

Abstract
The vast majority of the ca. 100 chemically distinct modified nucleosides in RNA appear to arise via the chemical transformation of a genetically encoded nucleoside. Two notable exceptions are queuosine and pseudouridine, which are incorporated into tRNA via transglycosylation. Transglycosylation is an extremely efficient process for incorporating highly modified bases such as queuine into RNA. Transglycosylation is also a requisite process for “isomerizing” an N-nucleoside into a C-nucleoside as is the case for pseudouridine formation. Finally, transglycosylation is an attractive possibility for certain RNA editing events (e.g., pyrimidine to purine conversions) that cannot occur via the known, more straightforward enzymatic reactions (e.g., deaminations). This review discusses what is known about the mechanisms of transglycosylation for the queuine and pseudouridine RNA modifications and will speculate about a potential role for transglycosylation in certain RNA editing events.
Keywords: RNA, Modification, Editing, Pseudouridine, Queuosine, Transglycosylation
1. Introduction
One characteristic of all RNAs is the occurrence of modified nucleosides throughout the primary structure. Modified nucleosides are incorporated into the polynucleotide chain by specific RNA-modifying enzymes during all stages of the post-transcriptional processing of nascent RNA transcripts. To date, ca. 100 chemically distinct nucleosides have been identified, spanning a wide range of chemical complexity [1]. “Simple” modifications (e.g., methylations, thiolations, etc.) are generally the result of a single enzymatic activity, while the more “complex” modifications are characterized by the presence of multiple modifications (e.g., methylation and thiolation) or the requirement for multi-step biosynthetic pathways to achieve the desired transformation. With few exceptions, modified nucleosides arise from the chemical transformation of a genetically encoded nucleoside [2]. One notable exception is queuosine, which is incorporated into tRNA via transglycosylation (in this case, cleavage/reformation of the glycosidic bond, exchanging one base for another). Pseudouridine is also introduced into various RNAs via intramolecular transglycosylation. We will review what is known about the mechanisms of transglycosylation for these two RNA modifications and will speculate about a potential role for transglycosylation in certain RNA editing events.
2. tRNA-guanine transgylcosylase
Of the known tRNA modifications, queuosine (Q, 7-(((4,5-cis-dihydroxy-2-cyclopenten-1-yl)amino)methyl)-7-deazaguanosine) and archaeosine (G+, 7-form-amidino-7-deazaguanosine) represent two of the most complex that have been identified thus far (Fig. 1). Both feature a heterocyclic base moiety that is structurally unique from other nucleosides [3,4]. In each case, the nucleosides consist of neither a purine nor pyrimidine but rather a pyrrolopyrimidine heterocyclic backbone. Furthermore, each of these modifications possesses an exocyclic side-chain extending from the 5-position (analogous to the purine 7-position) carbon of the pyrrolopyrimidine ring. Archaeosine appends an amidino functionality at this position, while queuosine possesses a much more elaborate aminomethyl cyclopentenyl diol side-chain (Fig. 1).
Fig. 1.
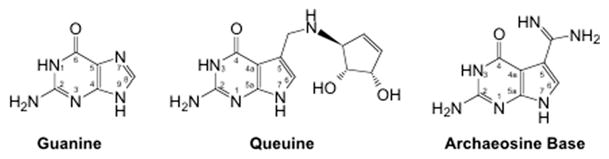
Structures of guanine, queuine, and archaeine (the base of archaeosine). Note that the N7 of the purine ring system corresponds to the C5 of the pyrrolopyrimidine ring system.
The mechanism of incorporation of both queuosine and archaeosine into tRNA is also relatively unique among those cases in which the modification mechanisms are known [5]. Rather than resulting from the direct transformation of a genetically encoded nucleoside, each of these modifications arises through the replacement of a genetically encoded guanine in a reaction that is catalyzed by the enzyme tRNA-guanine transglycosylase (TGT, E.C. 2.4.2.29) [6]. Interestingly, this base-exchange reaction does not require the cleavage of the phosphodiester backbone of the polynucleotide, nor does it require energy input (e.g., ATP hydrolysis) [7,8]. In both eubacteria and eukaryotes, with the exception of yeast [9], TGT activity ultimately leads to the incorporation of queuosine into the wobble position (position 34) of four cognate tRNAs (tRNAAsp,Asn,His,Tyr) [10,11]. In contrast, the archaeal TGT is involved in the incorporation of archaeosine at position 15 in the dihydrouridine (D) loop of many tRNAs [4,12].
While all three classes of TGTs (eubacteria, eukarya, and archaea) share a high degree of sequence similarity (~40%), domain-specific structural properties have been identified. For example, the eukaryotic TGT is a heterodimer that is comprised of a putative 60-kDa regulatory subunit and a 35-kDa catalytic subunit [13,14] while the eubacterial TGT has been considered to be monomeric [15,16] although a recent crystal structure shows the Zymomonas mobilis TGT to be a homodimer [17]. Additionally, the TGT from archaea is considerably larger, containing ca. 100–300 extra amino acids (species specific) extending from the C-terminus of the core catalytic domain. The reason for this C-terminal extension is still unclear; however, it is intriguing to speculate that it is involved in the conversion of preQ0-tRNA to archaeosine tRNA (Fig. 2).
Fig. 2.

Queuosine and archaeosine biosynthetic pathways. Archaeosine is found in the D-loop of many archaeal tRNAs. Queuosine occurs in the wobble position of the anticodon of tRNAs (Asp, Asn, His, and Tyr) in both eubacteria and eukarya.
