

Abstract
Spot detection and quantification for 2-DE are challenging and important tasks to fully extract the proteomic information from these data. Traditional analytical methods have significant weaknesses, including spot mismatching and missing data, which require time-consuming manual editing to correct, dramatically decreasing throughput and compromising the objectivity and reproducibility of the analysis. To address this issue, we developed Pinnacle, a novel, quick, automatic non-commercial method that borrows strength across gels in spot detection and has been shown to yield more precise spot quantifications than traditional methods. New commercial software, notably SameSpots, has also recently been developed as an improvement over traditional workflows. In this paper, we briefly describe Pinnacle and compare its performance to SameSpots in spot detection, spot quantification precision, and differential expression. Our analysis is performed in a rigorous fashion that, unlike other comparisons in the literature, summarizes performance across all spots detected on the gels, and we manually optimize SameSpots results while simply running Pinnacle with standard settings and no manual editing. While both methods showed marked improvement over a commercially available traditional method PG240, Pinnacle consistently yielded spot quantifications with greater validity and reliability, avoided spot delineation problems, and detected more differentially expressed proteins than SameSpots, and represents a significant non-commercial alternative for 2-DE processing.
Keywords: Two-dimensional polyacrylamide gel electrophoresis, Differential In-Gel Electrophoresis, False discovery rate, Differential expression, Bioinformatics, Spot detection, Image processing
Introduction
2-DE is an important method for proteomic analysis, allowing one to detect and measure thousands of proteins present within a biological sample. In 2-DE, proteins are physically separated within a polyacrylamide gel in two dimensions based on their pI and molecular weight. Subsequently, the gel is scanned to produce a digital image, in which the proteins appear as spots with high staining intensities. Each gel image must be processed to detect and quantify the intensities of the protein spots. Given a study with N gels and p protein spots detected across all of the gels, after this processing we are left with an N×p matrix Y of spot intensities. For comparative proteomics studies, this matrix can then be surveyed using standard statistical methods to determine which protein spots are differentially expressed across treatment groups, or are otherwise related to whatever factor is of interest in that study.
It is of crucial importance to effectively detect, match, and quantify the spots across gels. It is also important to have adequate sensitivity for spot detection, so subsequent analyses survey all of the detectable proteins in the sample and do not miss proteins that are differentially expressed. Accurate spot matching is critical, as mismatched spots across gels result in different proteins being matched together in the analysis, compromising the ability to find significant differences. Precise spot quantifications are important to reduce variability and thus maximize power for detecting significant differences.
Most traditional methods for gel processing involve first detecting spots, estimating spot boundaries, and computing spot volumes for each individual gel, then matching the detected spots across gels. This approach is problematic because it results in missing data in the N×p matrix Y for any spots not detected on all of the gels. It is also prone to matching errors because cognate spots may be in slightly different positions on each gel, and matching is only done after detection. In addition, spot boundaries are difficult to accurately estimate, especially when spots overlap, and so the measurement error in the spot quantification is increased by methods that estimate spot boundaries separately for each individual gel. Most currently available software packages permit manual editing of these errors which can be very time consuming (1-4 h per gel [1][2]) and compromises the objectivity and reproducibility of the analysis. Most problematically, all of these problems worsen as the number of gels N in the study increases. This encourages researchers to conduct studies with fewer samples, leading to insufficient statistical power to detect significant differences when multiple testing is appropriately taken into account. These issues compromise the ability to conduct automatic, objective, high-throughput, 2-DE analyses using traditional approaches [2].
Some recently developed 2-DE processing workflows mitigate some of these problems. We have introduced a novel non-commercial method Pinnacle [3] for spot detection and quantification. Pinnacle operates on pre-aligned gel images, and starts by computing an average gel. Unlike the composite gels used in many commercial packages, this average gel is an arithmetic average, which allows one to borrow strength across gels in performing spot detection. Wavelet denoising is then applied, and spots are detected on the average gel by identifying “pinnacles,” or staining intensities that are local maxima in both the horizontal and vertical dimensions. Quantifications for each individual gel are taken as the maximum pixel intensity in the neighborhood of the (x, y) coordinate for each pinnacle detected in the average gel. This method is quick and automatic, results in no missing data, and the spot detection accuracy should improve as the number of gels in the study increase. The use of pinnacles instead of spot volumes for quantification eliminates the need to estimate spot boundaries, resulting in improved reliability and validity [3].
After we developed Pinnacle and performed the studies in that paper, Nonlinear Dynamics (Newcastle upon Tyne, UK) released a new commercial 2-DE analysis product, Progenesis SameSpots, which provides another alternative processing workflow to the traditional approaches. The basic workflow has some similarities to Pinnacle: align the images before processing to avoid the problems resulting from spot matching after detection, and perform spot quantification in a more uniform way across gels to increase quantification precision. Also working on aligned gels, SameSpots starts by performing spot detection simultaneously across all of the images, obtaining a master list of detected spots and corresponding spot boundaries. Unlike Pinnacle, it does not compute an average gel or otherwise analytically borrow strength across gels in performing spot detection. Spot boundary masks are then overlaid on the aligned gels, and used to compute individual spot volumes, producing the N×p matrix Y. This method also results in no missing data, and its use of common spot boundaries for all of the gels should in principle increase the precision of spot volume estimates. Karp and colleagues [4] recently confirmed that the use of SameSpots reduced variability and in principle should increase power to detect differential expression when compared with traditional methods, and, in theory, should diminish the need for subjective editing. Note that unlike Pinnacle, which quantifies spots using pinnacle intensities, SameSpots quantifies spots using volumes after estimating spot boundaries.
Our previous study [3] compared the performance of Pinnacle with several available commercial packages, but all used the traditional workflow, which was all that was available at that time. In this study, we assess the performance of Pinnacle and SameSpots, representing a widely used commercial method with an alternative workflow. Our comparisons are done on four 2-DE data sets: two dilution series and two differential expression studies. Spot detection was evaluated by summarizing the number of spots automatically detected by each method, and by visually assessing the accuracy of detection and spot splitting. We separately investigated the two components of precision: validity and reliability. Validity was assessed by computing the linearity of the relationship between the spot quantification for detected spots and the protein loads in the two dilution series. Reliability was assessed by computing the %CV within each protein load group in the dilution series and within treatment groups in the comparative proteomics studies. Differential expression was evaluated by summarizing the number of spots in the comparative proteomics studies with p-values less than 0.05, 0.01, 0.005, and 0.001, and with local false discovery rates less than 0.15 and 0.20.
