

Abstract
Aims
Calmodulin (CaM) regulates Na+ channel gating through binding to an IQ-like motif in the C-terminus. Ca2+/CaM-dependent protein kinase II (CaMKII) regulates Ca2+ handling, and chronic overactivity of CaMKII is associated with left ventricular hypertrophy and dysfunction and lethal arrhythmias. However, the acute effects of Ca2+/CaM and CaMKII on cardiac Na+ channels are not fully understood.
Methods and results
Purified NaV1.5–glutathione-S-transferase fusion peptides were phosphorylated in vitro by CaMKII predominantly on the I–II linker. Whole-cell voltage-clamp was used to measure Na+ current (INa) in isolated guinea-pig ventricular myocytes in the absence or presence of CaM or CaMKII in the pipette solution. CaMKII shifted the voltage dependence of Na+ channel availability by ≈+5 mV, hastened recovery from inactivation, decreased entry into intermediate or slow inactivation, and increased persistent (late) current, but did not change INa decay. These CaMKII-induced changes of Na+ channel gating were completely abolished by a specific CaMKII inhibitor, autocamtide-2-related inhibitory peptide (AIP). Ca2+/CaM alone reproduced the CaMKII-induced changes of INa availability and the fraction of channels undergoing slow inactivation, but did not alter recovery from inactivation or the magnitude of the late current. Furthermore, the CaM-induced changes were also completely abolished by AIP. On the other hand, cAMP-dependent protein kinase A inhibitors did not abolish the CaM/CaMKII-induced alterations of INa function.
Conclusion
Ca2+/CaM and CaMKII have distinct effects on the inactivation phenotype of cardiac Na+ channels. The differences are consistent with CaM-independent effects of CaMKII on cardiac Na+ channel gating.
Keywords: Na-channel, Calcium, Calmodulin, Ca2+/CaM-dependent protein kinase II
1. Introduction
Voltage-gated Na+ channels are transmembrane proteins responsible for the action potential (AP) upstroke that initiates cell excitation and determines the velocity of impulse propagation.1 In humans, altered Na+ channel gating underlies some forms of long-QT syndrome (LQT3),2 Brugada's syndrome,3 and heritable cardiac conduction disease4,5 leading to life-threatening ventricular arrhythmias. Na+ channel gating is altered in acquired diseases such as cardiac ischaemia and heart failure, and aberrant Na+ channel regulation may contribute to ventricular arrhythmias and sudden cardiac death.
A number of signalling pathways that regulate Na+ channel function6 are altered in the diseased heart, such as cAMP-dependent protein kinase A (PKA)7,8 and PKC.9 Calcium ions (Ca2+) have a fundamental role in the coupling of cardiac myocyte excitation and contraction; however, mechanisms whereby intracellular Ca2+ may directly modulate Na+ channel function have yet to be fully clarified.10 Calmodulin (CaM) regulates Na+ channel gating through binding to an IQ-like motif in the C-terminus.11 Ca2+/CaM-dependent protein kinase II (CaMKII) is one of the most important transducers of Ca2+ signals in many cell types and is highly conserved across animal species.12 CaMKII plays an important role in cardiac physiology, and the specific effects of CaMKII in the myocardium may be difficult to identify because its targets converge with those of PKA, the key mediator of catecholamine responses in the heart. CaMKII regulates Ca2+ handling,13 and overexpression of CaMKII causes left ventricular hypertrophy and dysfunction14 and lethal arrhythmias.15 The effect of CaMKII on cardiac Na+ currents has been examined using adenovirus-mediated CaMKII overexpression.16 However, the direct physiological function of Ca2+/CaM and CaMKII or PKA on cardiac Na+ channels remains controversial. The purpose of this study is to investigate the acute effects of Ca2+/CaM, CaMKII, and PKA on native cardiac Na+ channels. We demonstrated that CaM and CaMKII have distinct effects on the inactivation phenotype of cardiac Na+ channels, consistent with CaM-independent effects of CaMKII on cardiac Na+ channel gating. These findings have been presented in preliminary form.17
2. Methods
The investigation confirms with the Guide for the Care and Use of Laboratory Animals published by US National Institution of Health (NIH Publication No. 85-23, revised 1996) and was approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions (which conform to National Institutes of Health guidelines).
2.1. Isolated guinea-pig left ventricular myocytes
Single ventricular myocytes were prepared from adult guinea-pig heart using enzymatic dissociation methods as described previously.18 After digestion, cells were stored at room temperature in a high potassium solution (mM: K-glutamate 120, KCl 25, MgCl2 1, glucose 10, HEPES 10, and EGTA 1; pH 7.4) until use.
2.2. Patch-clamp experiments
Whole-cell voltage-clamp was used to measure INa with an Axopatch 200A patch-clamp amplifier (Molecular Devices Corp.) at room temperature (22°C). Voltage command protocols were generated by custom-written software. Capacitance compensation was optimized and series resistance was compensated by 40–80%. Membrane currents were filtered at 5 kHz and digitized with 12-bit resolution through a DigiData-1200 interface (Molecular Devices Corp.). The patch pipettes had 1–1.5 MΩ tip resistances when filled with a pipette solution containing (in mM): NaCl 5, CsCl 40, glutamate 80, CsOH 80, Mg-ATP 5, EGTA 5, HEPES 10, and CaCl2 1.5 (free [Ca2+] = 100 nM, pH 7.2, with CsOH, liquid junction potential = +5 mV). The bath solution contained (in mM) NaCl 10, MgCl2 2, CsCl2 5, TEA-Cl 125, and HEPES 20 (pH 7.4 with CsOH). In all experiments, recording was begun 10–15 min after establishment of the whole-cell mode to permit stabilization of the voltage dependence and kinetics of gating.