Representative TGTs from all three classes have been studied; however, much of what is known regarding the TGT-catalyzed reaction has been gained through extensive study of the eubacterial enzyme. The crystal structure of the TGT from Z. mobilis has been solved at 1.85 Å, revealing a non-canonical (β/α)8-barrel fold [18]. Soaking of the crystals with the preQ1 substrate allowed for the first identification of the TGT active site [19]. Our laboratory, as well as others, has used this structure as the basis for mechanistic studies (Fig. 3). More recently, Xie et al. [17] have reported the elucidation of a TGT–RNA co-crystal structure, providing an enhanced view of the TGT active site during catalysis. To date, structural information has yet to be obtained for any TGT from the eukaryotic kingdom; however, a crystal structure of the TGT from the archaeaon Pyrococcus horikoshii has been solved, revealing the same (β/α)8-barrel fold displayed by the Z. mobilis enzyme [20,21]. This finding implies an evolutionary relationship between TGTs across the kingdoms.
Fig. 3.
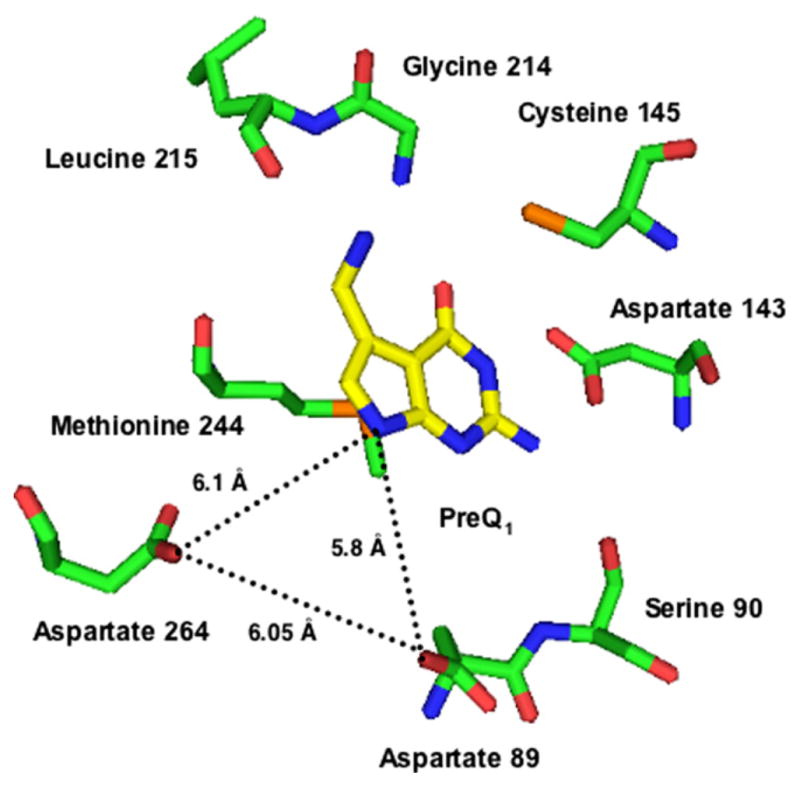
Active site of Z. mobilis TGT with PreQ1 bound. Aspartates 89 and 264 form a roughly equilateral triangle with N7 (corresponding to the purine N9) of preQ1 with sides ca. 6 Å. (Structure is from coordinates communicated by Dr. Ralf Ficner.)
Much of the biochemical characterization of the eubacterial TGT has been performed with the Escherichia coli enzyme. The recombinant TGT from E. coli is a 43-kDa enzyme that exists as a homotrimer in solution but dissociates into a monomer upon tRNA binding [22,23]. It is unclear if the trimeric structure is realized in vivo; however, it has been reported that the highly homologous TGT from Z. mobilis (ca. 60% similarity) exists as a monomer in the absence of tRNA [16]. Recently, studies in our laboratory have demonstrated that the E. coli TGT proceeds via a ping-pong kinetic mechanism whereby tRNA is the first substrate to bind to the enzyme [24]. This finding is consistent with the presence of a single binding site (as identified in the TGT-preQ1 crystal structure) that can accommodate either a guanine or preQ1 base and suggests that the preQ1 base can bind only after the displaced guanine diffuses from the active site.
In addition to a thorough understanding of the kinetics of the TGT reaction, studies over the last decade have helped define the recognition of both the tRNA [22,25–33] and heterocyclic base substrates [34–36] and have also helped unveil the roles that certain active-site amino acid residues serve in catalysis. Early work in our laboratory determined that TGT is a zinc metalloenzyme. Site-directed mutagenesis followed by biochemical characterization suggested four zinc ligands: cysteine 302, cysteine 304, cysteine 307, and histidine 317 [37]. The subsequent crystal structure of the TGT from Z. mobilis later confirmed that all three cysteine residues were coordinated to a zinc atom; however, the structure identified the fourth zinc ligand to be histidine 333 and not histidine 317 [19]. The role of the zinc atom appears to be in the maintenance of TGT structure and not in catalysis.
The roles of two additional cysteine residues have also been investigated in our laboratory. Both cysteine 265 and cysteine 145 are absolutely conserved in TGTs from eubacteria, suggesting they may be functionally important to this TGT class. Cysteine 265 was shown to exhibit sensitivity to thiol-modifying reagents (i.e., DTNB, MMTS, and N-ethylmaleamide) [38]. Subsequent mutagenesis experiments indicated that cysteine 265 did not serve a critical role in TGT catalysis. The X-ray crystal structure of the Z. mobilis TGT later indicated that the side chain of cysteine 265 is directed toward the surface of TGT in an area thought to bind tRNA [19]. Cysteine 145 has recently been the subject of extensive study in our laboratory (Goodenough and Garcia, unpublished); however, its exact role in catalysis remains yet unclear.