Our primary comparison is based on an automatic application of these methods to aligned gels with no manual editing. We also attempted to optimize SameSpots by evaluating alternative spot detection strategies and performing extensive manual editing of the spot detection and boundaries in SameSpots and compared these results. Using a graphical interface we are developing, it is quick and easy to edit Pinnacle's spot detection results while viewing the average gel if desired, but in this paper we only assess the automatic Pinnacle results with standard values for tuning parameters and no manual editing. To assess both of these methods relative to more traditional workflows, we also compared Pinnacle and SameSpots with Progenesis PG240 in terms of validity and reliability of spot quantification using the dilution series. We did not include PG240 in the differential expression comparisons, as it is unclear how to properly handle the missing data in that context, and using standard ad hoc approaches to deal with the missing data, we have already compared Pinnacle and PG240 in terms of differential expression in our previous study [3]. Unlike existing studies done in 2-DE, each analysis was done on all of the spots detected on the gels, not just a subset, to ensure that we provided a thorough assessment of the algorithms' performance.
Methods
Data Sets
Nishahara and Champion Study (NC)
These data were provided to us courtesy of Dr. Kathleen Champion-Francissen. The generation of this data set was described previously [5]. Briefly, these data consist of 28 gels from a dilution series using a sample of E. coli, with four replicate gels from each of seven different protein loads spanning a 100-fold range (0.5μg, 7.5μg, 10μg, 15μg, 30μg, 40μg, and 50μg).
SH-SY5Y Neuroblastoma Cell Dilution Series (SH)
These data were obtained from a dilution series performed by our lab. Details of the 2-DE, staining, and image capture were described previously [3]. These data consist of 18 gels from a dilution series created from SH-SY5Y neuroblastoma cells, with three replicate gels from each of 6 protein loads (5μg, 10μg, 25μg. 50μg, 100μg, 150μg).
D1 Differential expression study
These data were obtained by treating C6 glioma cells stably expressing the mu opioid receptor (mansour) with 1 μM of the agonist fentanyl or an equivalent volume of water for 12 hours. Four independent samples of cells in each treatment group were then collected and extracted and a sequential extraction was performed using the ProteoPrep (Sigma) kit as previously described (mouledous), except that after hypotonic lysis the remaining pellet was extracted in 2% CHAPS, 7M urea, 2M thiourea, and 20 mM Tris buffer. Protein loads were equalized and IEF was performed in a running buffer consisting of the above plus 30% (v/v) trifluoroethanol (TFE). Second-dimension SDS-PAGE, staining and image capture were performed as previously described [3].
D2 Differential expression study
C6 glioma cells as described above were also used to generate this data set. However, after drug treatment, cells were pelleted and 50 mM ammonium bicarbonate added at a 1:5 (w/v) ratio, and the pellet gently resuspended. TFE/Chloroform at a ratio of 2:1 (v/v) was then added to the sample in a ratio of 7:1 (v/v). The sample was then vortexed vigorously, sonicated on ice, and incubated on ice for one hour with gentle vortexing. The TFE fraction was collected and lyophilized. Proteins were then resuspended in 1% C7BzO, 7 M urea, 2M thiourea, and 30% (v/v) TFE for IEF. Second-dimension SDS-PAGE, staining and image capture were performed as previously described [3]. In this data set, there were 5 fentanyl gels and 4 control gels.
Processing Methods
Alignment and Cropping of Gels
All analyses were performed on gel images that had been aligned so that cognate spots are at corresponding locations across different gels, and cropped to remove the boundary regions without proteins. For the two dilution series, a reference gel was chosen, and all of the gel images in the set were aligned to the reference gel using the commercial software package TT900 (Nonlinear Dynamics, Newcastle Upon Tyne, UK), which provides an interface for the user to manually align cognate spots on each gel to the reference gel. This alignment process took approximately 30-60 minutes per gel. TT900 was originally purchased as a stand-alone package, but is now integrated with SameSpots. After alignment, we read the gel images into Matlab (Mathworks, Inc., Natick, MA USA) for cropping and resaved the cropped, aligned images as .tif files. These files were used in all subsequent analyses. For the differential expression studies, a preliminary alignment was done using the automatic 2d gel alignment package RAIN [6], which was then manually refined and improved as necessary using TT900, which took far less time (5-10 minutes/gel) than using TT900 alone. These gels were also read into Matlab, cropped, and saved as .tif files.
Pinnacle
Here we briefly summarize the Pinnacle method and describe its key features. Working on aligned images, Pinnacle first computes an “average gel,” which involves averaging the intensities pixel by pixel across all of the gels in the experiment. Next, Pinnacle applies wavelet denoising methods to denoise the average gel, followed by spot detection, which involves identifying “pinnacles” on the wavelet-denoised average gel. A pixel is said to be a pinnacle if it is a local maximum in both the horizontal and vertical directions, and has intensity greater than a specified threshold. By default, the threshold is the 75th percentile on the gel, although the user can change this parameter, if desired. Unlike most other methods, spot quantification in Pinnacle is not done by estimating spot boundaries and computing spot volumes, but instead the quantification is taken to be the pixel intensity at the pinnacle location. Specifically, the spot quantification for each individual gel is obtained by taking the maximum intensity within a rectangular window surrounding the pinnacle locations in the average gel. This window size is specified by the user, and by default, is 5 pixels by 5 pixels, accommodating slight remaining misalignment of spots between gels, but being small enough to avoid including pinnacles from any other cognate spots. Background correction can be done either globally or locally. We have found local background correction to work better for the data we have analyzed. Local background correction was done for each pinnacle by subtracting the minimum pixel intensity in a rectangular window surrounding the pinnacle. We find that large windows work well for background correction, so as to ensure that the window includes regions of the gel that are truly background, i.e. without protein content. By default, the window size for local background correction is 200 × 200, which is what we use here (for 1024 × 1024 images). Pinnacle can be used for DIGE data by treating the image from each channel as a separate image in the detection process, and then taking the simple ratio (or difference on the log scale) of corresponding pinnacle intensities across the two channels for quantification. No other background correction is necessary for DIGE.
Four key aspects of Pinnacle are that it (1) uses an average gel for spot detection, (2) uses wavelet denoising to remove noise from the average gel image before detection, (3) detects spots by their pinnacles, and (4) quantifies spots by pinnacle intensities rather than spot volumes. The use of the average gel for spot detection has several favorable characteristics. It provides unambiguous spot detection since a pinnacle is a well-defined quantity. Second, it allows borrowing of strength across gels in determining what is a spot, since spot signals are reinforced across gels, while noise and artifacts are down-weighted. This, in principal, should lead to improved sensitivity and specificity for spot detection, and spot detection accuracy that increases with sample size. A spot need not be present in all of the gels to be identified in the average gel: given N gels, by the central limit theorem of statistics, we expect that the background noise should be reduced by a factor of √N, leading to a higher signal-to-noise ratio on the average gel than individual gels (and thus greater sensitivity for spot detection) as long as the spot is present on at least 1/√N of the gels. In the context of MALDI-MS peak detection, this principle has been clearly demonstrated in a simulation study [7], and in the future we plan to validate this principle for 2-DE using simulation studies based on a 2-DE simulation tool we are developing. Another alternative that might lead to even more sensitivity (but also less robustness to artifacts) would be to use a weighted average gel [8].
