination, compritol 888 ATO were generated with drug/polymer ratios as shown in Table I. Batch G2 (1% w/w solution, 1:2 drug/gelatin) and CH3 (0.5% w/w solution, 1:2 drug/chitosan) were selected for which percent drug entrapment was about 89.0% and 86.0% w/w, respectively. During preparation of chitosan microspheres, initially 1% w/w solution was tried in batches CH1 and CH2 for spray drying; here, batch CH2 showed maximum entrapment efficiency, but still this batch was not further carried over because blocking of feed pipe was noticed sometimes during the spray-drying process due to viscosity of the solution. Further batches of chitosan microspheres were prepared with 0.5% w/w solution and drug/chitosan ratios of 1:2, 1:4, and 1:7; batch CH3 (1:2, drug/chitosan) was selected as amount of chitosan was less with percent drug entrapment of about 86.0% w/w, compared with other batches. Effect of HPβ-CD on entrapment efficiency and drug release was studied and it was found that addition of HPβ-CD increased entrapment efficiency of BUD but no change in release profile was observed. Combinations of gelatin–chitosan were tried out but not carried further as amount of polymer was more in these batches compared to batches prepared with gelatin and chitosan alone. Similar observations were noted in case of batches prepared using compritol 888 ATO.
Porous particles of gelatin and chitosan were prepared (Table I) with percent drug entrapment of about 98% and 96% w/w, respectively. Microspheres and porous particles were formulated with inhalable lactose (lactohale 300 M/pharmatose 150 M, 60:40) based on the entrapment efficiency such that 25 mg of formulation ≈ 200 µg of BUD.
In Vitro Assessment of Developed Aerosol Formulations
Selected formulations were characterized for in vitro deposition by TSI and ACI (54). Pulmosols and formulations of microspheres and porous particles prepared using gelatin showed FPF of 29.32%, 18.24%, and 12.18%, respectively, so these were not selected for further characterization. FPF for conventional (L2), microspheres (CH3), and porous particles formulation (PC1) was about 34%, 36%, and 47%, respectively, with standard deviation (SD) of ≤0.5 for all, which were very promising. Values were compared using normality (p = 0.159 > 0.05) and paired t test (p = 0.727 > 0.05). The difference is considered to be statistically significant (p < 0.001; one-way analysis of variance [ANOVA] test). MMAD, for conventional, microspheres, and porous particles formulation were 2.75, 4.60, and 4.30 µm, respectively. GSD for the above batches was 2.56, 1.75, and 2.54.
Drug Content, Content Uniformity, and In Vitro Release Studies
Drug content and content uniformity for all conventional formulations was in the range of 90–110% w/w. Drug entrapment for developed novel formulations varied from 10% to 110% w/w as given in Table I.
Mechanism of drug release was determined using various kinetic models. Coefficient of correlation were calculated from plots of Q vs t (cumulative percent drug release vs time), log Q vs log t, and Q vs square root of t (48). Regression coefficients (near to one) for zero-order, matrix, and Korsmeyer–Peppas kinetic equations confirmed release by slow zero-order kinetics through diffusion matrix. Korsmeyer–Peppas plot indicated good linearity (r2 = 0.9722).
Fourier Transform Infrared Spectroscopy
IR spectrum of BUD showed peaks at 3,378 cm−1 (O–H stretch), 2,935 cm−1 (C–H stretch), and 1,720 and 1,659 cm−1 (C=O stretch). Chitosan showed typical peaks at 3,446 and 1,633 cm−1 for N–H stretch and C=O stretch for free amine and amide carbonyl functionalities, respectively. In the IR spectrum of BUD chitosan microparticles, typical peaks for the N–H stretch of free NH2 of chitosan disappeared due to possible cross-linking of chitosan with drug via amine and hydroxyl functionalities while the broad peak ranging from 2,800 to 2,600 cm−1 observed as well as intensity of peaks in the range of 1,650–1,720 cm−1 was dramatically reduced which confirmed entrapment of BUD in chitosan.
Characterization of Particle Shape by Scanning Electron Microscopy
Morphology of microparticles was investigated and SEM micrographs are illustrated in Fig. 2. Figure 2a of pulmosols showed spherical particles with wide particle size distribution (1–20 µm) but uniform spherical microspheres and porous particles (Fig. 2b, c) were obtained with diameter ranging from 1 to 4 µm with similar particle morphology and size.
Characterization of Microparticles by Differential Scanning Calorimetry and Crystalline State by X-ray Powder Diffraction
DSC scans are shown in Fig. 3. Chitosan showed broad endotherm (Fig. 3d) at 109.13°C while BUD showed sharp endotherm (Fig. 3c) at 264.14°C. DSC spectra of microspheres revealed two endotherms (Fig. 3b) for chitosan and BUD at 105.18°C and 235.38°C, respectively. Again, in the case of porous particles, two endotherms (Fig. 3a) were observed for chitosan and BUD at 106.50°C and 222.98°C, respectively. BUD conventional formulation showed two sharp endotherms (Fig. 3e) for inhalable lactose and BUD at 148.44°C and 221.06°C, respectively. This confirmed no interaction between BUD and excipients occurred after the spray-drying process (52).
On the other hand, XRPD patterns (Fig. 4) showed that the spray-drying process did not completely affect the crystalline form of BUD (Fig. 4a). Peaks that represent the spray-dried samples (both microspheres and porous particles; Fig. 4b, c) correspond to those of chitosan (Fig. 4d) but differ in intensity, which means that the major component (in formulations) is partly amorphous.