The role of serine 90 was investigated by our laboratory after it was discovered that a phenylalanine substitution at this position resulted in an inactive TGT [39]. Site-directed mutagenesis studies determined that the observed loss of activity was catalytically significant, and not merely the result of a conformational change brought about by the non-conservative mutation [23]. This finding implicated an important role for serine 90 in TGT catalysis. Structural work by Grädler et al. [40] later confirmed the role of serine 90 to be substrate orientation.
Most recently, the role of aspartate 143 has been investigated both biochemically and computationally. Aspartate 143 has been proposed by Romier et al. [41] to aid in recognition of the preQ1 base via hydrogen bonds to N3 and the exocylic amine. The biochemical characterization of a series of TGT mutants has largely confirmed this role [42]. The mutants (alanine, asparagine, serine, and threonine) all exhibited ca. 10-fold reductions in kcat and an increasing trend in Km for guanine (in a guanine-exchange reaction) that spans ca. 3 orders of magnitude from the wild-type.
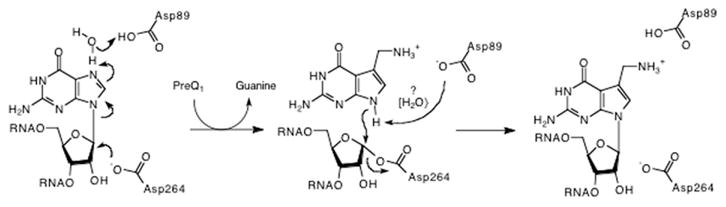
Two additional active-site aspartates have also been investigated for their role in the TGT catalysis. Interestingly, despite the substrate differences found across the three classes of TGT, both aspartate 89 and aspartate 264 are invariant in all known TGT sequences. Furthermore, based on the preQ1-bound TGT crystal structure (Fig. 3), both residues appear to be appropriately positioned in the active site to participate in TGT catalysis. Romier et al. [43] demonstrated that the replacement of aspartate 102 in the Z. mobilis TGT (corresponding to aspartate 89, E. coli numbering) with alanine leads to complete loss of catalytic activity; furthermore, this mutant is unable to form a covalent complex with substrate tRNA when observed under denaturing PAGE conditions. This prompted the proposal that this active-site aspartate was acting as an enzymic nucleophile in an SN2-type double displacement mechanism (“associative mechanism”) whereby its attack upon the 1′-ribosyl carbon leads to the displacement of the wobble position guanine from substrate tRNA and the formation of a covalent intermediate (Fig. 4).
Fig. 4.

Two potential chemical mechanisms for the TGT reaction involving two aspartates. In the “associative mechanism,” an aspartate acts as a nucleophile to displace guanine. In the “dissociative mechanism,” an aspartate stabilizes an oxocarbenium ion intermediate. In both mechanisms, a general acid (either enzymic or water) serves as a proton donor to facilitate the departure of guanine.
A similar associative mechanism has been reported for other enzymes involved in the cleavage of glycosidic bonds. For example, nucleoside 2′-deoxyribosyltransferase catalyzes the replacement of the base moiety of 2′-deoxynucleosides with a free purine or pyrimidine. This reaction strikingly resembles that which is catalyzed by TGT. It has been shown that catalysis by 2′-deoxyribosyltransferase proceeds via a deoxyribosylated-enzyme intermediate whereby an active-site glutamate residue is covalently linked to the 1′-carbon of deoxyribose [44,45]. Like TGT, detailed kinetic analysis of the 2′-deoxyribosyltransferase reaction has revealed that catalysis proceeds via a ping-pong kinetic mechanism [46]. Similarly, both α- and β-retaining glycosyl hydrolases proceed via an associative mechanism to achieve hydrolysis of glycosidic bonds with retention of configuration. These enzymes utilize the carboxylate moiety of either a glutamate [47–50] or aspartate [51,52] to nucleophilically attack the anomeric carbon, resulting in the formation of a glycosyl-enzyme covalent linkage. Lastly, pseudouridine synthase cleaves the C–N glycosidic bond of uridine during the synthesis of pseudouridine. As will be discussed later in this review, the mechanism for pseudouridine synthase appears to involve a covalent bond between the RNA and the enzyme via an aspartate residue.
Despite compelling experimental evidence supporting an associative mechanism, the results of Romier et al. are also consistent with a dissociative, SN1-type mechanism for TGT catalysis. In this alternative scenario, the displacement of the wobble position guanine would be facilitated through the development of an oxocarbenium ion intermediate. Stabilization of the positively charged intermediate could be achieved by the negatively charged active-site aspartate 89, previously demonstrated to be essential for activity. It is important to note that the observation of a covalent TGT–tRNA complex by Romier et al. [43] was not sufficient to rule out a dissociative-type mechanism for catalysis by TGT. This complex was observed by SDS–PAGE only after incubating the enzyme with a large excess of RNA for a long period of time. It was not impossible that, due to the experimental conditions, the enzyme–RNA complex is the result of the trapping of a long lived oxocarbenium ion by the active-site aspartate. Consistent with this hypothesis is the fact that an increased amount of complex formation is obtained using minihelical RNAs, even though these RNAs are poorer substrates (lower kcat) than their full-length tRNA counterparts [28].
A similar dissociative-type mechanism is utilized by several enzymes involved in the cleavage of C–N glycosidic bonds. For example, nucleosidases are responsible for hydrolyzing nucleosidic bonds. The transition state of inosine nucleosidase is characterized by formation of an oxocarbenium ion character that develops concurrently with the flattening of the ribofuranosyl ring [53]. In this case, the non-bonding electrons of the incoming water are proposed to stabilize the positive charge [53]. Likewise, the DNA repair enzyme uracil DNA glycosylase (UDG) catalyzes the hydrolytic cleavage of the N-glycosidic bond of deoxyuridine in DNA. Kinetic isotope effect experiments have demonstrated that catalysis by UDG proceeds via a dissociative transition state that is characterized by the formation of an oxocarbenium ion [54]. In addition to the flattening of the deoxyribosyl ring, stabilization of the positively charged oxocarbenium ion is thought to be achieved by an active-site aspartate residue [54]. Lastly, the hydrolysis of the nicotinamide-ribosyl bond of NAD+, catalyzed by NAD glycohydrolase, utilizes a dissociative-type mechanism that proceeds through an oxocarbenium ion transition state [55]. It should be noted that the leaving group potential of the nicotinamide pyrimidinium ring is such that activation via protonation would not be expected to be necessary.