Wavelet denoising has become a standard tool in denoising images in all areas of signal processing, including 2-DE [9][10][3], although Pinnacle is the first method to explicitly use the wavelet denoising to aid spot detection. This process smoothes out spurious bumps in the image that correspond to high-frequency noise without attenuating the dominant features of the gel too strongly, which allows sensitive pinnacle-based spot detection while reducing the number of spurious detected spots that are in fact noise. This can provide additional power in differential expression studies, since spurious spots increase the false discovery rate estimates for other spots, and thus reduce power to detect them.
The detection of spots by their pinnacles rather than their boundaries has the potential to improve the resolving of overlapping spots. Færgestad, et al. [11] outline the many shortcomings spot-based methods have in resolving overlapping spots. They point out that most spots on a gel overlap with another spot, and that frequently an “envelope” of overlapping spots is called a single spot by many spot detection methods, and in that case it can become nearly impossible to detect differential expression in one of the spots in the envelope. Further, they mention that in overlapping spots, the determination of boundaries between the spots is crucial, and the fact that these boundaries are estimated separately across gels introduces serious quantification errors into the data. By basing spot detection on the pinnacles, Pinnacle avoids many of these problems and permits the resolving of overlapping spots since even spots with a great degree of overlap typically have their own unique pinnacles. This can be seen in Figure 1 of Færgestad, et al.'s paper [11] illustrating an example with an “envelope of proteins” detected as a common spot by convention boundary-based spot detection methods. It is clear from that image that if an average were taken over the gels, two pinnacles would stand out distinctly from one another and be detected and quantified as separate spots.
The quantification of spots using pinnacle intensities rather than spot volumes precludes the estimation of spot boundaries. This has the potential to increase the reliability of spot quantifications, as spot boundary estimation is a difficult and error-prone process, especially when spots overlap. Even though spot volumes are traditionally used for quantification, if a given spot has a common shape across all of the gels, pinnacle intensities and true spot volumes should be strongly correlated with each other [3][12]. For quantification, the problem of overlapping spots is that the intensity at each pixel consists of a mixture of all spots spanning that pixel. By quantifying using only the pinnacle intensity, Pinnacle focuses on the part of the spot with maximum contribution from the spot of interest, the pinnacle, which potentially reduces the impact of the neighboring spots relative to volume-based methods whose quantification sums all pixel intensities within the estimated spot boundaries.
SameSpots
Working with aligned gels, Progenesis SameSpots starts by performing spot detection simultaneously across all of the images, obtaining a master list of detected spots and corresponding spot boundaries. These spot boundary masks are then overlaid on the individual gels, and are used to compute spot volumes, producing the N×p matrix Y.
The images for the four datasets were processed as single-stain/gel experiments. For the dilution series, the reference gel was selected from the highest protein load group. The reference gel remained constant throughout all analyses. As described previously, the images were aligned using TT900, therefore we proceeded to “analyze with SameSpots” without any further modifications. We did not employ the “mask of disinterest,” a tool used to exclude areas of the gel from analysis, because we had already cropped those regions from the gel images prior to analysis. Gels were grouped by protein load for the dilution series, and by treatment group for the comparative proteomics studies.
For the primary analysis, we evaluated SameSpots as an automatic method, with no manual editing of spots or spot boundaries. All detected spots were selected for analysis using the “spot review tool.” For background correction, we used the default “lowest on boundary,” which computes the spot background as the minimum pixel intensity adjacent to the spot boundary. Previous versions of Progenesis recommended the “Progenesis background subtraction” algorithm. We also evaluated this background correction method, but found that it resulted in considerably larger %CVs than “lowest on boundary.” In order to preserve the relationship between protein loads and spot volumes used to assess validity, normalization was not applied to the dilution series, but was applied to the differential expression data. The spot volumes for p spots for each of the N gels were exported as an N×p spreadsheet for further analysis.
We found that the spot detection algorithm within SameSpots at times resulted in a smaller spot count than expected, with the merging of numerous visibly distinct spots. In an attempt to improve spot count, we reapplied spot detection on the selected reference gel using the conventional (previous generation) Progenesis platform. The new spot boundaries were applied using the “SameSpots” option available within Progenesis (PG) to quantify the remaining gels in the experiment. This approach mimics the step-wise approach in SameSpots by detecting proteins on the reference gel and then applying the spot boundary “mask” to all of the gels in the experiment. This increased spot detection by an average of 27.6%, and improved spot splitting. No manual editing was performed for the primary comparisons.
As a secondary analysis, we edited the spots (split, merged, added, selected for deletion) found by SameSpots using the “spot review tool.” All of the edits were done by one author (BNC) to minimize user-induced variation. Spot volumes were then exported into an Excel spreadsheet. We found that the user interface in the conventional portion of the Progenesis platform (PG) had enhanced contrast and zooming capabilities, which permitted a better visual assessment of the spots than the interface in SameSpots. When viewing the SameSpots-edited spot boundaries in PG, we noticed that some of the spot boundaries could be further improved by additional manual editing in PG. Necessary edits were applied, and the SameSpots option was used to transfer the new spot boundaries to quantify cognate spots in the remaining gels in the experiment. This editing process removed the default settings applied by SameSpots (i.e., clearing all normalization from the experiment and background values from each edited gel). Therefore, the “lowest on boundary” background subtraction was re-applied, and normalization was reapplied for the differential expression studies, but not the dilution series. Spot volumes were exported into an Excel spreadsheet. The time required for manual editing was recorded.
In summary, we obtained spot volumes from SameSpots using four different methods, SameSpots alone (SameSpots), SameSpots aided by conventional Progenesis spot detection (SameSpots+PG), SameSpots with manual editing of spots (SameSpots+edits), and SameSpots with further manual editing in the conventional Progenesis platform (SameSpots+edits (PG)).
Progenesis PG240
We also applied a previous version of Progenesis (PG240, version 2006, Build 2405.1), which is based on the traditional approach, to the NC and SH dilution series so that we could determine whether newer methods (SameSpots and Pinnacle) showed improvements over traditional algorithms. We applied PG240 (without SameSpots) to the aligned, cropped gel images previously described. The analysis wizard was used to set up the experiments. Gels were grouped by dilution series. Spot criteria for the reference gel included all of the spots present on at least one gel. No manual editing of the spots was performed, and the spot volumes were exported into an Excel spreadsheet. We excluded from the quantitative analysis spots that were not present in 3 of 4 replicates for at least 1 protein load group in the NC study, or in 3 of 3 replicates for at least 1 protein load group in the SH study. Spot volumes of zero were substituted for spots present on other gels that did not have a match on the current gel.