Evaluation of Other Physical Characteristics
The EI of microspheres (54.48; Table I) and porous particle formulations (64.22) was found to be better compared to conventional formulation (49.21), suggesting more effective microparticulate drug deposition into lungs (36).
Carr's index and Hausner ratio, which are considered as appropriate methods of evaluation of the flow properties of solids, were also determined from tapped and bulk density values (46). Carr's index values of <25 are usually taken to indicate good flow characteristics; values beyond 40 indicate poor powder flowability. Carr's index values (Table I) for all formulations were found to be in the range of 20–30% which indicated good powder flow properties.
Hausner ratio is measure of flowability of powder. A low Hausner ratio means that the powder has a high flowability (but it should be >2.0). For all formulations, this ratio was in the range of 1.2–1.7 (Table I), indicating good flowability.
Percent porosity for chitosan microspheres and porous particles formulation was found to be 30.76 and 26.66, respectively, which was good compared to developed conventional formulations (6–20%). Moisture content for all formulations was <1%.
HPLC Method Development for Analysis of Budesonide Metabolite in Plasma
A simple, rapid, and stability-indicating HPLC method for BUD was developed. The method was linear over a concentration range of 10 ng/ml–100 µg/ml (r2 = 0.9990; Fig. 5). The limit of detection of BUD was 25 ng/ml and the limit of quantitation was 100 ng/ml (57). Plasma/BALF/tissue samples were analyzed using the developed method. The HPLC chromatogram of all samples showed the presence of a common peak at tR = 4.10 min while the peak of BUD (tR = 4.36 min) was not observed, which confirmed the transformation of BUD into one of its metabolite. In literature, BUD is reported to get transformed into three active metabolites as given below (1).
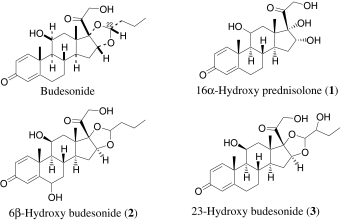
As expected from structural changes in reported metabolites, the new peak appeared at lesser retention time (tR = 4.10 min; Fig. 5b1, b2) compared to BUD peak (tR = 4.36 min; Fig. 5a1, a2). Another interesting change was noticed; the peak at tR = 4.10 min showed an λmax of 261 nm (Fig. 5b2). A 17-nm bathochromic shift in the λmax of the metabolite could be attributed to the addition of extra substitution (OH) in the chromophore region or near the chromophore region. By analyzing reported metabolite structures by Woodward–Fieser rules, structure 2 (6β-hydroxy budesonide) could be assigned which has the λmax of 262 nm (increment for OH at γ position = 18 nm) while the other two metabolites (structures 1 and 3) has the λmax of 244 nm. Thus, from reported metabolites, most likely 6β-hydroxy budesonide (structure 2) seems to be formed as a major metabolite which appeared at tR = 4.10 min. This metabolite was formed in major amount and was present in all chromatograms, therefore, was used to study pharmacokinetic parameters of BUD formulations.
In Vivo Studies After Intratracheal Administration of Developed Formulations
Mean plasma/BALF/tissue concentration vs time profiles after administration of developed formulations are shown in Fig. 6. All calculated pharmacokinetic parameters such as Ka, Ke, Tmax, Cmax, AUC (using trapezoidal rule), and Vd are listed in Table II. The higher the absorption rate constant, the faster the absorption of the drug. Thus, porous particles formulations would give the fastest onset of effect since it has the highest Ka value of 0.66 in the lungs compared with conventional and microspheres formulations with Ka values of 0.07 and 0.33 in the lungs, respectively (Table II). Peak concentration is a function of both the rate of absorption and the rate of elimination. Since the elimination rate was almost same for all three formulations, as the absorption rate increases, the peak concentration increases. The elimination half life of the formulations was calculated and found to be 9.04, 9.42, and 14.00 h for conventional, microspheres, and porous particles formulations, respectively. This indicated that the novel porous particles formulation extended the release of the drug and thus retention of drug particles in the lungs.
The formulation with the lowest peak concentration would give the lowest intensity of pharmacologic effects. From the Cmax values of the formulations, it was found that the porous particle formulation has higher Cmax value (5.43 ng/ml) in the lungs compared to conventional (1.54 ng/ml) and microspheres formulation (3.04 ng/ml), indicating higher lung deposition. Conventional formulation (L2) exhibited the lowest intensity of effect as Cmax of microspheres formulation L2 was much lower than the Cmax of conventional formulation CH3 and porous particles formulation PC1 (Table II). Area under the plasma concentration time curve (AUC) is very useful for calculating the relative bioavailability of different formulations. The AUC of formulation PC1 and CH3 was comparatively higher than formulation L2. The AUC values for porous particles formulation in lungs and plasma were higher than AUC values of conventional and microspheres formulation, indicating higher local and systemic bioavailability of the drug after administration of novel formulations. The relative bioavailability of novel formulations that is microspheres and porous particle formulations was found to be onefold to fourfold higher than the conventional formulation. All listed pharmacokinetic parameters were analyzed statistically using SigmaStat-2.0, Jandel Scientific Software. Data was analyzed using the Kolmogorov–Smirnov test (KS). It was found that there was no significant change in p values (0.21–0.38; p > 0.05). The results of the pharmacokinetic study indicated that the developed novel formulations extended the half life (14.00 h) compared to the conventional formulation (9.04 h) with onefold to fourfold improved local and systemic bioavailability. Estimates of relative bioavailability (Table II) suggested that developed formulations had excellent lung deposition characteristics with extended T1/2 from 9.4 to 14 h compared to conventional formulation. From DPI formulation PC1, concentration of drug was found to be increased fourfold in the lungs, indicating pulmonary targeting potential.