In an effort to distinguish between these two differing mechanisms (Fig. 4) and to gain a better understanding of the role of this active-site aspartate, we undertook a more comprehensive mutagenesis study whereby aspartate 89 of the E. coli TGT was mutated to alanine, asparagine, cysteine, and glutamate [56]. While all mutants were able to non-covalently bind tRNA, only the glutamate mutant was able to form a covalent complex with the RNA substrate. Furthermore, only the conservative glutamate mutant displayed significant catalytic activity, with kinetic parameters that were comparable to wild-type. Given these findings, it was reasoned that the TGT active site is considerably tolerant of the positioning of the carboxylate residue at position 89 and that TGT catalysis likely proceeds via an associative chemical mechanism whereby aspartate 89 acts as an enzymic nucleophile, consistent with the proposal of Romier et al.
Another important aspect of TGT catalysis is the requirement for the activation of the leaving group and of the incoming substrate via proton transfer. Specifically, the guanine of substrate tRNAs must be protonated either prior to or concomitant with its displacement. Likewise, the incoming heterocycle (guanine, preQ1, etc.) must be deprotonated prior to its attack upon the 1′-ribosyl carbon. Indeed, structure–activity studies on a series of 5-substituted preQ1 analogs have suggested the importance of deprotonation of N7 (pKa = 15) during the TGT reaction [34].
Enzymes utilize a variety of strategies to achieve proton transfer in catalyzed reactions. For example, the inosine-uridine nucleoside hydrolase from Crithidia fasciculata utilizes an active-site histidine residue to activate the leaving group base through proton donation [57]. Additionally, an active-site calcium ion coordinates the incoming water molecule and activates it, via reduction of pKa, for nucleophilic attack upon the C1′-ribosyl carbon [58]. Alternatively, hypoxanthine–guanine phosphoribosyltransferase utilizes an active-site aspartate as a catalytic acid/base, responsible for protonation/deprotonation of N7 on the purine ring [59]. Likewise, the glycosyl hydrolases also utilize either an aspartic or glutamic acid side-chain to facilitate proton transfer [60–62]. This active-site residue serves the dual function of general acid and general base. As such, it is responsible for the activation of the leaving group and the deprotonation of the incoming water. Typically, the general acid/general base residue in β-retaining glycosyl hydrolases is approximately 5 Å from the active-site nucleophile [60]. Interestingly, the X-ray crystal structure of TGT bound to preQ1 indicates that aspartates 89 and 264 form a roughly equilateral triangle with N7 (corresponding to the purine N9) of preQ1 with sides ca. 6 Å (Fig. 3). Based on this, we speculated that aspartate 264 may serve a role in proton transfer in the TGT-catalyzed reaction.
To explore the significance of aspartate 264 in the TGT chemical mechanism, we mutated this residue to alanine, asparagine, glutamate, glutamine, lysine, and histidine [63]. Biochemical characterization of each these TGT mutants revealed that only the conservative glutamate mutant retained catalytic activity. Furthermore, only the conservative glutamate mutant was capable of forming a TGT–RNA covalent intermediate. Studies designed to probe the covalent intermediate showed that, unlike wild-type TGT, neither excess guanine nor excess preQ1 is capable of eliminating the TGT–RNA covalent complex formed with the glutamate mutant. In fact, only treatment with excess hydroxylamine afforded complete cleavage of the covalent complex. This result is consistent with aspartate 264 playing a role in deprotonating the incoming base. In an effort to better understand the unique biochemical properties of the glutamate mutant at position 264, we solved the crystal structure of the Z. mobilis TGT with the analogous mutation (D280E). A hydrogen-bonding network linking the carboxylate of aspartate 264, the carboxylate of aspartate 89, and the N9 of preQ1 and a bound water molecule was observed, suggesting the possibility that the protonation of the displaced guanine and the deprotonation of the incoming heterocycle is mediated via an active-site water molecule.
As previously mentioned, the X-ray crystal structure of the Z. mobilis TGT bound to tRNA was recently reported by Xie et al. [17], thus providing a better framework with which to interpret biochemical data regarding the TGT mechanism. This structure does indeed confirm that a TGT–RNA covalent complex can form. Additionally, they were able to demonstrate that this covalent complex is chemically competent via the observation that, upon soaking preQ1 into the crystals, the product, preQ1-tRNA is formed. However, contrary to the interpretation of our biochemical studies, it is aspartate 264 that is attached to the 1′ ribosyl carbon (Fig. 5). Based on this new structural data, it appears that aspartate 264, and not the previously proposed aspartate 89, acts as the nucleophile that is responsible for the displacement of wobble position guanine base from substrate tRNA. Interestingly, the positioning of most active-site residues proximal to the preQ1 binding pocket does not change in response to RNA binding. However, this co-crystal structure shows that the position of aspartate 89 is rotated approximately 90° toward the heterocyclic binding pocket. Given this, it is intriguing to speculate that aspartate 89 is appropriately positioned to serve as the general acid/base in the TGT reaction. Yet, such a role cannot be assigned from structural data alone.
Fig. 5.