For the measurement of APs, whole-cell current-clamp was performed at 37°C; the patch pipettes had 3–4 MΩ tip resistances when filled with pipette solution containing (in mM): 120 K+-glutamate, 10 KCl, 10 HEPES, 5 EGTA, and 5 Mg-ATP adjusted to pH 7.2 with KOH and standard Tyrode's bath solution. The stimulation frequency was varied over a range of pacing intervals from 0.5–4.0 s, and the steady-state APs were recorded and analysed at least 1 min after the initiation of pacing at each pacing interval.
2.3. Solutions/pharmacology
CaMKII (CaMKIIα, Biolabs, NE, USA) was activated by diluting the desired amount of enzyme in a reaction buffer containing (in mM): Tris–HCl 50, MgCl2 10, dithiothreitol 2, Na2EDTA 0.1 (pH 7.5) with ATP 100 µM, CaM 1.2 µM, and CaCl2 2 mM, then incubating for 10 min at 30°C. Activated CaMKII was diluted in the pipette solution (1:10) such that the final CaMKII concentration in the pipette was 5000 U/mL. The CaMKII-containing pipette solution which contains both CaMKII and CaM is referred to as the CaMKII condition. CaM (in the absence of CaMKII), the CaMKII-specific inhibitor autocamtide-2-related inhibitory peptide (AIP, Calbiochem), PKA inhibitor peptide (PKI, Millipore), and PKA catalytic subunit (New England Biolabs) were added to the pipette solution to final concentrations of 110, 200 nM, 50 µM, and 5000 U/mL, respectively.
2.4. Voltage protocols
The protocols used for the assessment of the voltage dependence of activation/inactivation, recovery from inactivation, and entry into inactivation are provided as insets in the relevant figures. To determine the V1/2 and slope factor k, steady-state inactivation data were fit with a Boltzmann function of the form: I/Imax = {1 + exp[(V− V1/2)/k]}−1. Recovery from inactivation data were fit with a bi-exponential function of the form: I(t)/Imax = A1 exp(−t/τ1) + A2 exp(−t/τ2) + A3, using a non-linear least squares minimization. The decay phase of the current during a voltage step was fit with a bi-exponential function of the form: I(t) = Af exp(−t/τf) + As exp(−t/τs), where Af and As are the fractions of fast and slow inactivating components, respectively. The persistent inward (late) Na+ current was the tetrodotoxin (TTX)-sensitive current (30 µM) measured at 100–500 ms after the depolarizing voltage step. Data analysis was performed with custom-written software (Ionview) and Origin (Microcal).
2.5. Western blotting and phosphorylation
PCR amplified cDNAs encoding regions of NaV1.5 were cloned into the EcoRI site of pGEX-6P1 (GenBank ID: U78872). The resulting glutathione-S-transferase (GST) fusion constructs were confirmed for proper orientation and sequence by automated DNA sequencing. These plasmids were used to transform BL21 Escherichia coli. The proteins were expressed by isopropylthiogalactoside induction of cultures grown to an absorbance at 600 nm (A600) between 0.6–1.0, followed by culture for another 1–2 h. Cells were pelleted and resuspended in ice-cold phosphate-buffered saline (PBS) with protease inhibitors and then lysed by sonication. The sonicate was centrifuged and the soluble fraction was passed over a GSTrap column (GE Healthcare). The column was washed with more than 10 column volumes of PBS and eluted with 10 mM glutathione. The purified fragments were separated by electrophoresis on 4–12% polyacrylamide gel electrophoresis (PAGE). The purified fragments were incubated with 5000 IU CaMKIIα in the presence of γ-32P-labelled ATP. The labelled peptides were run on the same percentage gel and exposed to film.
2.6. Statistics
The results are presented as mean ± SD or SEM. Statistical comparisons were made using a one-way ANOVA followed by Bonferroni/Dunn tests for multiple comparisons. Differences in serial studies were assessed by repeated measures ANOVA. In some cases, an unpaired Student's t-test was used to evaluate the significance of the difference between means. Statistical significance was assumed at P < 0.05.
3. Results
3.1. CaMKII-dependent phosphorylation on Na+ channel
We first demonstrated that the α-subunit of the cardiac Na+ channel (NaV1.5) is a substrate for CaMKII-dependent phosphorylation. We prepared GST fusion peptides of the N- and C-termini and all of the interdomain linkers of NaV1.5. Purified NaV1.5 peptides incubated with activated CaMKII and ATP-γ-32P were phosphorylated predominantly on the I–II linker and to a lesser extent in the C-terminus (Figure 1).
Figure 1.
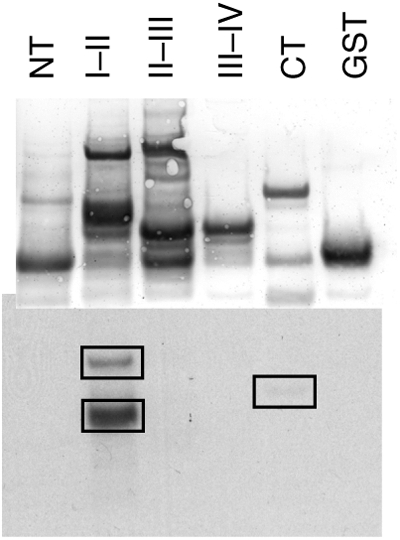
In vitro phosphorylation of the Na channel by CaMKII. Purified GST fusion proteins of the intracellular domains of NaV1.5 were phosphorylated in vitro with CaMKII in the presence of γ-32P-labelled ATP. Proteins were separated by SDS–PAGE and transferred to nitrocellulose. Total protein was visualized by Ponceau-S stain (top) and incorporated 32P was visualized by autoradiography (bottom). Phosphorylation occurred predominantly on the I–II linker, with a smaller amount occurring on the CT domain. NT, amino terminus; CT, carboxyl terminus. I–II, II-III, and III–IV denotes the I–II, II–III, and III–IV interdomain linkers, respectively.