Cytological analysis of BALF was performed and differential count was found to be around 90% epithelial cells, 2–6% neutrophils, 1–3% lymphocytes, and 0–1% both eosinophils and macrophages in all samples viz. control, conventional, microspheres, porous particles formulation, and sample collected after acute toxicity study which indicated that cell count remained unchanged. The higher percentage of epithelial cells in BALF may be because of pressing the lungs during BALF collection.
Histopathological slides of trachea and lungs (at the end of 4 h) did not show any histopathological changes or sign of irritation or inflammation in the tissues which indicated safety of developed novel formulation (Fig. 7).
Acute toxicity study was performed for 14 days after single intratracheal administration of formulation PC1. Histopathological slides indicated no structural changes in the heart, brain, adrenals, lungs, esophagus, stomach, duodenum, small intestine, large intestine, and kidney which proved the safety of the formulation after intratracheal administration. Hematological analysis was conducted and it was found that there were no abnormalities with respect to WBC, RBC, platelet count, and hemoglobin percentage compared to the control.
In Vitro Anti-Inflammatory Activity and Cytotoxicity Study of Developed Formulations
Initially, BUD along with all developed formulations was evaluated for anti-inflammatory activity against TNF-α and IL-6 in LPS-induced cytokine production in THP-1 cells. In preliminary screening, pure BUD along with all formulations showed 75–87% inhibition of IL-6 production at 10 µM while they did not show TNF-α inhibition up to the concentration of 30 µM (Table III). Data was statistically analyzed using Bartlett statistics (TNF-α = 0.0889; IL-6 = 0.03943) and normality test (KS coefficient for TNF-α = 0.3524 > 0.10 and IL-6 = 0.3699 > 0.10, therefore passes the normality test). ANOVA assumes that the data are sampled from populations that follow Gaussian distributions. This assumption was tested using the KS method. As KS coefficients for TNF-α and IL-6 were 0.3524 and 0.3699 > 0.10, respectively, it passes the normality test.
Furthermore, these samples were evaluated in human monocytes (hPBMC assay) as mentioned earlier. BUD along with all developed formulations exhibited promising TNF-α as well as IL-6 inhibitory activity with IC50s of 0.03–0.035 and 0.09–011 µM for TNF-α and IL-6, respectively (Table III). The values for bioactivity of BUD alone and in formulation remained unchanged, indicating that therapeutic activity of BUD in the formulation was the same as that of BUD. Data was statistically compared using KS normality test which indicated that there was no significant difference (TNF-α: p = 0.710; IL-6: p = 0.289).
The reduction of TNF-α and IL-6 induced by BUD alone and in the form of formulations was same which proved that the spray-drying process, polymers, and excipients did not alter therapeutic activity of the drug.
MTT is reduced metabolically by active cells via the action of dehydrogenase enzymes resulting in intracellular purple formazan. Hence, the difference in cell proliferation rate in control compared to cells treated with developed BUD formulations could be estimated using MTT assay. Placebo formulations at different dilutions and BUD formulations in different concentrations exhibited insignificant change in OD (Table IV).
Table IV.
Results of MTT Cytotoxicity Study
| Concentration (µM) | Percent cell viability after 24 h treatment | Percent cell viability after placebo formulations treatment | ||
|---|---|---|---|---|
| Conventional formulations (L2) | Microspheres formulations (CH3) | Porous formulations (PC1) | ||
| 0.01 | 97.5 ± 5.8 | 107 ± 9.5 | 100.5 ± 7.6 | 106 ± 3.3 |
| 0.1 | 96.9 ± 5.7 | 100 ± 1.2 | 100 ± 8.4 | 102 ± 6.9 |
| 1 | 95.9 ± 5.9 | 96.9 ± 2.8 | 114 ± 10.2 | 96 ± 6.4 |
| 10 | 96.3 ± 3.0 | 95.8 ± 3.5 | 96.7 ± 5.7 | 93 ± 4.3 |
| 100 | 93.6 ± 6.25 | 96.7 ± 3.5 | 96.5 ± 7.1 | 99 ± 2.2 |
| 500 | 94.3 ± 4.0 | 91.4 ± 5 | 95.3 ± 4.8 | 96 ± 1.9 |
Percent cell viability after exposing cells to developed formulations, namely conventional, microspheres, and porous particles was in the range of 95–100% indicated that these formulations were not toxic to alveolar epithelial cells.
Results clearly indicated that the placebo formulation was nontoxic to alveolar epithelial cells, whereas developed formulations of BUD did not exhibit significant cytotoxicity.
In Vivo Pulmonary Deposition Pattern of Developed Conventional Formulation in Human Volunteers by Gamma Scintigraphy
The gamma scintigraphy images obtained with BUD conventional DPI formulation for one representative volunteer were depicted in Fig. 8. The ROI was drawn on the figure using a computer software attached to the camera. Counts were corrected for attenuation correction factor, lung clearance, and time for elimination of radioactivity from the lungs. The amount of drug deposited into the lungs was calculated based upon counts of the radioactivity in the formulation. The maximum respirable fraction obtained from radiolabeled BUD formulation was 22.16% with a standard deviation of 4.31.