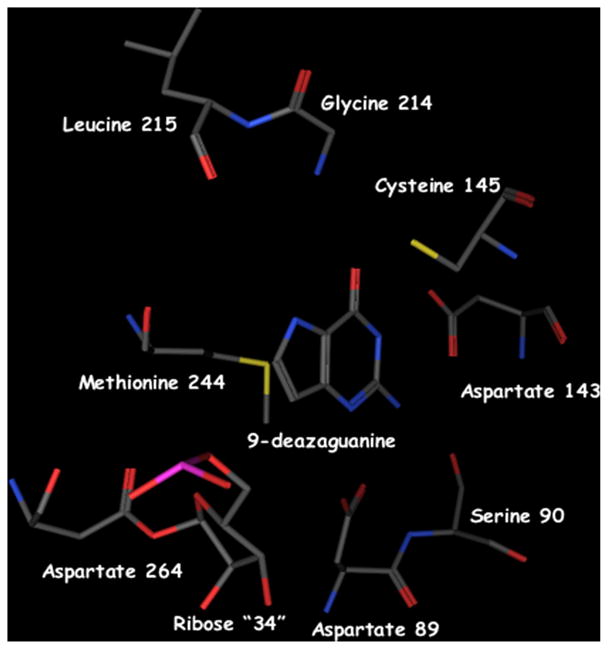
Crystal structure of the covalent complex between the Z. mobilis TGT and RNA. This structure (PDB Accession No. 1Q2R) was generated from crystals of the Z. mobilis TGT complexed with a minihelical RNA and 9-deazaguanine [17]. Aspartate 264 forms a covalent bond with the 1′ position of the ribose corresponding to #34. Guanine 34 has been displaced and 9-deazaguanine is occupying the guanine/preQ1 site.
Our biochemical data indicate that there is a strict requirement for a carboxylate residue at both positions 89 and 264 in the TGT active site. In each case, only the aspartate to glutamate substitution yielded an enzyme with catalytic activity, suggesting that the enzyme is able to tolerate a larger side-chain at either position. Consistent with this activity, only the glutamate substitution at either position was able to form a covalent TGT–RNA complex as judged by SDS–PAGE. Based on the ping-pong kinetic mechanism for TGT catalysis, whereby it is known that tRNA is the first substrate to bind to the enzyme, followed immediately by guanine displacement, it can be deduced that two active-site carboxylate/carboxylic acids are necessary for formation of the TGT–tRNA intermediate. This highlights the importance of both the nucleophilic carboxylate and the second, carboxylic group (presumably acting as a general acid) in the formation of the covalent intermediate.
Kinetic analysis of the glutamate substitutions at each position reveals subtle differences between the two residues. Both enzymes displayed Km values for tRNA that were similar to that of wild-type TGT. The most obvious difference between the two enzymes is found in the Km value of guanine. The guanine Km for the D89E mutant is 10-fold greater than that of the D264E mutant, and approximately 25-fold greater than that for wild-type TGT. This was rationalized to be due to steric interactions between the larger side-chain at position 89 and the ribose of the wobble nucleoside, indirectly altering the conformation of the heterocyclic binding pocket. Within the context of the newly solved TGT–RNA co-crystal structure, this discrepancy in the guanine Km between the two mutant TGTs seems more likely to be due to the larger side-chain (D89E) directly affecting access to the heterocyclic binding pocket.
Data regarding the role of aspartate 264 is less straightforward to understand. This mutant displayed Km values for both substrates which were approximately 3- to 4-fold higher than those of wild-type TGT. The kcat was depressed by an order of magnitude, presumably a direct result of the sub-optimal orientation of the carboxylate in the active site. The crystal structure of the corresponding mutation in the Z. mobilis TGT suggested that this residue, with the aid of an intermediary water molecule, formed a hydrogen bond with N7 of preQ1. This suggested that this residue might function in a proton transfer with the heterocyclic base. This role was consistent with experiments in which we investigated the collapse of the covalent intermediate by exogenous nucleophiles. Unlike what was observed with the wild-type TGT, as well as the D89E mutant, the covalent intermediate formed by D264E and RNA could not be completely eliminated by excess preQ1over a long time course. This result was consistent with the crystal structure and implicated aspartate 264 being at least partially responsible for deprotonation of the incoming preQ1. However, it is inconsistent with the recently reported TGT–RNA co-crystal structure. It is possible that the additional methylene in the D264E mutant could shift the placement of the ribose such that it is more difficult for preQ1 to bind and attack the 1′ carbon of the ribosyl moiety.
Most recently, we have kinetically characterized the wild-type TGT, as well as the two glutamate mutants at positions 89 and 264, with the alternative RNA substrate dG34-ECYMH (Kittendorf and Garcia, unpublished). This RNA contains a 2′-deoxyguanosine at the wobble position (position of TGT catalysis) and has been previously reported to be a substrate in the TGT reaction [30]. Interestingly, the Km value for this substrate is approximately 3- to 5-fold lower for the D264E mutant than for wild-type or the D89E mutant, respectively. The most logical explanation for this is that the larger glutamate side-chain is better accommodated by an RNA substrate that lacks the 2′ hydroxyl. Such a scenario is consistent with aspartate 264 interacting with the ribose of the RNA substrate and acting as the enzymic nucleophile.
The recently reported crystal structure of TGT bound with RNA has provided new information regarding the molecular details of the TGT reaction. This has prompted us to re-examine our previously proposed chemical mechanism for TGT. While it should be recognized that a structure cannot provide definitive proof of an enzyme mechanism, it is apparent that aspartate 264 can form a covalent linkage with the ribosyl moiety of the wobble position guanosine of tRNA. This strongly suggests that, contrary to our previously reported conclusions [56,63], aspartate 264 functions as an enzymic nucleophile in the TGT-catalyzed reaction. While a role for aspartate 89 cannot be assigned based on this structure alone, it is positioned in a logical orientation to play a role in protonation of the displaced guanine and/or deprotonation of the incoming preQ1 base (possibly via an intermediary water molecule). Such a role is consistent with our biochemical data and our conclusion that this residue serves a critical role in the formation of the TGT–tRNA intermediate.