Statistical Comparisons
For the dilution series, we assessed the two different components of precision, reliability and validity, separately. To assess reliability of spot quantifications, we computed the %CV across replicates for each detected spot and each protein load group for each method. We considered %CV a better measure than standard deviation for reliability, as the standard deviation is typically correlated to the mean, while the coefficient of variation is not. For the comparative proteomics study, we computed %CV for each treatment group. As different numbers of spots were detected by the different methods, we compared methods by summarizing the distribution of %CV across treatment groups, computing the mean and 5-number summary (5th percentile, 25th percentile, 50th percentile, 75th percentile, 95th percentile) for each protein load group. We also combined them over the protein load groups to get a single set of summary statistics for each study.
To assess validity, for each method we computed the coefficient of variation (R2) for the linear regression of the spot quantification vs. the protein load for each detected spot in the respective dilution series. Without normalization, more reliable spot quantification methods should yield stronger linear relationships, and thus greater R2, between spot quantifications and protein loads. This measure can also be interpreted as the square of the Pearson correlation coefficient between the spot quantifications and protein loads. We summarized again the distribution of R2 across spots within an experiment using the mean and 5-number summary.
The measures R2 and %CV provide a reasonable summary of the precision of a spot quantification method, and increased precision of spot quantifications should correspond to increased power in differential expression studies. However, they are not in themselves sufficient measures for summarizing performance. For example, neither measure penalizes a method for merging several distinct spots. That type of error will not decrease the R2 because each distinct spot should have the same ratio across gels in a dilution series; we may even expect the R2 to be higher and the %CV to be lower due to the reduced variability of any inherent technical noise from summing the volumes across spots. However, a spot quantification method that merges spots will tend to mask differential expression in comparative proteomics studies.
Thus, we also compared the methods in terms of differentially expressed proteins using two studies, both of which were expected to show evidence of some differentially expressed proteins.
For the differential expression studies, we first log-transformed spot quantifications to stabilize the variance [13] and then performed two-sample t-tests for each spot to detect differences in protein expression between treatment groups. We summarized the number of spots with p-values <0.05, 0.01, 0.005, and 0.001. When searching for differentially expressed proteins over several hundred or thousand spots, it is essential to do some sort of multiple comparison adjustment to ensure that the differences observed are not simply due to random chance alone [14]. For example, with 1000 spots, even if there is in fact no true difference between the protein expression in the treatment groups, by basic statistical principles it is expected that roughly 50, 10, 5, and 1 spots should have p-values less than 0.05, 0.01, 0.005, and 0.001, respectively. One way to adjust for multiple comparisons is to consider the false discovery rate (FDR), which is the probability that a spot determined to be significantly different is, in fact, a false discovery [14][15]. We applied the method fdrtool in R [16] to the p-values for each spot, and computed the corresponding q-values, which indicate the probability of a given spot being a false positive assuming it is called significant [17]. We summarized the number of spots with q-values<0.20 and 0.15, spots for which the probability of being a false discovery is <0.20 and 0.15, respectively, if flagged as differentially expressed.
Results
Spot Detection
NC Study
Using the traditional Progenesis PG240 method, a total of 1422 spots were detected in at least one gel in the NC study. Of these, only 312 spots (22%) were detected on all of the gels. There were 996 spots detected on at least 3 of 4 replicates in at least one protein load group. These spots were used for validity and reliability analyses.
Progenesis SameSpots detected 688 spots, and Pinnacle found 937 spots (see Figure 1d). The low number of spots detected by SameSpots appeared to be due to a merging of distinct spots in dense regions of the gel (Figure 1a). This problem has been previously observed [4]. Pinnacle did not have trouble resolving overlapping spots or defining spots in regions of high density because even overlapping spots have distinct pinnacles.
Figure 1. Spot Detection, NC Study.

Results of spot detection on Nishahara and Champion study for (a) SameSpots, (b) SameSpots with PG spot detection, (c) SameSpots with manual editing, and (d) Pinnacle. For (a)-(c), the image is from gel 22; spots are marked by their estimated boundaries. For (d), the image is the average gel used for spot detection; pinnacles for detected spots are marked by a circled x.
When PG was used for spot detection after gel alignment by SameSpots, 1034 spots were detected (see Figure 1b). This improvement appeared to be due to improved splitting of spots. After 240 minutes of manual spot editing of the reference gel using SameSpots, the number of spots detected increased from 688 to 900 spots (see Figure 1c). After 60 additional minutes of manual spot editing in PG after SameSpots alignment, the spot count increased from 900 to 1082 spots.
SH Study
For PG240, a total of 1344 spots were detected on at least one gel. Of these, just 99 were detected in all of the gels, and 1094 were detected in all 3 replicates for at least one protein load group. SameSpots detected a total of 1037 spots, and again spot merging appeared to be an issue. The number increased to 1305 spots when PG was used to perform spot detection on the back end. After 310 minutes of manual editing, SameSpots detected 1244 spots, and with an additional 180 minutes of manual editing in PG, detected 1383 spots. Pinnacle found a total of 1322 spots.
D1 Study
SameSpots detected 1094 spots, with 1319 detected when PG was used on the back end of SameSpots to enhance detection. After 180 minutes of manual editing in SameSpots, 1153 spots were detected, and after an additional 25 minutes of manual editing in PG, 1255 spots were detected. Pinnacle found a total of 719 spots with automatic settings and no manual editing.
D2 Study
SameSpots detected 1410 spots, with 1928 detected when PG was used on the back end. After 180 minutes of manual editing in SameSpots, 1390 spots were detected, and after 25 additional minutes editing in PG, 1458 spots were detected. Pinnacle found a total of 1136 spots with automatic settings and no manual editing.
Discussion
We found SameSpots at times experienced problems with merging distinct spots in dense regions of the gel. This problem was largely mitigated when detection was done in PG after alignment using SameSpots. Extensive manual editing was required to correct errors in spot splitting and detection. Pinnacle appeared to do a good job of detecting spots without any manual editing, although if desired, the user can edit spots after detection in Pinnacle or try different tuning parameters to optimize results. We did not apply this option, so all of the results include only the spot lists automatically obtained by the Pinnacle algorithm using standard tuning parameter settings.
Validity
Validity of spot quantifications was summarized by the R2 of a linear regression of the spot quantifications on the protein loads. Not all of the methods identified the same spots; therefore, we summarized the results by their distributions across all of the detected spots for each method, as shown in Table 1.
Table 1. Validity of Quantifications for Detected Spots.