CONCLUSION
Given the experimental evidence reported in the sections above, the use of microparticles offers an opportunity with sustained-release profile and improved delivery of the drug to the pulmonary tract. Microparticulates extended the drug release up to 4 h. Twin impinger and ACI analysis demonstrated high respirable fractions for microparticles, which have potential for pulmonary delivery. Microparticulates appear to be a promising new pharmaceutically acceptable filler and/or carrier which have controlled-release properties for DPI products. Results of pharmacokinetic study indicated that developed novel formulations showed higher lung deposition and extended release of drug, thereby retention of drug particles in the lungs. These formulations showed improved local and systemic bioavailability (onefold to fourfold higher relative bioavailability than the conventional formulation) indicating pulmonary targeting potential of developed formulations as well. In vitro anti-inflammatory activity and cytotoxicity of developed formulations proved that the spray-drying process, polymers, and excipients did not alter therapeutic activity of the drug as well as did not exhibit significant cytotoxicity in alveolar epithelial cell line. Scintigraphy results indicated potential in vitro–in vivo correlation in the performance of conventional BUD DPI formulation. This data can be accepted as proof of bioavailability of the drug after pulmonary delivery.
Abbreviations
- ACFs
attenuation correction factors
- ACI
Anderson cascade impactor
- AUC
area under the curve
- BALF
bronchoalveolar lavage fluid
- BUD
budesonide
- CI
Carr's index
- COPD
chronic obstructive pulmonary disease
- DCM
dichloromethane
- DPI
dry powder inhaler
- DSC
differential scanning calorimetry
- ED
emitted dose
- EI
effective index
- FPF
fine particle fraction
- GSD
geometric standard deviation
- hPBMCs
human peripheral blood mononuclear cells assay
- HPMC
hydroxypropyl methyl cellulose
- ICSs
inhaled corticosteroids
- IL-6
interleukin-6
- Ka
absorption rate constant
- Ke
elimination rate constant
- MEK
methyl ethyl ketone
- LPS
lipopolysaccharide
- MMAD
mass median aerodynamic diameter
- OECD
Organization for Economic Co-operation and Development
- SEM
scanning electron microscope
- TNF-α
tumor necrosis factor-alpha
- tR
retention time
- TSI
twin stage impinger
- Vd
apparent volume of distribution
- XRPD
X-ray powder diffraction
References
- 1.Szefler SJ. Pharmacodynamics and pharmacokinetics of budesonide: a new nebulized corticosteroid. J Allergy Clin Immunol. 1999;104:S175–S183. doi: 10.1016/S0091-6749(99)70059-X. [DOI] [PubMed] [Google Scholar]
- 2.Basyigit I, Yildiz F, Ozkara SK, Yildirim E, Boyaci H, Ilgazli A. Addition of inhaled corticosteroid on combined bronchodilator therapy in patients with COPD. Pulm Pharmacol Ther. 2005;20:1–5. doi: 10.1016/j.pupt.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Cazzola M. Single inhaler budesonide/formoterol in exacerbations of chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2006;19:79–89. doi: 10.1016/j.pupt.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Thorsson L, Borga O, Edsbacker S. Systemic availability of budesonide after nasal administration of three different formulations: pressurized aerosol, aqueous pump spray, and powder. Br J Clin Pharmacol. 1999;47:619–624. doi: 10.1046/j.1365-2125.1999.00956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Learoyd TP, Burrows JL, French E, Seville PC. Chitosan-based spray-dried respirable powders for sustained delivery of terbutaline sulfate. Eur J Pharm Biopharm. 2008;68:224–234. doi: 10.1016/j.ejpb.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 6.Fu J, Fiegel J, Krauland E, Hanesa J. New polymeric carriers for controlled drug delivery following inhalation or injection. Biomaterials. 2002;23:4425–4433. doi: 10.1016/S0142-9612(02)00182-5. [DOI] [PubMed] [Google Scholar]
- 7.Koushik K, Kompella UB. Particle & device engineering for inhalation drug delivery. Drug Deliv Technol. 2004;4:40–50. [Google Scholar]
- 8.Ribeiro AJ, Silva C, Ferreira D, Veiga F. Chitosan-reinforced alginate microspheres obtained through the emulsification/internal gelation technique. Eur J Pharm Biopharm. 2005;25:31–40. doi: 10.1016/j.ejps.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 9.Sanna V, Kirschvink N, Gustin P, Gavini E, Roland I, Delattre L, et al. Preparation and in vivo toxicity study of solid lipid microparticles as carrier for pulmonary administration. AAPS PharmSciTech. 2003;5:1–7. doi: 10.1208/pt050227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irngartinger M, Camuglia V, Damm M, Goede J, Frijlink HW. Pulmonary delivery of therapeutic peptides via dry powder inhalation: effects of micronisation and manufacturing. Eur J Pharm Biopharm. 2004;58:7–14. doi: 10.1016/j.ejpb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 11.Jashnani RN, Byron PR, Dalby RN. Testing of dry powder aerosol formulations in different environmental conditions. Int J Pharm. 1995;113:123–130. doi: 10.1016/0378-5173(94)00197-D. [DOI] [Google Scholar]