The newly solved crystal structure, combined with our previously reported biochemical data, serves as the basis for a revised model for TGT catalysis (Fig. 6). Upon binding of substrate tRNA, aspartate 264 attacks the 1′ ribosyl carbon of guanosine34, resulting in the displacement of guanine base and the formation of a TGT–tRNA covalent intermediate. The displaced guanine is most likely protonated by an enzymic general acid (presumably aspartate 89) either prior to or concomitant with attack of the ribose by aspartate 264. Upon its diffusion from the active site, the incoming preQ1 base is able to bind in the active site, where it is deprotonated by a general base (perhaps aspartate 89 with the aid of water) prior to its nucleophilic attack to the TGT–tRNA covalent intermediate.
Fig. 6.
Chemical mechanism for the TGT reaction that is consistent with biochemical and structural data. Two active-site aspartate residues serve critical roles in catalysis by TGT. Aspartate 264 nucleophilically attacks the 1′-ribosyl carbon, resulting in a TGT–RNA covalent linkage and the displacement of the guanine base. It is hypothesized that aspartate 89 might be responsible for protonation of the guanine base and/or deprotonation of the incoming preQ1 (perhaps through the intermediacy of water). While guanine is shown being protonated at N7, protonation could be directly at N9.
3. Pseudouridine synthase
Pseudouridine (5-β-D-ribofuranosyluracil, Ψ) is the most abundant modified nucleoside found in RNA. To date, pseudouridine has been located in transfer RNA [64], ribosomal RNA [65], and nuclear RNA [66]. Naturally occurring C-nucleosides are not uncommon, however, pseudouridine and its 1- and 2′-substituted analogs are the only C-nucleosides that have been identified in RNA [67]. The biosynthesis of pseudouridine is carried out via post-transcriptional modification of the substrate RNAs by a class of enzymes called pseudouridine synthases (E.C. 5.4.99.12). The pseudouridine synthase reaction (Fig. 7) has been widely referred to as an “isomerization.” While this is not incorrect, the reaction can also be viewed as a transglycosylation where the glycosidic bond to N1 of the uracil is broken and a new glycosidic bond is formed to C5.
Fig. 7.
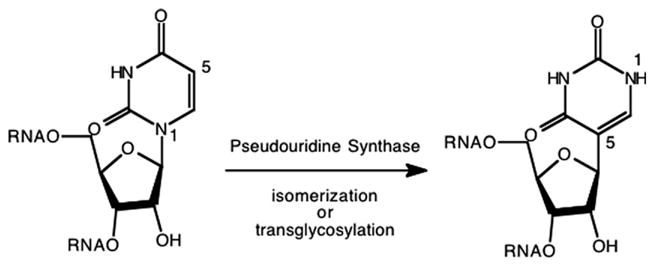
Pseudouridine synthase reaction.
To date, five families of pseudouridine synthases have been isolated [68–73]. Two families of enzymes appear to catalyze formation of pseudouridine in tRNA, while the others appear to be responsible for pseudouridine formation in rRNA. However, dual-specificity pseudouridine synthases, acting upon rRNA and tRNA in E. coli [70], snRNA and tRNA in yeast [74], and mitochondrial and cytoplasmic tRNAs in yeast [75] have been identified. The pseudouridine synthases that act upon tRNA appear to recognize their substrates in a more classical fashion of a specific enzyme recognizing a specific substrate. Consequently, there are different pseudouridine synthases in these families that are responsible for pseudouridines at different, specific positions within tRNAs. The dual-specificity pseudouridine synthases are an exception to this “rule,” although it is likely that the recognition motifs in the tRNA are reproduced in the rRNA or snRNA. In contrast, it has recently been shown that the pseudouridine synthases that act upon ribosomal RNAs utilize guide RNAs to direct the formation of pseudouridine at the 50–100 positions within rRNAs that contain pseudouridine [76,77]. Other aspects of pseudouridine chemistry, biochemistry, and physiology have been recently reviewed [78]. Here, we will focus on the molecular mechanism of pseudouridine formation.
The E. coli pseudouridine synthase I (PSU-I) has been shown to bind both substrate and non-substrate tRNA [68]. Although no exogenous ions are necessary for binding, a monovalent cation (preferably or K+) is required for catalysis [79]. Furthermore, no cofactors or energy sources are required [68]. PSU-I was also analyzed for the ability to catalyze the exchange of exogenous uracil into wild-type and hisT tRNA, as well as the ability to catalyze 3H release from uracil, uridine, and UTP [68]. No activity was detected in any of these studies supporting direct isomerization of the RNA-incorporated uridine and a requirement for at least some portion of the tRNA structure. Therefore, based on the known information concerning the E. coli PSU-I, a minimal mechanism for pseudouridine formation must involve four steps: (a) binding of substrate tRNA to the enzyme; (b) cleavage of the glycosidic bond; (c) a 180° rotation of the base with respect to the ribose; and (d) reformation of the glycosidic bond through a carbon–carbon linkage. Consequently, the pseudouridine synthase reaction can be thought of as an intramolecular transglycosylation.