Summary of R2 measuring linearity of quantification method across protein loads within dilution series for all spots automatically detected by the various preprocessing methods, including (1) Pinnacle, (2) SameSpots, (3) SameSpots with PG spot detection (SameSpots +PG), (4) SameSpots with manual editing (SameSpots+edits), (5) SameSpots with manual editing in PG (SameSpots+edits (PG)), and (6) Progenesis PG240 (PG240).
| Study | Method | Number of Spots | R2 (Validity) | |||||
|---|---|---|---|---|---|---|---|---|
| Mean | Q05 | Q25 | Median | Q75 | Q95 | |||
| NC | Pinnacle | 937 | 0.965 | 0.899 | 0.966 | 0.980 | 0.987 | 0.992 |
| SameSpots | 688 | 0.956 | 0.889 | 0.934 | 0.977 | 0.984 | 0.992 | |
| SameSpots +PG | 1034 | 0.948 | 0.818 | 0.959 | 0.976 | 0.984 | 0.992 | |
| SameSpots+edits | 900 | 0.958 | 0.892 | 0.963 | 0.977 | 0.984 | 0.992 | |
| SameSpots+edits (PG) | 1082 | 0.956 | 0.878 | 0.960 | 0.976 | 0.984 | 0.992 | |
| PG240 | 996 | 0.900 | 0.571 | 0.909 | 0.957 | 0.980 | 0.990 | |
| SH | Pinnacle | 1322 | 0.879 | 0.734 | 0.840 | 0.900 | 0.954 | 0.986 |
| SameSpots | 1037 | 0.804 | 0.087 | 0.780 | 0.895 | 0.959 | 0.990 | |
| SameSpots +PG | 1305 | 0.867 | 0.598 | 0.821 | 0.910 | 0.962 | 0.991 | |
| SameSpots+edits | 1244 | 0.876 | 0.622 | 0.836 | 0.917 | 0.964 | 0.990 | |
| SameSpots+edits (PG) | 1383 | 0.870 | 0.610 | 0.827 | 0.909 | 0.962 | 0.990 | |
| PG240 | 1094 | 0.698 | 0.039 | 0.601 | 0.815 | 0.920 | 0.979 | |
NC Study
Boxplots of R2 for detected spots are contained in Figure 2(a). For the 937 spots detected by Pinnacle, the mean R2 was 0.965, which corresponds to a correlation of 0.982 between the spot quantifications and the true protein loads. Of the 688 spots detected by SameSpots, the mean R2 was 0.956. With PG spot detection and no manual editing, the mean R2 for the 1034 detected spots was 0.948. After manual editing, the mean R2 across the 900 detected spots was 0.958. After spot editing using PG, the mean R2 was 0.956. Comparing the distributions of R2 by looking at the 5-number summaries and boxplots, we see that the differences between Pinnacle and SameSpots was strongest at the lower and middle part of the distribution, i.e. for the less reliable spots. Pinnacle and each of the SameSpots methods greatly outperformed the traditional PG240, which resulted in spot quantifications with a mean R2 of 0.900 across the 996 detected spots, and with dramatic differences at the low end of the R2 distribution.
Figure 2. Boxplots of Validity and Reliability of Detected Spots, NC and SH Dilution Series.
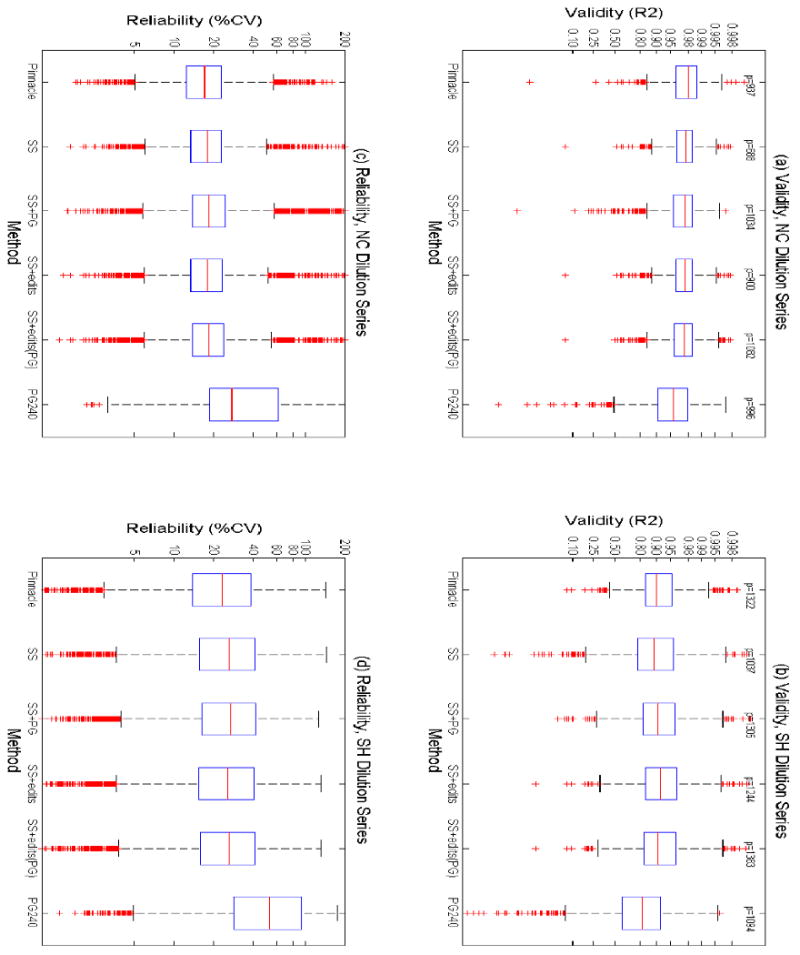
Boxplot of (a) validity (R2), Nishahara and Champion Study (NC), (b) validity (R2), SH-SY5Y Neuroblastoma Cells Dilution Series (SH), (c) reliability (%CV), NC study, (d) reliability (%CV), SH study, for all spots detected by various methods, including Pinnacle, SameSpots (SS), SameSpots+PG (SS+PG), SameSpots+edits (SS+edits), SameSpots+edits-PG (SS+edits-PG), and PG240, with the numerical results plotted on a log axis for clarity of presentation.
SH Study
Boxplots of R2 for detected spots are contained in Figure 2(b). For the 1322 spots detected by Pinnacle, the mean R2 was 0.879. This was much higher than the mean R2 for the 1037 spots detected by SameSpots, which was 0.804. When SameSpots alignment was followed by spot detection using PG, the mean R2 for 1305 spots improved to 0.867. After manual editing, the R2 across 1244 detected spots was 0.876. With additional manual editing in PG, the mean R2 was 0.870. Comparing the distributions of R2 between the methods, we see that the lower quantiles of R2 are considerably larger for Pinnacle than the SameSpots methods, and the upper quantiles were similar, with the SameSpots methods slightly higher. Again, PG240 performed much worse than either Pinnacle or any variation of SameSpots, with a mean R2 of 0.698 across 1094 detected spots, and many spots with low R2 values.