- 12.Sommerville ML, Hickey AJ. Aerosol generation by metered-dose inhalers containing dimethyl ether/propane inverse microemulsions. AAPS PharmSciTech. 2003;4:1–7. doi: 10.1208/pt040458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leach CL, Davidson PJ, Boudreau RJ. Improved airway targeting with the CFC-free HFA–beclomethasone metered-dose inhaler compared with CFC–beclomethasone. Eur Respir J. 1998;12:1346–1353. doi: 10.1183/09031936.98.12061346. [DOI] [PubMed] [Google Scholar]
- 14.Chougule MB, Padhi BK, Jinturkar KA, Misra A. Development of dry powder inhalers. Recent Patents on Drug Delivery and Formulation. 2007;1:11–21. doi: 10.2174/187221107779814159. [DOI] [PubMed] [Google Scholar]
- 15.Johnson KA. Preparation of peptide and protein powders for inhalation. Adv Drug Deliv Rev. 1997;26:3–15. doi: 10.1016/S0169-409X(97)00506-1. [DOI] [PubMed] [Google Scholar]
- 16.Yu Z, Rogers TL, Hu J, Johnston KP, Williams RO. Preparation and characterization of microparticles containing peptide produced by a novel process: spray freezing into liquid. Eur J Pharm Biopharm. 2002;54:221–228. doi: 10.1016/S0939-6411(02)00050-4. [DOI] [PubMed] [Google Scholar]
- 17.Zeng XM, Martin GP, Tee SK, Marriott C. The role of fine particle lactose on the dispersion and deaggregation of salbutamol sulphate in an air stream in vitro. Int J Pharm. 1998;176:99–110. doi: 10.1016/S0378-5173(98)00300-7. [DOI] [Google Scholar]
- 18.Larhrib H, Martin GP, Marriott C, Prime D. The influence of carrier and drug morphology on drug delivery from dry powder formulations. Int J Pharm. 2003;257:283–296. doi: 10.1016/S0378-5173(03)00156-X. [DOI] [PubMed] [Google Scholar]
- 19.Smith IJ, Billings MP. The inhalers of the future? A review of dry powder devices on the market today. Pulm Pharmacol Ther. 2003;16:79–95. doi: 10.1016/S1094-5539(02)00147-5. [DOI] [PubMed] [Google Scholar]
- 20.Schnurch AB. Chitosan and its derivatives: potential excipients for peroral peptide delivery systems. Int J Pharm. 2000;194:1–13. doi: 10.1016/S0378-5173(99)00365-8. [DOI] [PubMed] [Google Scholar]
- 21.Cook RO, Pannu RK, Kellaway IW. Novel sustained release microspheres for pulmonary drug delivery. J Control Release. 2005;104:79–90. doi: 10.1016/j.jconrel.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Spoelstra FM, Postma DS, Hovenga H, Noordhoek JA, Kauffman HF. Budesonide and formoterol inhibit ICAM-1 and VCAM-1 expression of human lung fibroblasts. Eur Respir J. 2000;15:68–74. doi: 10.1183/09031936.00.15106800. [DOI] [PubMed] [Google Scholar]
- 23.Uings I, Puxeddu I, Temkin V, Smith SJ, Fattah D, Ray KP, et al. Effects of dexamethasone on TNF-alpha-induced release of cytokines from purified human blood eosinophils. Clin Mol Allergy. 2005;3:5–9. doi: 10.1186/1476-7961-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sorbera LA, Martin L, Leeson PA. Treatment of IBD: anti-TNF-α MAb. Drugs Future. 2000;25:669–673. doi: 10.1358/dof.2000.025.07.585694. [DOI] [Google Scholar]
- 25.Kang BS, Chung EY, Yun YP, Lee MK, Lee YR, Lee KS, et al. Inhibitory effects of anti-inflammatory drugs on interleukin-6 bioactivity. Biol Pharm Bull. 2001;24:701–703. doi: 10.1248/bpb.24.701. [DOI] [PubMed] [Google Scholar]
- 26.Escotte S, Tabary O, Dusser D, Teboul CM, Puchelle E, Jacquot J. Fluticasone reduces IL-6 and IL-8 production of cystic fibrosis bronchial epithelial cells via IKK-β kinase pathway. Eur Respir J. 2003;21:574–581. doi: 10.1183/09031936.03.00031803. [DOI] [PubMed] [Google Scholar]
- 27.Smirnov IM, Bailey K, Flowers CH, Garrigues NW, Wesselius LJ. Effects of TNF-α and IL-1β on iron metabolism by A549 cells and influence on cytotoxicity. Am J Physiol. 1999;277:L257–L263. doi: 10.1152/ajplung.1999.277.2.L257. [DOI] [PubMed] [Google Scholar]
- 28.Toogood JH, Baskerville J, Jennings B, Lefcoe NM, Johansson SA. Bioequivalent doses of budesonide and prednisone in moderate and severe asthma. J Allergy Clin Immunol. 1989;84:688–700. doi: 10.1016/0091-6749(89)90297-2. [DOI] [PubMed] [Google Scholar]
- 29.Kamada AK, Szefler SJ, Martin RJ, Boushey HA, Chinchilli VM, Drazen JM, et al. Issues in the use of inhaled glucocorticoids. The Asthma Clinical Research Network. Am J Respir Crit Care Med. 1996;153:1739–1748. doi: 10.1164/ajrccm.153.6.8665030. [DOI] [PubMed] [Google Scholar]
- 30.Miyamoto T, Takahashi T, Nakajima S, Makino S, Yamakido M, Mano K. A double-blind, placebo-controlled dose-response study with budesonide Turbuhaler in Japanese asthma patients. Respirology. 2000;5:247–256. doi: 10.1046/j.1440-1843.2000.00256.x. [DOI] [PubMed] [Google Scholar]