Mullenbach et al. observed that treatment of PSU-I with thiol reactive reagents (e.g., p-chloromercuribenzoate and iodoacetic acid) completely inactivates the E. coli PSU-I [68,79]. This observation suggested the involvement of one or more cysteine residues in the pseudouridine synthase reaction. This apparent role of one or more cysteine residues, as well as the potent inhibitory action of 5-fluorouridine-containing tRNA [80], led Kammen et al. [68] to propose a conceptual model for uridine isomerization based on the mechanisms of known pyrimidine C5-modifying enzymes such as thymidylate synthase [81] and RUMT [82]. Kammen et al. suggested that the initiating step is attack at C6 by a nucleophilic group, possibly one of the cysteine residues, and proceeding though a transient dihydropyrimidine-enzyme adduct (Fig. 8). The glycosidic bond is then broken in a unimolecular decomposition yielding an oxocarbenium ion intermediate. This intermediate is then trapped by the C5 of the uridine-enzyme adduct, forming pseudouridine after elimination of the enzymic nucleophile (presumably a cysteine). Support for this mechanism comes from the report that a 6-hydroxy-5,6-dihydrouridine intermediate is kinetically competent in the acid-catalyzed hydrolysis of uridine to uracil [83].
Fig. 8.
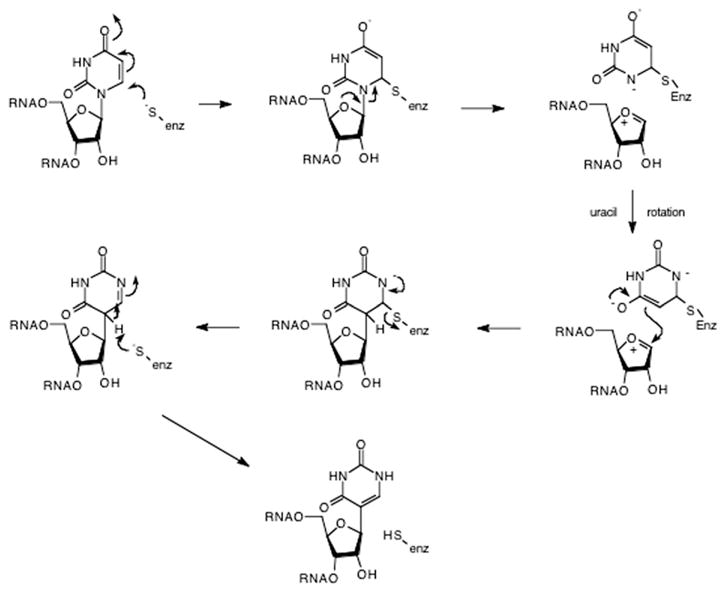
Postulated pseudouridine mechanism involving a cysteine nucelophile. The cysteine thiolate attacks C6 forming a dihydrouridine-like complex. Scission of the glycosidic bond yields an oxocarbenium ion intermediate. Attack of C5 upon the 1′ position following rotation of the uracil reforms the glycosidic bond. Elimination of the enzymic thiolate, followed by tautomerization yields pseudouridine.
However, mutagenesis studies of the three cysteine residues found in E. coli PSU-I rule out the possibility of a mechanism involving a cysteine nucleophile [84]. Studies involving the mutants C154A and C169A, as well as a triple mutant, in which all three cysteine residues were converted to alanine, revealed that all three mutants exhibited less than 10% reduction in activity compared to the wild-type enzyme [84]. Similar results (e.g., unaltered activities upon mutagenesis of conserved cysteines) were reported for members of two other families of pseudouridine synthases represented by RluA and TruA by Mueller and co-workers [85]. Based on these observations, one can conclude that the pseudouridine synthase reaction does not involve a covalent cysteine intermediate, nor are any cysteine residues necessary for catalysis. In fact, only a 2-fold decrease in Vmax/Km was observed for the alanine triple mutant of PSU-I [84]. It has been demonstrated that the interaction of thiol reactive reagents with cysteine residues located in or near an enzyme active site can inhibit catalysis, despite the lack of a direct catalytic role for the cysteine [38,86]. It is possible that this accounts for the loss of activity observed by Mullenbach et al. for PSU-I. These results do not rule out a nucleophilic mechanism for the pseudouridine synthase reaction, rather they only eliminate the possibility of cysteine acting as the nucleophile.
While there is very little sequence homology across the four families of pseudouridine synthases, Santi and co-workers [87] were able to identify a single, conserved aspartic acid (D60 in PSU-I). The conservation of this aspartate was confirmed by the comparison of subsequent crystal structures of representatives of three of the five pseudouridine synthase families (reviewed by Mueller [88]). Mutation of this aspartate to a number of different amino acids (A, S, E, N, and K) yielded inactive enzymes that retained the ability to bind tRNA. The authors observed that 5-fluorouridine-containing tRNA caused a time-dependent inactivation of PSU-I and formed a covalent complex with the enzyme. Interestingly, none of the D60 mutants were able to form this covalent complex with 5-fluorouridine-containing tRNA. Mutagenesis experiments on the corresponding aspartic acid in representatives of pseudouridine synthases from two other families by Mueller and co-workers support the critical nature of the conserved aspartate [89]. These results prompted the postulation of two potential mechanisms for the pseudouridine synthase reaction (Fig. 9). The first mechanism (Fig. 9A) is similar to that previously postulated for pseudouridine synthase except that aspartate 60 is the nucleophile that attacks C6 of the uridine. The second mechanism (Fig. 9B) involves aspartate 60 attacking the 1′ position of the uridine ribose, displacing a uracil anion and forming a covalent intermediate virtually identical to the intermediate in the tRNA-guanine transglycosylase reaction. Displacement of aspartate 60 by C5 of the uracil (after uracil rotation) followed by tautomerization yields the products. Further characterization of the product of the reaction of 5-fluorouridine-containing tRNA with PSU-I indicated that the 5-fluorouridine was converted to 5,6-dihydro-6-hydroxy-5-fluorouridine [90]. This was reasoned to be indicative of the intermediacy of a covalent bond between aspartate 60 and C6 of 5-fluorouridine tRNA. The authors concluded that the ester of this bond must have been hydrolyzed to give the observed product.
Fig. 9.