3.2. Current–voltage relationship and steady-state gating
The current–voltage (I–V) relationship of cardiac INa was influenced by CaMKII and CaM (Figure 2A and B). The peak current density was significantly larger with CaMKII (−45 to +10 mV) or CaM (−50 to +5 mV) in the pipette compared with that in control solution, and the peak of the I–V relationship was shifted −4 mV in CaM (−44 ± 4 mV, P < 0.05 vs. control) but not in CaMKII (−39 ± 4 mV) compared with control conditions (−40 ± 3 mV) (Figure 2B). The CaM- or CaMKII-induced increase in the peak current density was abolished by AIP. No significant difference was found in the reversal potentials in the presence of CaM or CaMKII (see Supplementary material online, Table S1).
Figure 2.

Ca2+/CaM and CaMKII increase Na+ currents in guinea-pig ventricular myocytes. (A) Representative families of Na+ currents (INa) are shown with control and CaMKII-containing pipette solutions. Cells were held at −120 mV and currents were elicited by 50 ms test pulses to potentials ranging from −100 to +40 mV. (B) Peak current–voltage relationships of cardiac INa with pipette solutions containing either CaM or CaMKII with or without AIP. (C) The voltage dependence of activation (G/Gmax) of INa in guinea-pig myocytes with the same pipette solutions. The data are fit to a Boltzmann distribution. CaM and CaMKII had no effect on the voltage dependence of activation. The number of cells is shown next to the symbols in the legend in this and all subsequent figures. †P < 0.05 vs. control and *P < 0.05 vs. CaM.
Neither CaM nor CaMKII altered the voltage dependence of Na+ channel activation as illustrated in the conductance (GNa) curves (Figure 2C and Table 1). In contrast, the voltage dependence of steady-state inactivation was influenced by CaMKII/CaM signalling. Representative Na+ currents (INa) and steady-state inactivation curves are shown in Figures 3A and B. To assess the voltage dependence of steady-state inactivation, INa was measured during test pulses to −20 mV (50 ms) after 500 ms pre-pulses from −140 to −20 mV (in 5 mV increments). CaMKII in the pipette produced a +5 mV shift in INa steady-state inactivation compared with the control condition (V1/2: −72.7 ± 8.4 vs. −79.7 ± 5.2 mV, P < 0.05; Table 1). The CaMKII-activating buffer contains CaM, and CaM alone produced the same voltage shift in INa steady-state inactivation (V1/2: −71.8 ± 6.7 mV, P < 0.05 vs. control). However, CaMKII-activating buffer in the absence of both CaM and CaMKII (vehicle) had no significant effect on the voltage dependence of INa availability (see Supplementary material online, Figure S1). To assess whether CaM or CaMKII modulates Na+ channels, we added the specific CaMKII inhibitor AIP to the pipette solution with CaM or CaMKII. AIP completely abolished not only the CaMKII-induced voltage shift in steady-state inactivation (−79.8 ± 7.0 mV CaMKII + AIP, P < 0.05 vs. CaMKII) but also the CaM-induced alteration of steady-state inactivation (−79.9 ± 6.0 mV CaM + AIP, P < 0.05 vs. CaM), suggesting that the CaM effect is mediated by endogenous CaMKII which is blocked by AIP. Furthermore, to assess whether the CaM/CaMKII-induced effect is mediated by PKA, we added the PKI to the pipette solution with CaM or CaMKII. PKI did not affect the CaM/CaMKII-induced change of INa steady-state inactivation (Table 1). Therefore, the CaM/CaMKII-induced voltage shift of Na+ channels is mediated by CaMKII-induced phosphorylation of the Na+ channel or other targets that influence the channel gating, but is not the direct result of CaM binding to the channel. This is not a general effect of channel phosphorylation as PKA altered neither steady-state inactivation nor activation (Table 1, see Supplementary material online, Figure S2).
Table 1.
Functional effects of CaM/CaMKII on Na+ current recorded from guinea-pig ventricular myocytes
| Steady-state inactivation (mV) |
Decay time constant (ms) at −20 mV |
Recovery from inactivation (ms) |
Activation (mV) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| V1/2 (k) | n | τfast | τslow | n | τfast | τslow | n | V1/2 (k) | n | |
| Control | −79.7 ± 5.2 (6.2 ± 0.6) | 15 | 1.57 ± 0.20 | 9.1 ± 9.1 | 10 | 5.4 ± 0.9 | 28.3 ± 14.7 | 9 | −54.3 ± 3.2 (3.0 ± 0.7) | 12 |
| AIP | −78.5 ± 5.9 (6.2 ± 0.6) | 7 | 1.34 ± 0.13 | 9.1 ± 6.8 | 7 | 6.4 ± 3.0 | 46.7 ± 31.2 | 5 | −56.3 ± 4.4 (3.2 ± 0.8) | 6 |
| PKI | −78.6 ± 6.1 (6.3 ± 0.8) | 9 | 1.55 ± 0.21 | 31.3 ± 8.7 | 7 | 6.2 ± 3.1 | 71.7 ± 43.2† | 5 | −54.1 ± 6.9 (3.0 ± 0.6) | 5 |
| CaMKII | −72.7 ± 8.4† (5.6 ± 0.8) | 11 | 1.47 ± 0.21 | 12.3 ± 9.4 | 7 | 3.8 ± 1.2† | 20.8 ± 4.6 | 7 | −55.1 ± 6.3 (3.0 ± 0.8) | 7 |
| CaMKII + AIP | −79.8 ± 7.0# (5.6 ± 0.9) | 7 | 1.49 ± 0.15 | 13.4 ± 6.7 | 7 | 6.5 ± 2.5# | 54.1 ± 37.0# | 6 | −52.6 ± 7.8 (3.7 ± 1.1) | 7 |