- 31.Busse W, Chervinsky P, Condemi J, Lumry WR, Petty TL, Rennard S. Budesonide delivered by Turbuhaler is effective in a dose-dependent fashion when used in the treatment of adult patients with chronic asthma. J Allergy Clin Immunol. 1998;101:457–463. doi: 10.1016/S0091-6749(98)70353-7. [DOI] [PubMed] [Google Scholar]
- 32.van den Brink KI, Boorsma M, den Brekel AJ Staal-van, Edsbäcker S, Wouters EF, Thorsson L. Evidence of the in vivo esterification of budesonide in human airways. Br J Clin Pharmacol. 2008;66:27–35. doi: 10.1111/j.1365-2125.2008.03164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edsbacker S, Brattsand R. Budesonide fatty-acid esterification: a novel mechanism prolonging binding to airway tissue. Review of available data. Ann Allergy Asthma Immunol. 2002;88:609–616. doi: 10.1016/S1081-1206(10)61893-5. [DOI] [PubMed] [Google Scholar]
- 34.Tronde A, Gillen M, Borgstrom L, Lotvall J, Ankerst J. Pharmacokinetics of budesonide and formoterol administered via 1 pressurized metered-dose inhaler in patients with asthma and COPD. J Clin Pharmacol. 2008;48:1300–1308. doi: 10.1177/0091270008322122. [DOI] [PubMed] [Google Scholar]
- 35.Joshi M, Misra A. Dry powder inhalation of liposomal ketotifen fumarate: formulation and characterization. Int J Pharm. 2001;223:15–27. doi: 10.1016/S0378-5173(01)00705-0. [DOI] [PubMed] [Google Scholar]
- 36.Shah SP, Misra A. Liposomal amikacin dry powder inhaler: effect of fines on in vitro performance. AAPS PharmSciTech. 2004;5:1–7. doi: 10.1208/pt050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naikwade SR, Meshram RN, Bajaj AN. Preparation and in vivo efficacy study of pancreatin microparticles as enzyme replacement therapy for pancreatitis. Drug Dev Ind Pharm. 2009;35:417–432. doi: 10.1080/03639040802422104. [DOI] [PubMed] [Google Scholar]
- 38.Wakode R, Bhanushali R, Bajaj A. Development and evaluation of push–pull based osmotic delivery system for pramipexole. PDA J Pharm Sci Tech. 2008;62:22–31. [PubMed] [Google Scholar]
- 39.Amrutiya N, Bajaj A, Madan M. Development of microsponges for topical delivery of mupirocin. AAPS PharmSciTech. 2009;10:402–409. doi: 10.1208/s12249-009-9220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naikwade SR, Kulkarni PP, Jathar SR, Bajaj AN. Development of time and pH dependent controlled release colon specific delivery of tinidazole. DARU. 2008;16:119–127. [Google Scholar]
- 41.Naikwade SR, Bajaj AN. Development and evaluation of once a day oral controlled multiparticulate drug delivery systems of cefixime trihydrate. Indian J Pharm Educ Res. 2008;42:283–294. [Google Scholar]
- 42.Patel V, Patel M, Patel R. Chitosan: a unique pharmaceutical excipient. Drug Deliv Technol. 2005;5(6). http://www.drugdeliverytech.com.
- 43.Baxter A, Dillon M, Taylor KDA, Roberts GAF. Improved method for I.R. determination of the degree of N-acetylation of chitosan. Int J Biol Macromol. 1992;14:166–169. doi: 10.1016/S0141-8130(05)80007-8. [DOI] [PubMed] [Google Scholar]
- 44.Zijlstra GS, Rijkeboer M, Drooge DJ, Sutter M, Jiskoot W, Weert MV, et al. Characterization of a cyclosporine solid dispersion for inhalation. AAPS J. 2007;9:E190–E199. doi: 10.1208/aapsj0902021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang YC, Chiang CH, Yeh MK. Optimizing formulation factors in preparing chitosan microparticles by spray-drying method. J Microencapsul. 2003;20:247–260. doi: 10.1080/02652040210162522. [DOI] [PubMed] [Google Scholar]
- 46.Sebti T, Amighi K. Preparation and in vitro evaluation of lipidic carriers and fillers for inhalation. Eur J Pharm Biopharm. 2006;63:51–58. doi: 10.1016/j.ejpb.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 47.Heng PWS, Chan LW, Lim LT. Quantification of the surface morphologies of lactose carriers and their effect on the in vitro deposition of salbutamol sulphate. Chem Pharm Bull. 2000;48:393–398. doi: 10.1248/cpb.48.393. [DOI] [PubMed] [Google Scholar]
- 48.Corrigan DO, Healy AM, Corrigan OI. Preparation and release of salbutamol from chitosan and chitosan co-spray dried compacts and multiparticulates. Eur J Pharm Biopharm. 2006;62:295–305. doi: 10.1016/j.ejpb.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 49.Corrigan DO, Healy AM, Corrigan OI. The effect of spray drying solutions of bendroflumethiazide/polyethylene glycol on the physicochemical properties of the resultant materials. Int J Pharm. 2003;262:125–137. doi: 10.1016/S0378-5173(03)00338-7. [DOI] [PubMed] [Google Scholar]
- 50.Prinn KB, Costantino HR, Tracy M. Statistical modeling of protein spray drying at the lab scale. AAPS PharmSciTech. 2002;3:1–6. doi: 10.1208/pt030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirst PH, Pitcairn GR, Weers JG, Tarara TE, Clark AR, Dellamary LA, et al. In vivo lung deposition of hollow porous particles from a pressurized metered dose inhaler. Pharm Res. 2002;19:258–264. doi: 10.1023/A:1014482615914. [DOI] [PubMed] [Google Scholar]