Discussion
The gels in the NC dilution series were exceptionally clean and well run. This was reflected by the excellent validity measures found for this data set for all of the methods. The SH dilution series may be more representative of gels commonly seen in practice. That dilution series resulted in smaller, but still reasonable, R2 values. However, the relative performance of the different processing methods was similar for both data sets. Both Pinnacle and SameSpots provided spot quantifications with much higher validity than PG240. This may be due to the missing data problem inherent in the traditional approaches, and the extra measurement variability induced by estimating separate spot boundaries for cognate spots in each gel in PG240. The results produced by SameSpots were improved when manual editing was performed to correct a number of spots that were inaccurately detected or split. In both dilution series, Pinnacle appeared to compare favorably to the SameSpots methods, with higher mean R2, improved lower quantiles, and similar higher quantiles in the R2 distribution.
Reliability
Reliability of spot quantifications was summarized by the %CV for each spot within each group. The different methods did not detect the same sets of spots; therefore, we summarized the reliability of spot quantifications by their distributions across all of the detected spots for each method. Summary values for each protein load group are found in the supplementary material, including the mean %CV, 5-number summary, plus the number and proportion of reliable spots, i.e. with %CV>20. The summary values combining the results for all of the protein load groups are found in Table 2.
Table 2. Reliability of Quantifications for Detected Spots.
Summary of %CV for spots detected by various preprocessing methods, across all dilution series and treatment groups for differential expression studies. Methods include (1) Pinnacle, (2) SameSpots, (3) SameSpots with PG spot detection (SameSpots+PG), (4) SameSpots with manual editing with SameSpots (SameSpots+edits), (5) SameSpots with manual editing with PG (SameSpots+edits (PG)), (6) Progenesis PG240 (PG240).
| Study | Method | Number of Spots | %CV (Reliability) | |||||
|---|---|---|---|---|---|---|---|---|
| Mean | Q05 | Q25 | Median | Q75 | Q95 | |||
| NC | Pinnacle | 937 | 18.8 | 7.4 | 12.5 | 17.1 | 22.8 | 33.9 |
| SameSpots | 688 | 20.2 | 8.2 | 13.4 | 17.9 | 22.8 | 36.2 | |
| SameSpots +PG | 1033 | 22.7 | 8.0 | 13.7 | 18.5 | 24.4 | 44.4 | |
| SameSpots +edits | 900 | 20.2 | 7.9 | 13.4 | 17.9 | 23.0 | 35.7 | |
| SameSpots +edits (PG) | 1082 | 20.9 | 7.8 | 13.7 | 18.3 | 23.9 | 38.1 | |
| PG240 | 566 | 49.7 | 10.5 | 18.6 | 27.4 | 61.5 | 200.0 | |
| SH | Pinnacle | 1322 | 27.5 | 5.2 | 13.7 | 23.2 | 38.3 | 61.0 |
| SameSpots | 1037 | 29.9 | 5.9 | 15.5 | 26.4 | 41.1 | 64.4 | |
| SameSpots +PG | 1305 | 30.4 | 6.3 | 16.2 | 27.1 | 41.8 | 64.9 | |
| SameSpots +edits | 1244 | 29.4 | 5.7 | 15.4 | 25.6 | 40.5 | 64.0 | |
| SameSpots +edits (PG) | 1383 | 30.0 | 6.1 | 15.9 | 26.3 | 41.2 | 65.1 | |
| PG240 | 391 | 66.9 | 11.1 | 28.4 | 53.0 | 92.3 | 173.2 | |
| D1 | Pinnacle | 719 | 25.6 | 11.0 | 16.5 | 22.4 | 30.1 | 53.0 |
| SameSpots | 1094 | 43.8 | 22.5 | 30.7 | 37.8 | 51.6 | 85.6 | |
| SameSpots +PG | 1319 | 44.0 | 23.6 | 31.5 | 38.3 | 51.1 | 79.2 | |
| SameSpots +edits | 1153 | 40.5 | 22.5 | 31.0 | 36.8 | 46.6 | 69.3 | |
| SameSpots +edits (PG) | 1255 | 40.3 | 22.5 | 31.0 | 36.8 | 46.5 | 68.5 | |
| D2 | Pinnacle | 1136 | 28.8 | 10.4 | 16.2 | 21.9 | 31.4 | 75.0 |
| SameSpots | 1410 | 36.8 | 14.7 | 21.6 | 29.1 | 44.3 | 86.0 | |
| SameSpots +PG | 1928 | 36.7 | 15.3 | 23.4 | 32.2 | 44.7 | 72.0 | |
| SameSpots +edits | 1390 | 30.2 | 14.3 | 20.7 | 27.2 | 35.7 | 56.0 | |
| SameSpots +edits (PG) | 1458 | 30.3 | 14.2 | 20.8 | 27.5 | 36.1 | 55.6 | |
NC Study
Boxplots of %CV for the detected spots are contained in Figure 2(c). The mean %CV for Pinnacle was 18.8; for SameSpots it was 20.2, and 22.7 when PG was used for spot detection. After manual editing, the mean %CV was 20.2 after SameSpots editing, and 20.9 after also editing with PG. Looking at the distribution of %CV, we see that Pinnacle tended to have slightly smaller %CV quantiles throughout the distribution. For PG240 alone, the mean %CV across the detected spots was 49.7.
SH Study
Boxplots of %CV for detected spots are contained in Figure 2(d). The mean %CV for Pinnacle was 27.5; for SameSpots it was 29.9, and 30.4 when PG was used for spot detection. After manual editing, the mean %CV was 29.4 after SameSpots editing, and 30.0 after also editing with PG. Again, the differences between Pinnacle and SameSpots were consistent throughout the distribution of spot %CV. The mean %CV for PG240 alone was 66.9.
D1 Study
The mean %CV for Pinnacle was 25.6; for SameSpots it was 43.8, and 44.0 when PG was used for spot detection. After manual editing, the mean %CV was 40.5 after SameSpots editing, and 40.3 after also editing with PG. The differences in %CV distribution were seen across the entire distribution of spot %CV.
D2 Study
The mean %CV for Pinnacle was 28.8; for SameSpots it was 36.8, and 36.7 when PG was used for spot detection. After manual editing, the mean %CV was 30.2 after SameSpots editing, and 30.3 after also editing with PG. Again, we see consistent differences across the entire range of %CV.
Discussion
For both dilution series, SameSpots and Pinnacle had much smaller %CV than PG240, which is likely due to the missing data and extra technical variability from estimating separate spot boundaries for cognate spots on each gel. The %CV for SameSpots improved after manual editing, but Pinnacle still had considerably smaller %CV than any of the SameSpots methods across all of the studies. In the supplemental tables split out by protein load, we see that use of Pinnacle tended to find a higher number and higher proportion of spots that were reliable, defined as %CV<20. We propose that the use of pinnacles rather than spot volumes in quantification is a key factor in the improved reliability. Increased reliability is important in differential expression studies, as lower %CV should lead to increased power to detect differentially expressed proteins.