| CaMKII + PKI | −70.1 ± 3.6† (5.1 ± 0.6) | 4 | 1.60 ± 0.15 | 12.3 ± 11.5 | 5 | 5.5 ± 4.2 | 46.6 ± 28.1 | 4 | −53.6 ± 4.3 (2.6 ± 0.4) | 5 |
| CaM | −71.8 ± 6.7† (6.1 ± 0.7) | 11 | 1.53 ± 0.09 | 5.5 ± 4.9 | 9 | 7.1 ± 3.1 | 28.0 ± 14.7 | 6 | −57.3 ± 5.4 (2.7 ± 0.4) | 9 |
| CaM + AIP | −79.9 ± 6.0# (5.7 ± 0.6) | 8 | 1.67 ± 0.17 | 9.6 ± 9.1 | 6 | 5.1 ± 1.8 | 28.4 ± 6.9 | 5 | −52.3 ± 5.9 (3.0 ± 0.6) | 6 |
| CaM + PKI | −71.5 ± 6.1† (5.6 ± 0.4) | 5 | 1.60 ± 0.27 | 13.9 ± 9.4 | 5 | 5.3 ± 1.9 | 86.1 ± 37.7† | 4 | −52.0 ± 6.1 (2.4 ± 0.6) | 6 |
| PKA | −81.3 ± 9.3 (5.2 ± 0.7) | 6 | 1.36 ± 0.31 | 3.5 ± 3.4 | 8 | 3.9 ± 1.7† | 40.6 ± 34.5 | 5 | −55.6 ± 6.9 (2.8 ± 0.6) | 6 |
Mean ± SD. CaMKII, calcium/calmodulin-dependent protein kinase II; CaM, calmodulin; PKA, cyclic AMP-dependent protein kinase A; AIP, autocamtide-2-related inhibitory peptide; PKI, PKA inhibitor.
†P < 0.05 vs. control.
#P < 0.05 vs. AIP (−) by ANOVA with Bonferroni/Dunn test.
Figure 3.

CaM and CaMKII modulate steady-state inactivation of Na+ current in guinea-pig ventricular myocytes. (A) Representative whole-cell currents are shown in the absence (control) or presence of CaMKII in the pipette solution. Cells were held at −120 mV and currents were elicited by 50 ms test pulses at −20 mV after 500 ms pre-pulses ranging from −140 to −20 mV. (B) The voltage dependence of steady-state inactivation (I/Imax) of INa in the previously described pipette solutions. The data are fit to a Boltzmann distribution. CaM and CaMKII shifted the voltage dependence of steady-state inactivation by approximately +5 mV, whereas AIP abolished both CaM- and CaMKII-induced shift of voltage-dependent steady-state inactivation.
3.3. Recovery from inactivation
Inactivation and recovery from inactivation are closely correlated and critically regulate channel function and cardiac electrophysiology. Recovery from inactivation was investigated using a conventional two-pulse protocol. We used a sustained depolarization to −20 mV for 300 ms (P1) followed by a variable recovery interval and subsequent −20 mV test pulse (P2). As shown in Figure 4, CaMKII hastened the recovery from inactivation compared with control conditions. The fast time constant of recovery from inactivation (τfast) was significantly smaller in CaMKII than that in control (τfast: 3.8 ± 1.2 ms, CaMKII vs. 5.4 ± 0.9 ms, control; P < 0.05). The CaMKII-induced faster recovery from inactivation was abolished by the addition of AIP to the pipette (τfast: 6.5 ± 2.5 ms, and τslow: 54.1 ± 37.0 ms, CaMKII + AIP; P < 0.05 vs. CaMKII). On the other hand, neither CaM alone nor CaMKII-activating buffer (vehicle) altered the recovery from inactivation (Figure 4, see Supplementary material online, Figure S1). In contrast to steady-state inactivation, PKA also hastened the recovery from inactivation compared with control (τfast: 3.9 ± 1.7 ms, P < 0.05 vs. control) (Table 1, see Supplementary material online, Figure S2); however, PKI did not abolish the CaMKII-induced faster recovery from inactivation (Table 1).
Figure 4.

Recovery from inactivation of INa in guinea-pig ventricular myocytes. (A) Representative current recordings in control and CaMKII-containing pipette solutions. The currents were elicited by 300 ms pulses to −20 mV (P1) with varying inter-pulse durations at a recovery potential of −140 mV and subsequent −20 mV test pulses (P2). (B) The time course of recovery from inactivation of INa. Recovery data are fit to double exponential functions. CaMKII hastened the recovery from inactivation compared with control, and AIP suppressed the CaMKII-induced change in recovery from inactivation of INa.
3.4. Entry into inactivation
Figure 5 and Table 2 summarize the effect of CaM and CaMKII on the entry into inactivated states of cardiac Na+ channels. Entry into inactivation was measured using depolarizations of variable duration (P1) followed by a 20 ms recovery period at −140 mV, allowing for recovery from fast inactivation but not from intermediate or slower inactivation (IM). CaMKII decreased the fraction of INa that undergo slow inactivation (y0: 0.78 ± 0.05 CaMKII vs. 0.73 ± 0.07 control, P = 0.08), and CaM alone produced the same decrease in the fraction of channels undergoing slow inactivation (0.79 ± 0.06 CaM, P < 0.05 vs. control). AIP completely abolished not only the CaMKII-induced decrease in the entry into INa inactivation but also the CaM-induced alteration of entry into inactivation (0.69 ± 0.05 CaMKII + AIP and 0.71 ± 0.08 CaM + AIP, respectively), suggesting that the CaM effect is mediated by endogenous CaMKII. On the other hand, PKI did not abolish the CaMKII- and CaM-induced alteration of IM. Thus, Ca2+/CaM and CaMKII decreased the fraction of the current undergoing intermediate inactivation.