- 52.Ungaro F, Rosa GD, Miro A, Quaglia F, Rotonda MIL. Cyclodextrins in the production of large porous particles: development of dry powders for the sustained release of insulin to the lungs. Eur J Pharm Sci. 2006;28:423–432. doi: 10.1016/j.ejps.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 53.Skiba M, Bounoure F, Barbot C, Arnaud P, Skiba M. Development of cyclodextrin microspheres for pulmonary drug delivery. J Pharm Pharmaceut Sci. 2005;8:409–418. [PubMed] [Google Scholar]
- 54.Boer AHD, Hagedoorn P, Gjaltema D, Goede J, Frijlink HW. Air classifier technology (ACT) in dry powder inhalation part 1: introduction of a novel force distribution concept (FDC) explaining the performance of a basic air classifier on adhesive mixtures. Int J Pharm. 2004;260:187–200. doi: 10.1016/s0378-5173(03)00250-3. [DOI] [PubMed] [Google Scholar]
- 55.Cerchiara T, Luppi B, Chidichimo G, Bigucci F, Zecchi V. Chitosan and poly(methyl vinyl ether-co-maleic anhydride) microparticles as nasal sustained delivery systems. Eur J Pharm Biopharm. 2005;61:195–200. doi: 10.1016/j.ejpb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 56.Jain SK, Agrawal GP, Jain NK. Evaluation of porous carrier-based floating orlistat microspheres for gastric delivery. AAPS PharmSciTech. 2006;7:E1–E9. doi: 10.1208/pt070490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Naikwade SR, Bajaj AN. Development of validated specific HPLC method for budesonide and characterization of its alkali degradation product. Can J Anal Sci Spect. 2008;53:113–122. [Google Scholar]
- 58.Jansson AH, Eriksson C, Wang X. Effects of budesonide and N-acetylcysteine on acute lung hyperinflation, inflammation and injury in rats. Vascul Pharmacol. 2005;43:101–111. doi: 10.1016/j.vph.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 59.Kouno T, Katsumata N, Mukai H, Ando M, Watanabe T. Standardization of the body surface area (BSA) formula to calculate the dose of anticancer agents in Japan. Jpn J Clin Oncol. 2003;33:309–313. doi: 10.1093/jjco/hyg062. [DOI] [PubMed] [Google Scholar]
- 60.Landry ML, Cohen S, Huber K. Comparison of EDTA and acid-citrate-dextrose collection tubes for detection of cytomegalovirus antigenemia and infectivity in leukocytes before and after storage. J Clin Microbiol. 1997;35:305–306. doi: 10.1128/jcm.35.1.305-306.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leung TF, Wong CK, Lam CWK, Li AM, Wong GWK, Fok TF. Plasma TARC concentration may be a useful marker for asthmatic exacerbation in children. Eur Respir J. 2003;21:616–620. doi: 10.1183/09031936.03.00083303. [DOI] [PubMed] [Google Scholar]
- 62.Baek M, Chung HS, Kim Y, Kim DH. Disposition and metabolism of 2-(2″(1″,3″-dioxolan-2-yl)-2-methyl-4-(2′-oxopyrrolidin-1-yl)-6-nitro-2h-1-benzopyran (Skp-450) in rats. Drug Metab Dispos. 1999;27:510–516. [PubMed] [Google Scholar]
- 63.Kronkvist K, Gustavsson M, Wendel AK, Jaegfeldt H. Automated sample preparation for the determination of budesonide in plasma samples by liquid chromatography and tandem mass spectrometry. J Chromatogr. 1998;823:401–409. doi: 10.1016/S0021-9673(98)00592-5. [DOI] [PubMed] [Google Scholar]
- 64.Deventer K, Mikulkova P, Hoecke HV, Eenoo PV, Delbeke FT. Detection of budesonide in human urine after inhalation by liquid chromatography–mass spectrometry. J Pharm Biomed Anal. 2006;42:474–479. doi: 10.1016/j.jpba.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 65.Kaiser H, Aaronson D, Dockhorn R, Edsbacker S, Korenblat P, Kallen A. Dose-proportional pharmacokinetics of budesonide inhaled via Turbuhaler. Br J Clin Pharmacol. 1999;48:309–316. doi: 10.1046/j.1365-2125.1999.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Faouzi MA, Dine T, Luyckx M, Brunet C, Gressier B, Cazin M, et al. High-performance liquid chromatographic method for the determination of budesonide in bronchoalveolar lavage of asthmatic patients. J Chromatogr. 1995;664:463–467. doi: 10.1016/0378-4347(94)00473-I. [DOI] [PubMed] [Google Scholar]
- 67.Larsson AM, Jansson P, Runström A, Brattsand R. Prolonged airway activity and improved selectivity of budesonide possibly due to esterification. Am J Respir Crit Care Med. 2000;162:1455–1461. doi: 10.1164/ajrccm.162.4.9806112. [DOI] [PubMed] [Google Scholar]
- 68.Vyve TV, Chanez P, Lacoste JY, Bousquet J, Michel FB, Godard P. Comparison between bronchial and alveolar samples of bronchoalveolar lavage fluid in asthma. Chest. 1992;102:356–361. doi: 10.1378/chest.102.2.356. [DOI] [PubMed] [Google Scholar]
- 69.Jendbro M, Johansson CJ, Strandberg P, Falknilsson H, Edsbacker S. Pharmacokinetics of budesonide and its major ester metabolite after inhalation and intravenous administration of budesonide in the rat. Drug Metab Dispos. 2001;29:769–776. [PubMed] [Google Scholar]