Differential Expression
The differential expression results for the comparative proteomics studies are in Table 3.
Table 3. Differential Expression Comparison, Comparative Proteomics Studies.
Summary of performance of preprocessing methods in detecting differential expression in comparative proteomics studies. The p-values are from two-sample t-tests performed on the log-transformed spot quantifications for the various methods. The q-values are local FDR estimates, and measure the probability that a given spot would be a false positive if it is declared differentially expressed.
| Method | Number of Spots | q-values | p-values | |||||
|---|---|---|---|---|---|---|---|---|
| <0.15 | <0.20 | <0.001 | <0.005 | <0.01 | <0.05 | |||
| D1 | Pinnacle | 719 | 2 | 3 | 3 | 6 | 17 | 76 |
| SameSpots | 1094 | 0 | 0 | 0 | 0 | 1 | 17 | |
| SameSpots +PG | 1319 | 0 | 0 | 0 | 1 | 3 | 24 | |
| SameSpots +edits | 1153 | 0 | 0 | 0 | 0 | 1 | 11 | |
| SamesSpots +edits (PG) | 1255 | 0 | 0 | 0 | 0 | 1 | 13 | |
| D2 | Pinnacle | 1136 | 5 | 7 | 5 | 10 | 20 | 107 |
| SameSpots | 1410 | 0 | 0 | 2 | 11 | 24 | 123 | |
| SameSpots +PG | 1928 | 0 | 0 | 2 | 16 | 35 | 151 | |
| SameSpots +edits | 1390 | 0 | 0 | 1 | 14 | 32 | 157 | |
| SamesSpots +edits (PG) | 1458 | 0 | 0 | 0 | 12 | 27 | 159 | |
D1 Study
Although Pinnacle with default tuning parameters found fewer overall spots for this data set (719 for Pinnacle vs. 1094-1319 for the various SameSpots methods), it found more spots with p-values <0.05 (76 vs. 11-24), <0.01 (17 vs. 1-3), <0.005 (6 vs. 0-1), and <0.001 (3 vs. 0). Flagging spots with local false discovery rates <0.15 or <0.20 as differentially expressed, Pinnacle found 2 or 3 differentially expressed proteins, respectively, while none of the SameSpots methods found any.
D2 Study
Of the 1136 spots detected, Pinnacle flagged 5 or 7 spots as significantly differentially expressed with a local FDR of <0.15 or 0.20, respectively, while none were found using any of the SameSpots methods. Pinnacle found more spots with p-values <0.001 (5 vs. 1-2) than the SameSpots methods. In this study, SameSpots methods found more spots with p-values <0.005 (11-16 vs. 10), <0.01 (24-35 vs. 20), and <0.05 (123-159 vs. 107), with the difference roughly proportional to the total number of spots detected (1410-1928 vs. 1136).
Discussion
Even with smaller spot counts in these two studies, compared to SameSpots, Pinnacle found more spots with very small p-values. After adjusting for multiple testing using FDR methods, Pinnacle found significant spots, whereas the SameSpots methods found none. These results were not surprising given that spot quantifications using Pinnacle had higher validity and reliability than any of the SameSpots methods.
Conclusions
The newer preprocessing methods, SameSpots and Pinnacle, both show tremendous improvement over PG240, a method based on a traditional analytical approach, in terms of validity and reliability of spot quantifications for 2-DE. Both SameSpots and Pinnacle avoid missing data, and reduce technical variability because they do not try to estimate different spot boundaries for corresponding spots on each gel, which is a difficult and error-prone step that increases variability. SameSpots still estimates spot boundaries, but estimates a single boundary for each spot that is applied to quantify that spot for all of the aligned gels. This eliminates missing data, and reduces technical variability in the spot quantifications.
Pinnacle bases the entire spot detection and quantification process on the pinnacles of the spots, and so avoids estimating spot boundaries altogether. This may seem controversial to some, as spot volumes are often viewed as the “correct” quantifications for proteins in a gel. However, given the characteristics of pinnacles—they highly correlate with volumes [3], are much easier to estimate, introduce less technical variability, and are less influenced by overlap from neighboring spots—an analytic plan using pinnacles seems to be a reasonable approach.
As far as we know, there have been no definitive studies demonstrating that estimated spot volumes yield better quantifications than pinnacle intensities and, in fact, our results here and in our previous study [3] suggest the opposite. We have demonstrated that Pinnacle on average yields spot quantifications with higher validity than volume-based SameSpots, even after SameSpots results are improved by hours of manual editing. Pinnacle-based spot quantifications were also consistently more reliable than SameSpots, yielding lower %CV in four different data sets. The significance of these improvements in spot quantification precision was strikingly demonstrated by the differential expression analysis. Using Pinnacle resulted in spots flagged as differentially expressed using FDR-based methods, while even extensive manual editing of the SameSpots analysis did not yield significant differential expression in our two examples.
The spot detection algorithm utilized by the SameSpots interface in the Progenesis package appears, at times, to have difficulty splitting overlapping spots in dense regions of the gel. The negative effects of the merging of distinct spots were not reflected in their validity and reliability results. This is because the merged “superspots” would still have reasonable R2 and %CV in the dilution series due to the spots having the same ratio of volumes between gels of differing protein loads. In the differential expression setting, in which we expect different expression ratios between distinct spots on different gels, such merging of spots would in principle mask differential expression if one but not all of the proteins in the “superspot” were in fact differentially expressed [11]. This might also partially explain Pinnacle's better performance in the differential expression studies.
Since the completion of the studies reported in this paper, other companies have updated their 2-DE processing software to use a similar workflow as SameSpots, aligning the gels and using common spot outlines across gels for quantification, including REDFIN Solo (Ludesi; Malmo Sweden) and Delta2D (Decodon; Greifswald, Germany). These commercial packages were not yet available at the time this study was done, so were not compared in this study.
One situation in which we do not expect Pinnacle, or any other 2-DE analysis package, to work well is when pixel intensities on the gel image reach saturation, disrupting the linear relationship between pinnacle intensities and spot volumes, and potentially obscuring differential expression [18]. We avoid saturating any spots by adjusting the contrast settings when imaging gels, and we recommend that approach for all methods.
In conclusion, we have demonstrated that new analysis algorithms based on image alignment and standardized spot boundaries (or pinnacles) between gels markedly improve the robustness of analysis when compared with previous methods involving conventional spot detection and individual matching techniques. Thus, these new methods more accurately extract the proteomic information from the gel images, overcoming some of technical problems that in the past have prevented 2DGE and DIGE from realizing their full potential for biomedical research.