Figure 5.

Kinetics of entry into slow inactivated states of guinea-pig ventricular INa. The voltage protocol is shown in the inset and the mean data are fit to a single exponential function. CaM and CaMKII decreased the extent of slow inactivation without changing the rate of entry into slow inactivation. AIP completely abolished the CaM/CaMKII-induced change of entry into slow inactivation. *P < 0.05 by repeated ANOVA.
Table 2.
Effects of CaM/CaMKII on entry into inactivation of Na+ current
| y0 | A | τ | n | |
|---|---|---|---|---|
| Control | 0.73 ± 0.07 | 0.28 ± 0.08 | 222 ± 98 | 8 |
| AIP | 0.72 ± 0.09 | 0.29 ± 0.09 | 169 ± 48 | 5 |
| PKI | 0.72 ± 0.11 | 0.26 ± 0.11 | 222 ± 197 | 4 |
| CaMKII | 0.78 ± 0.05 (P = 0.08 vs. control) | 0.19 ± 0.07† | 256 ± 137 | 8 |
| CaMKII + AIP | 0.69 ± 0.05# | 0.28 ± 0.09 (P = 0.06 vs. CaMKII) | 344 ± 85 | 5 |
| CaMKII + PKI | 0.79 ± 0.01 | 0.18 ± 0.04 | 232 ± 192 | 4 |
| CaM | 0.79 ± 0.06† | 0.18 ± 0.08† | 230 ± 85 | 9 |
| CaM + AIP | 0.71 ± 0.08# | 0.28 ± 0.10 (P = 0.06 vs. CaM) | 274 ± 106 | 6 |
| CaM + PKI | 0.82 ± 0.06† | 0.13 ± 0.02† | 341 ± 169 | 4 |
| PKA | 0.69 ± 0.05 | 0.31 ± 0.01 | 178 ± 78 | 4 |
Mean ± SD. Abbreviations are as given in Table 1. The development of intermediate inactivation was fitted with single exponential functions: y(t) = y0 + Aexp(–t/τ).
†P < 0.05 vs. control.
#P < 0.05 vs. AIP (−) by ANOVA with Bonferroni/Dunn test.
3.5. Decay time constant
The initial (τfast) and late (τslow) time constants of INa decay are shown in Table 1. Neither the initial nor the slow time constants of INa decay were altered by CaM, CaMKII, or PKA in the pipette. Furthermore, the addition AIP or PKI to the pipette either with or without CaM/CaMKII had no effect on INa decay.
3.6. Late current
Figure 6A shows the superimposed persistent (late) INa in control conditions and with CaMKII in the pipette. Late INa was elicited by 800 ms depolarizations to −20 mV (from −140 mV), and TTX-sensitive currents were normalized to peak INa. The current integral was calculated between 100 and 500 ms after the depolarizing pulse. As shown in Figure 6, CaMKII significantly increased late INa compared with control (0.95 ± 0.47 vs. 0.23 ± 0.10%, P < 0.05), and the CaMKII-induced increase in persistent INa was completely abolished by AIP (0.29 ± 0.11% CaMKII + AIP, P < 0.05 vs. CaMKII) (Figure 6B). On the other hand, CaM and PKA did not increase the late INa compared with control. These findings demonstrate that the CaMKII-induced increase in late INa could not be replicated by CaM alone but instead required CaMKII and presumably phosphorylation of a Na+ channel subunit or regulatory protein. Moreover, phosphorylation by PKA does not reproduce the effect of CaMKII on the late Na+ current.
Figure 6.

CaMKII enhanced late INa. Late INa was elicited by 800 ms depolarizations to −20 mV (from −140 mV), and TTX-sensitive currents were normalized to peak INa. (A) Representative normalized TTX-sensitive current in control and CaMKII-containing pipette solution. (B) The average amplitude of the late current between the 100 and 500 ms was normalized to the peak INa. CaMKII (n = 7) significantly increased the late INa compared with control (n = 7), and the addition of AIP (n = 4) abolished the CaMKII-induced increase in late INa. CaM (n = 5) and PKA (n = 5) did not change the late INa.
3.7. CaMKII-dependent alterations on AP
A number of ionic currents in the heart are modulated by CaMKII, yet it is still not clear how acute increases in the activity of CaMKII affect the cardiac AP. Therefore, we examined the effects of CaMKII and its antagonists KN-93 or AIP on the AP duration (APD). As shown in the representative APs superimposed at various pacing intervals (Figure 7A), KN-93 (10 µmol/L in the bath solution) significantly abbreviated the APD compared with control (P < 0.01), whereas CaMKII in pipette prolonged the APD (P = 0.05) compared with control. CaMKII increased the APD over a range of pacing cycle lengths and steepened the APD–pacing rate relationship particularly at rapid rates of stimulation (Figure 7B). The pacing cycle length–APD relation curves show attenuation of CaMKII-induced APD prolongation and flattening of the curves by both KN-93 and AIP compared with both CaMKII and control. Furthermore, the maximum dV/dt of phase 0 of the APs were significantly decreased by KN-93 and AIP, but not significantly changed by CaMKII over a range of stimulation frequencies compared with control (Figure 7C).
Figure 7.

The effect of CaMKII on the APD and dV/dtmax. (A) Representative APs in the absence (control) or presence of CaMKII in the pipette and bath-applied KN-93 paced at cycle lengths (CL) of 500, 1000, 2000, and 4000 ms. (B) The relationship between pacing CL and APD in control, CaMKII-, AIP-, and KN-93-containing pipette solutions. The data are fit by a hyperbolic relation of the form: APD = CL/[(aCL) + b]. (C) Relationship between pacing CL and dV/dtmax in control, CaMKII-, AIP- and KN-93-containing pipette solutions. †P < 0.05 vs. control by repeated ANOVA.