- 70.Gandhi R, Kaul CL, Panchagnula R. Pharmacokinetic evaluation of an azithromycin controlled release dosage form in healthy human volunteers: a single dose study. Int J Pharm. 2004;270:1–8. doi: 10.1016/j.ijpharm.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 71.Lewis S, Subramanian G, Pandey S, Udupa N. Design, evaluation and pharmacokinetic study of mucoadhesive buccal tablets of nicotine for smoking cessation. Indian J Pharm Sci. 2006;68:829–831. doi: 10.4103/0250-474X.31030. [DOI] [Google Scholar]
- 72.Donnelly R, Seale JP. Clinical pharmacokinetics of inhaled budesonide. Clin Pharmacokinet. 2001;40:427–440. doi: 10.2165/00003088-200140060-00004. [DOI] [PubMed] [Google Scholar]
- 73.Yadav PN, Liu Z, Rafi MM. A diarylheptanoid from lesser galangal (Alpinia officinarum) inhibits proinflammatory mediators via inhibition of mitogen-activated protein kinase, p44/42, and transcription factor nuclear factor-{kappa}B. J Pharmacol Exp Ther. 2003;305:925–931. doi: 10.1124/jpet.103.049171. [DOI] [PubMed] [Google Scholar]
- 74.Henry JR, Rupert KC, Dodd JH, Turchi IJ, Wadsworth SA, Cavender DE, et al. Potent inhibitors of the MAP kinase p38. Bioorg Med Chem Lett. 1998;8:3335–3340. doi: 10.1016/S0960-894X(98)00589-7. [DOI] [PubMed] [Google Scholar]
- 75.Wilson BMG, Severn A, Rapson NT, Chana J, Hopkins P. A convenient human whole blood culture system for studying the regulation of tumor necrosis factor release by bacterial lipopolysaccharide. J Immunol Methods. 1991;139:233–240. doi: 10.1016/0022-1759(91)90193-J. [DOI] [PubMed] [Google Scholar]
- 76.Monn C, Becker S. Cytotoxicity and induction of proinflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse Particles (PM10-2.5) in outdoor and indoor air. Toxicol Appl Pharmacol. 1999;155:245–252. doi: 10.1006/taap.1998.8591. [DOI] [PubMed] [Google Scholar]
- 77.Kompella UB, Bandi N, Ayalasomayajula SP. Subconjunctival nano- and microparticles sustain retinal delivery of budesonide, a corticosteroid capable of inhibiting VEGF expression. Invest Ophthalmol Vis Sci. 2003;44:1192–1201. doi: 10.1167/iovs.02-0791. [DOI] [PubMed] [Google Scholar]
- 78.Greenberg AK, Hu J, Basu S, Hay J, Reibman J, Yie T, et al. Glucocorticoids inhibit lung cancer cell growth through both the extracellular signal-related kinase pathway and cell cycle regulators. Am J Respir Cell Mol Biol. 2002;27:320–328. doi: 10.1165/rcmb.4710. [DOI] [PubMed] [Google Scholar]
- 79.Fujitani Y, Trifilieff A. In vivo and in vitro effects of SAR 943, a rapamycin analogue, on airway inflammation and remodeling. Am J Respir Crit Care Med. 2003;167:193–198. doi: 10.1164/rccm.200205-455OC. [DOI] [PubMed] [Google Scholar]
- 80.Newman SP, Pitcairn GR, Hirst PH, Rankin L. Radionuclide imaging technologies and their use in evaluating asthma drug deposition in the lungs. Adv Drug Deliv Rev. 2003;55:851–867. doi: 10.1016/S0169-409X(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 81.Ball DJ, Hirst PH, Newman SP, Sonet B, Streel B, Vanderbist F. Deposition and pharmacokinetics of budesonide from the Miat Monodose inhaler, a simple dry powder device. Int J Pharm. 2002;245:123–132. doi: 10.1016/S0378-5173(02)00338-1. [DOI] [PubMed] [Google Scholar]
- 82.Pitcairn GR, Hooper G, Luria X, Rivero X, Newman SP. A scintigraphic study to evaluate the deposition patterns of a novel antiasthma drug inhaled from the Cyclohaler dry powder inhaler. Adv Drug Deliv Rev. 1997;26:59–67. doi: 10.1016/S0169-409X(97)00511-5. [DOI] [PubMed] [Google Scholar]
- 83.Bondesson E, Bengtsson T, Borgström L, Nilsson LE, Norrgren K, Trofast E, et al. Planar gamma scintigraphy—points to consider when quantifying pulmonary dry powder aerosol deposition. Int J Pharm. 2003;258:227–240. doi: 10.1016/S0378-5173(03)00148-0. [DOI] [PubMed] [Google Scholar]
- 84.Lee Z, Ljungberg M, Muzic RF, Berridge MS. Usefulness and pitfalls of planar γ-scintigraphy for measuring aerosol deposition in the lungs: a Monte Carlo investigation. J Nucl Med. 2001;42:1077–1083. [PubMed] [Google Scholar]
- 85.Bondesson E, Bengtsson T, Borgstrom L, Nilsson LE, Norrgren K, Trofast E, et al. Planar gamma scintigraphy—points to consider when quantifying pulmonary dry powder aerosol deposition. Int J Pharm. 2003;251:33–47. doi: 10.1016/S0378-5173(02)00579-3. [DOI] [PubMed] [Google Scholar]
- 86.Myers MJ, Lavender JP, Deoliviera JB, Maseri A. A simplified method of quantitating organ uptake using a gamma camera. Br J Radiol. 1981;54:1062–1067. doi: 10.1259/0007-1285-54-648-1062. [DOI] [PubMed] [Google Scholar]
- 87.Pitcairn GR, Newman SP. Tissue attenuation corrections in gamma scintigraphy. J Aerosol Med. 1997;10:187–198. doi: 10.1089/jam.1997.10.187. [DOI] [Google Scholar]