Pinnacle is a non-commercial method that differs from the available commercial packages in its use of pinnacle detection on wavelet-denoised average gels for spot detection, and its use of pinnacle intensities in lieu of spot volumes for quantification. In the studies presented here, Pinnacle demonstrated greater measurement precision, permitting the identification of many more differentially expressed spots, than prominent commercial software SameSpots, even after extensive hand editing was done to optimize the SameSpots results. Further, Pinnacle is automatic, eliminating the potential for inadvertent bias, and did not experience the problems with spot-merging of overlapping spots that was sometimes seen with SameSpots. It represents a significant non-commercial alternative for preprocessing 2-DE images.
Supplementary Material
Acknowledgments
We thank Dr. Kathy Champion-Francissen for the use of gel images from her 2002 study. We also thank Miguel Diaz, Nicole Bjorklund, and Gaelle Parsons for excellent technical assistance. This work was supported by grants from the National Cancer Institute (CA107304) to JSM and from the National Institute on Drug Abuse (DA18310) and the National Institute on Alcohol Abuse and Alcoholism (AA16157) to HBG.
Abbreviations Used
- FDR
false discovery rate
- SH
SH-SY5Y neuroblastoma cell dilution series
- MALDI-MS
Matrix assisted laser desorption ionization time-of-flight mass spectrometry
- NC
Nishihara and Champion dilution series
- pI
isoelectric point
- R2
Coefficient of variation (squared correlation) for validity
- 2d
two-dimensional
- 2-DE
two-dimensional polyacrylamide gel electrophoresis
- D1
First differential expression study
- D2
Second differential expression study
- %CV
Coefficient of variation (percentage) for reliability
- DIGE
Differential In-Gel Electrophoresis
Footnotes
Conflict of Interest Statement
The authors do not have any financial or commercial conflicts of interest regarding this paper.
References
- 1.Cutler P, Heald G, White IR, Ruan J. A novel approach to spot detection for two-dimensional gel electrophoresis images using pixel value collection. Proteomics. 2003;3:392–401. doi: 10.1002/pmic.200390054. [DOI] [PubMed] [Google Scholar]
- 2.Clark BN, Gutstein HB. The myth of automated, high-throughput two-dimensional gel electrophoresis. Proteomics. 2008;8:1197–1203. doi: 10.1002/pmic.200700709. [DOI] [PubMed] [Google Scholar]
- 3.Morris JS, Clark BN, Gutstein HB. Pinnacle: A fast, automatic and accurate method for detecting and quantifying protein spots in 2-dimensional gel electrophoresis data. Bioinformatics. 2008;24:529–536. doi: 10.1093/bioinformatics/btm590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karp NA, Feret R, Rubtsov DV, Lilley KS. Comparison of DIGE and post-stained gel electrophoresis with both traditional and SameSpots analysis for quantitative proteomics. Proteomics. 2008;8:948–960. doi: 10.1002/pmic.200700812. [DOI] [PubMed] [Google Scholar]
- 5.Nishihara JC, Champion KM. Quantitative evaluation of proteins in one- and two-dimensional polyacrylamide gels using a fluorescent stain. Electrophoresis. 2002;23:2203–2215. doi: 10.1002/1522-2683(200207)23:14<2203::AID-ELPS2203>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 6.Dowsey AW, Dunn MJ, Yang GZ. Automated image alignment for 2D gel electrophoresis in a high-throughput proteomics pipeline. Bioinformatics. 2008;24:950–957. doi: 10.1093/bioinformatics/btn059. [DOI] [PubMed] [Google Scholar]
- 7.Morris JS, Coombes KR, Koomen J, Baggerly KA, Kobayashi R. Feature extraction and quantification for mass spectrometry in biomedical applications using the mean spectrum. Bioinformatics. 2005;21:1764–1775. doi: 10.1093/bioinformatics/bti254. [DOI] [PubMed] [Google Scholar]
- 8.Luhn S, Berth M, Hecker M, Bernhardt J. Using standard positions and image fusion to create proteome maps from collections of two-dimensional gel electrophoresis images. Proteomics. 2003;3:1117–1127. doi: 10.1002/pmic.200300433. [DOI] [PubMed] [Google Scholar]
- 9.Kaczmarek K, Walczak B, de Jong S, Vandeginste BGM. Preprocessing of two-dimensional gel electrophoresis images. Proteomics. 2004;4:2377–2389. doi: 10.1002/pmic.200300758. [DOI] [PubMed] [Google Scholar]
- 10.Daszykowski M, Stanimirova I, Bodzon-Kulakowska A, Silberring J, Lubec G, Walczak B. Journal of Chromatography A. 2007;1158:306–317. doi: 10.1016/j.chroma.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Færgestad EM, Rye M, Walczak B, Gidskehaug L, Wold JP, Grove H, Jia X, Hollung K, Indahl UG, Westad F, van den Berg F, Martens H. Pixel-based analysis of multiple images for the indentification of changes: A novel approach applied to unravel patterns of 2-D electrophoresis gel images. Proteomics. 2007;7:3450–3461. doi: 10.1002/pmic.200601026. [DOI] [PubMed] [Google Scholar]
- 12.Mahon P, Dupree P. Quantitative and reproducible two-dimensional gel analysis using Phoretix 2D Full. Electrophoresis. 2001;22:2075–2085. doi: 10.1002/1522-2683(200106)22:10<2075::AID-ELPS2075>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 13.Dutt MJ, Lee KH. The scaled volume as an image analysis variable for detecting changes in protein expression levels by silver stain. Electrophoresis. 2001;22:1627–1632. doi: 10.1002/1522-2683(200105)22:9<1627::AID-ELPS1627>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 14.Gutstein HB, Morris JS. Laser capture sampling and analytical issues in proteomics. Expert Reviews in Proteomics. 2007;4:627–637. doi: 10.1586/14789450.4.5.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gutstein HB, Morris JS, Annangudi SP, Sweedler JV. Microproteomics: Analysis of protein diversity in small samples. Mass Spectrometry Reviews. 2008;27:316–330. doi: 10.1002/mas.20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strimmer K. fdrtool: a versitile R package for estimating local and tail area-based false discovery rates. Bioinformatics. 2008;24:1461–1462. doi: 10.1093/bioinformatics/btn209. [DOI] [PubMed] [Google Scholar]
- 17.Storey JD. The positive false discovery rate: A Bayesian interpretation and the q-value. The Annals of Statistics. 2003;31(6):2013–2035. [Google Scholar]
- 18.Almeida JS, Stanislaus R, Krug E, Arthur J. Normalization and analysis of residual variation in 2D gel electrophoresis for quantitative differential proteomics. Proteomics. 2005;5(5):1242–1249. doi: 10.1002/pmic.200401003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.