4. Discussion
The present study demonstrates the acute effect of CaM and CaMKII on cardiac Na+ channel function in isolated guinea-pig ventricular myocytes. We demonstrated that CaMKII shifted the voltage dependence of Na+ channel availability to more positive membrane potentials, hastened recovery from inactivation, decreased fraction of channels undergoing slow inactivation, and increased the persistent (late) current. CaM alone reproduced the CaMKII-induced changes on INa availability and the fraction of channels undergoing slow inactivation, but did not alter recovery from inactivation or late current. Furthermore, the CaM-induced changes on the I–V relationship, channel availability, and entry into slow inactivation were also completely abolished by AIP. Thus CaM and CaMKII have distinct effects on the inactivation phenotype of cardiac Na+ channels, consistent with both interdependent and independent CaM and CaMKII regulation of cardiac Na+ channel gating. Furthermore, these effects are distinct from those mediated by activation of PKA.
4.1. Na+ channel regulation by CaM and CaMKII
The effects of CaM and CaMKII on Na+ channels remain controversial. Tan et al.11 showed that CaM binds to the carboxy-terminal ‘IQ’ domain of expressed human cardiac Na+ channels (NaV1.5) in a Ca2+-dependent manner. This binding significantly enhanced slow inactivation, shifted the steady-state inactivation curve to more negative potentials, and slowed fast inactivation and recovery from inactivation. It is important to recognize that other studies have shown that CaM effects on expressed Na+ currents are both isoform and cell expression system-dependent.19,20 It has been suggested that CaM modulates the interaction of the C-terminus of NaV1.5 channels with the III–IV linker, stabilizing the inactivation gate in the closed position, minimizing the persistent INa.21–23 In general, the effects of CaM and CaMKII are closely correlated;24 therefore, we attempted to study the effects of CaM alone and CaM–CaMKII on native cardiac Na+ channels. CaM alone (or in the presence of CaMKII inhibitors, AIP) did not alter the Na+ channel voltage-dependent steady-state availability, recovery from inactivation, and entry into slow or intermediate inactivation in normal guinea-pig ventricular myocytes.
CaM activates the multifunctional CaMKII, which targets several critical calcium homeostatic proteins, including L-type Ca2+ channels.25 The cardiac (and other isoforms) Na+ channel is equipped with functional Ca2+ sensing sites in the C-terminus including a Ca2+-binding EF-hand-like domain10 and a CaM-binding IQ domain.11,26 Pharmacological CaMKII inhibition slows fast inactivation, delays entry into inactivation, and shifts channel availability to depolarized potentials of NaV1.5 channels expressed in HEK cells.20 However, in cerebral granule cells in culture, CaMKII inhibitors shifted the voltage dependence of Na+ channels to more hyperpolarized potentials and reduced current density.27 These data highlight the importance of isoform-specific and native compared with expressed current regulation by CaMKII.
In this study, CaMKII shifted the voltage dependence of Na+ channel availability to more positive membrane potentials, hastened recovery from inactivation, and increased late current in normal guinea-pig ventricular myocytes. These results are distinct from (except for the increase in late INa) a recent study that examined the effects of chronic overexpression by adenovirus-mediated CaMKIIδc infection of rabbit ventricular myocytes and CaMKIIδc-TG mice.16 CaMKIIδc overexpression shifted the voltage dependence of Na+ channel availability to more negative membrane potentials, enhanced intermediate inactivation, slowed recovery from inactivation, and increased persistent INa.16 The divergence of findings is not surprising given the methodological differences between the studies. First, it is notable that chronic cardiac CaMKII overexpression causes ventricular hypertrophy and dilated cardiomyopathy28 with associated mechanical, electrical, and cellular Ca2+ remodelling responsible for prolongation of APD and susceptibility to arrhythmias. In cells isolated from the failing canine heart, CaMKII signalling slows inactivation of Na+ current and increases late current.29 The data are consistent with both direct and indirect effects of CaMKII overexpression on Na+ channel function. The indirect effects are a consequence of CaMKII-induced cardiac hypertrophy and heart failure and the associated electrical remodelling. An increase in late INa is consistently observed with both acute and chronic increases in CaMKII activity. The differences in the voltage dependence and kinetics of gating with CaMKII overexpression are more difficult to interpret given the structural and functional remodelling that occurs with chronic increases in CaMKII activity that are absent in this acute overexpression study.
4.2. Comparison of CaMKII with PKA effects on INa
Stimulation of β-adrenergic receptors activates the PKA signalling cascade, which plays a pivotal role in the modulation of cardiac function not only in normal but also in failing hearts. The functional effects of PKA on cardiac INa have been debated.30–33 Two serine residues (Ser532 and Ser529) in the I–II cytoplasmic linker of cardiac Na+ channels are substrates for phosphorylation by PKA.7 Upon phosphorylation by PKA, Na+ channel gating is unchanged, but whole-cell conductance increases,33 which is neither due to increased single channel amplitude nor altered mean open or closed times but due to an increase in the number of functional channels.34 Stimulation of PKA increases expressed NaV1.5 current by increasing trafficking of cardiac Na+ channels to the plasma membrane.8 These findings are consistent with our results, in that PKA did not alter the voltage dependence of INa availability but hastened recovery from inactivation, and increased peak current (see Supplementary material online, Figures S2 and S3).
Chronic β-adrenergic receptor stimulation in the heart is intimately linked to CaMKII signalling.35,36 PKA and CaMKII have overlapping targets for phosphorylation including L-type Ca2+ channels,37,38 ryanodine receptors (RyR2), and the SR Ca2+-ATPase regulator phospholamban.39 We compared the effects of PKA and CaMKII regulation on another common effector, cardiac INa. Our data demonstrates that the cardiac Na+ channel is a substrate for phosphorylation by CaMKII predominantly in the I–II linker (Figure 1). Both kinases increased the current density, but the increase was much larger with PKA compared with CaMKII (−53.2 ± 9.0 pA/pF at −45 mV vs. −39.7 ± 6.9 pA/pF at −40 mV, P < 0.05). CaMKII but not PKA shifted the steady-state inactivation of INa positively and increased late INa; in contrast, both kinases hastened recovery from inactivation. CaMKII-induced changes in Na+ current in part overlap with the effects of PKA, but also exhibit distinct phenotypic features, some of which may be highly arrhythmogenic. It is uncertain whether CaMKII and PKA phosphorylate common sites on the channel or other regulatory proteins accounting for the partially overlapping functional effects.
4.3. CaMKII alters APD and dynamics
Previous studies have demonstrated that increased CaMKII activity is linked to not only cardiomyopathy14 but also fatal arrhythmias due to prolongation of AP and development of early afterdepolarizations. CaMKII-mediated prolongation of the AP may primarily result from an increase in L-type Ca2+ current (ICa) density.12,13,15 However, CaMKII affects a number of ion channels, SR Ca2+-handling proteins, and the other intracellular proteins; it is difficult to discern the relative effects of CaMKII modulation of INa, ICa, or delayed rectifier K+ current (IK) on the ventricular AP. Recently, Grandi et al.40 simulated the effect of CaMKII overexpression on ventricular APs and suggested that CaMKII-induced alteration of the APD was dependent on the effect on transient outward current (Ito) because CaMKII enhanced not only inward current but also outward K+ current. In the present study, we directly recorded the acute changes in the AP by CaMKII and its specific inhibitors using normal guinea-pig myocytes, showing that CaMKII prolonged AP, whereas CaMKII inhibitors significantly abbreviated APD and decreased maximum dV/dt at phase 0. The prolongation of APD by CaMKII in this model is almost certainly caused by an increased ICa, but an enhanced persistent INa may also play a role. In guinea-pig ventricular myocytes, Ito is not significantly contributing to the AP changes. In addition, guinea-pig AP simulations support a significant role for CaM/CaMKII modulation of INa on the prolongation of ventricular AP, primarily because of increased late INa.41 The increased late INa by CaMKII is consistent with previous overexpression models,16 but contrasts with the CaM effect on expressed NaV1.5, which minimizes the persistent INa.21 Thus, CaMKII suppression might be a useful therapeutic target for some forms of congenital LQT3. Notably, these APD changes were recorded in normal but not failing myocytes; therefore, further studies to evaluate the effect of CaMKII or its inhibitors on APD in failing myocytes are necessary.
4.4. Study limitations
In this study, we used CaMKIIα (mainly expressed in the nerve cells) not δ, which is prominently expressed in the heart. However, the CaMKII isoforms have highly homologous functional motifs (catalytic, regulatory, and association), sharing ∼80–90% identity across these core domains,12,42 thus we believe that the difference in CaMKII isoforms did not significantly alter the regulation of cardiac Na+ channels, but cannot exclude this possibility definitively. As in all studies of signalling in native cardiac myocytes, the interpretation of the data regarding the effects on INa is complicated by the existence of multiple effectors in these signalling pathways. This is especially true of Ca2+/CaM/CaMKII which prominently influences intracellular [Ca2+] which has direct and indirect effects on cardiac INa.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: G.F.T. holds the Michel Mirowski MD Professorship in Cardiology at Johns Hopkins University.
Funding
The work was supported by NIH RO1 HL50411 (G.F.T.) and PO1 HL077180.
Supplementary Material
References
- 1.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett PB, Yazawa K, Makita N, George AL., Jr Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 3.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 4.Schott JJ, Alshinawi C, Kyndt F, Probst V, Hoorntje TM, Hulsbeek M, et al. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet. 1999;23:20–21. doi: 10.1038/12618. [DOI] [PubMed] [Google Scholar]
- 5.Tan HL, Bink-Boelkens MT, Bezzina CR, Viswanathan PC, Beaufort-Krol GC, van Tintelen PJ, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 2001;409:1043–1047. doi: 10.1038/35059090. [DOI] [PubMed] [Google Scholar]
- 6.Abriel H, Kass RS. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc Med. 2005;15:35–40. doi: 10.1016/j.tcm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Murphy BJ, Rogers J, Perdichizzi AP, Colvin AA, Catterall WA. cAMP-dependent phosphorylation of two sites in the alpha subunit of the cardiac sodium channel. J Biol Chem. 1996;271:28837–28843. doi: 10.1074/jbc.271.46.28837. [DOI] [PubMed] [Google Scholar]
- 8.Zhou J, Yi J, Hu N, George AL, Jr, Murray KT. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ Res. 2000;87:33–38. doi: 10.1161/01.res.87.1.33. [DOI] [PubMed] [Google Scholar]
- 9.Murray KT, Hu NN, Daw JR, Shin HG, Watson MT, Mashburn AB, Jr, et al. Functional effects of protein kinase C activation on the human cardiac Na+ channel. Circ Res. 1997;80:370–376. doi: 10.1161/01.res.80.3.370. [DOI] [PubMed] [Google Scholar]
- 10.Wingo TL, Shah VN, Anderson ME, Lybrand TP, Chazin WJ, Balser JR. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat Struct Mol Biol. 2004;11:219–225. doi: 10.1038/nsmb737. [DOI] [PubMed] [Google Scholar]
- 11.Tan HL, Kupershmidt S, Zhang R, Stepanovic S, Roden DM, Wilde AA, et al. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415:442–447. doi: 10.1038/415442a. [DOI] [PubMed] [Google Scholar]
- 12.Maier LS, Bers DM. Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J Mol Cell Cardiol. 2002;34:919–939. doi: 10.1006/jmcc.2002.2038. [DOI] [PubMed] [Google Scholar]
- 13.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 14.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 15.Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, et al. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 16.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aiba T, Hesketh GG, Biswas S, Liu T, Carlisle R, O'Rourke B, et al. Na+ channel modification by Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Heart Rhythm. 2007;4:149. (Abstract) [Google Scholar]
- 18.Hoppe UC, Johns DC, Marban E, O'Rourke B. Manipulation of cellular excitability by cell fusion: effects of rapid introduction of transient outward K+ current on the guinea pig action potential. Circ Res. 1999;84:964–972. doi: 10.1161/01.res.84.8.964. [DOI] [PubMed] [Google Scholar]
- 19.Young KA, Caldwell JH. Modulation of skeletal and cardiac voltage-gated sodium channels by calmodulin. J Physiol. 2005;565:349–370. doi: 10.1113/jphysiol.2004.081422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deschenes I, Neyroud N, DiSilvestre D, Marban E, Yue DT, Tomaselli GF. Isoform-specific modulation of voltage-gated Na(+) channels by calmodulin. Circ Res. 2002;90:E49–E57. doi: 10.1161/01.res.0000012502.92751.e6. [DOI] [PubMed] [Google Scholar]
- 21.Kim J, Ghosh S, Liu H, Tateyama M, Kass RS, Pitt GS. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. 2004;279:45004–45012. doi: 10.1074/jbc.M407286200. [DOI] [PubMed] [Google Scholar]
- 22.Motoike HK, Liu H, Glaaser IW, Yang AS, Tateyama M, Kass RS. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol. 2004;123:155–165. doi: 10.1085/jgp.200308929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glaaser IW, Bankston JR, Liu H, Tateyama M, Kass RS. A carboxyl-terminal hydrophobic interface is critical to sodium channel function. Relevance to inherited disorders. J Biol Chem. 2006;281:24015–24023. doi: 10.1074/jbc.M605473200. [DOI] [PubMed] [Google Scholar]
- 24.Pitt GS. Calmodulin and CaMKII as molecular switches for cardiac ion channels. Cardiovasc Res. 2007;73:641–647. doi: 10.1016/j.cardiores.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 25.Kohlhaas M, Zhang T, Seidler T, Zibrova D, Dybkova N, Steen A, et al. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ Res. 2006;98:235–244. doi: 10.1161/01.RES.0000200739.90811.9f. [DOI] [PubMed] [Google Scholar]
- 26.Biswas S, Disilvestre D, Tian Y, Halperin VL, Tomaselli GF. Calcium-mediated dual-mode regulation of cardiac sodium channel gating. Circ Res. 2009;104:870–878. doi: 10.1161/CIRCRESAHA.108.193565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carlier E, Dargent B, De Waard M, Couraud F. Na(+) channel regulation by calmodulin kinase II in rat cerebellar granule cells. Biochem Biophys Res Commun. 2000;274:394–399. doi: 10.1006/bbrc.2000.3145. [DOI] [PubMed] [Google Scholar]
- 28.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, et al. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 29.Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol. 2008;294:H1597–H1608. doi: 10.1152/ajpheart.00484.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schubert B, Vandongen AM, Kirsch GE, Brown AM. Inhibition of cardiac Na+ currents by isoproterenol. Am J Physiol. 1990;258:H977–H982. doi: 10.1152/ajpheart.1990.258.4.H977. [DOI] [PubMed] [Google Scholar]
- 31.Matsuda JJ, Lee H, Shibata EF. Enhancement of rabbit cardiac sodium channels by beta-adrenergic stimulation. Circ Res. 1992;70:199–207. doi: 10.1161/01.res.70.1.199. [DOI] [PubMed] [Google Scholar]
- 32.Ono K, Fozzard HA, Hanck DA. Mechanism of cAMP-dependent modulation of cardiac sodium channel current kinetics. Circ Res. 1993;72:807–815. doi: 10.1161/01.res.72.4.807. [DOI] [PubMed] [Google Scholar]
- 33.Frohnwieser B, Chen LQ, Schreibmayer W, Kallen RG. Modulation of the human cardiac sodium channel alpha-subunit by cAMP-dependent protein kinase and the responsible sequence domain. J Physiol. 1997;498:309–318. doi: 10.1113/jphysiol.1997.sp021859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu T, Lee HC, Kabat JA, Shibata EF. Modulation of rat cardiac sodium channel by the stimulatory G protein alpha subunit. J Physiol. 1999;518:371–384. doi: 10.1111/j.1469-7793.1999.0371p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao RP, et al. Sustained beta1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ Res. 2004;95:798–806. doi: 10.1161/01.RES.0000145361.50017.aa. [DOI] [PubMed] [Google Scholar]
- 36.Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Brown JH, et al. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest. 2003;111:617–625. doi: 10.1172/JCI16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol. 2000;2:173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- 38.Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, et al. L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 39.Anderson ME. Multiple downstream proarrhythmic targets for calmodulin kinase II: moving beyond an ion channel-centric focus. Cardiovasc Res. 2007;73:657–666. doi: 10.1016/j.cardiores.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 40.Grandi E, Puglisi JL, Wagner S, Maier LS, Severi S, Bers DM. Simulation of Ca/Calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophys J. 2007;93:3835–3847. doi: 10.1529/biophysj.107.114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hashambhoy YL, Winslow RL, Greenstein JL. A mechanistic, minimal model of Ca2+/calmodulin dependent kinase II signaling in the cardiac myocytes. Biophys J. 2009;96:540a. (Abstract) [Google Scholar]
- 42.Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005;123:849–860. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.