Postulated pseudouridine mechanisms involving an aspartic acid nucleophile. (A) “C6 Attack” or “Michael addition” mechanism. (B) “1′ Attack” or “Acylal” mechanism.
Even stronger evidence supporting the C6-addition mechanism came from a co-crystal structure of the E. coli TruB pseudouridine synthase with 5-fluorouridine-containing RNA minihelix [91]. This structure shows that the RNA is non-covalently bound to the enzyme and that the 5-fluorouracil has formed a C5–C1′ covalent bond. Further, C5 retains the fluorine and a hydroxyl group is found at C6. The carboxyl group of aspartate 48 (corresponding to aspartate 60 of PSU-I) was found directly adjacent to the hydroxyl at C6, but not covalently bound. This processing of 5-fluorouracil to the hydrated C-nucleoside was also reported by Spedaliere and Mueller where they found that not all pseudouridine synthases are inhibited by 5-fluorouridine-containing tRNA [92]. To further investigate the pseudouridine synthase mechanism, Mueller and co-workers [93] carried out the TruB reaction with 5-fluorouridine-containing tRNA in the presence of 50% O18 water. Surprisingly, they found that the O18 did not end up in the aspartate, but rather was found in the hydroxylated 5-fluorouracil. They concluded that “…hydrolysis of an ester adduct with Asp-48 of TruB does not give rise to the observed product of f5U…” and that “The hydration product of f5U cannot, therefore, be construed to support the Michael (C6 addition) mechanism.” They proposed that the reaction could proceed through an “acylal” mechanism (Fig. 9B) in which the aspartate displaces uracil (as the monoanion) from the 1′ position. The uracil rotates and displaces the aspartate forming the C-nucleoside. The aspartate can then deprotonate the C5 position leading to the pseudouridine product. In the case of 5-fluorouridine, the freed aspartate cannot deprotonate C5, and therefore is proposed to activate a water molecule to attack C6 forming the hydrated product. An alternative mechanism proposed by Spedaliere and Mueller is that the C-nucleoside could be released from the enzyme and be hydrated in solution; however, this is not consistent with the observation of the hydrated product bound to the enzyme in the crystal structure (unless the product re-bound to the free enzyme and then crystallized).
While there is no detectable overall sequence homology between pseudouridine synthases and tRNA-guanine transglycosylases (Garcia, unpublished), the consensus sequences around the catalytically critical aspartates are not entirely dissimilar (Fig. 10). Additionally, the yeast pseudouridine synthase I has recently been shown to contain a structural zinc ion [94], reminiscent of tRNA-guanine transglycosylase [19,37,95].
Fig. 10.
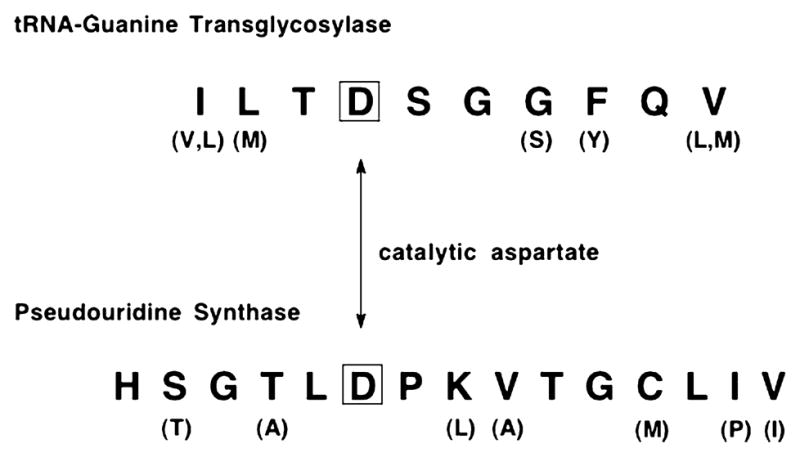
Consensus sequences around catalytically important aspartic acids in tRNA-guanine transglycosylase and pseudouridine synthase. For those residues that vary somewhat, the variant amino acids are shown in parentheses below the consensus amino acid in the figure.
4. RNA editing
In the early to mid 1980s, it was discovered that RNA sequences could contain nucleosides that are different from those encoded in the genome [96]. These differences come about via a process subsequently termed “RNA editing” [97]. In the ensuing years, a large number of RNA editing events have been identified in different types of RNAs, from various organelles, spanning the three kingdoms of life (including some viral RNAs). The field of RNA editing has been the subject of numerous reviews [97–103]. There appear to be two general, overarching classes of RNA editing events: “insertion/deletion” where nucleosides that are not present in the DNA gene are either inserted into or removed from the RNA (excluding splicing) and “substitution” where a nucleoside that is contained in the DNA gene is converted into another canonical nucleoside (or one that is functionally equivalent to one such as inosine/guanosine). The insertion process utilizes guide RNAs to serve as templates for and to direct the specific insertion of the nucleosides via a scission/ligation activity. The substitution editing events involve the conversion of one nucleoside to another. This process can be thought of as a special case of RNA modification in which the “modified” base product is simply another canonical base (or one that functions as a canonical base, e.g., inosine acting as guanosine).
The vast majority of the known substitution editing events consist of “C to U” or “A to I” conversions. (Note that the “A to I” conversion is functionally equivalent to an “A to G” conversion as inosine base pairs in a similar fashion to guanosine.) Much work from many laboratories has shown that these conversions arise via deamination mechanisms, catalyzed by enzymes similar to adenosine and cytidine deaminases (Fig. 11) [99,102]. One other substitution editing event is the “U to C” conversion [102]. While the theory of microscopic reversibility dictates that the cytidine deaminase reaction could “go backward” to generate this conversion, it has also been proposed that a CTP synthetase-type activity could be responsible [98].
Fig. 11.
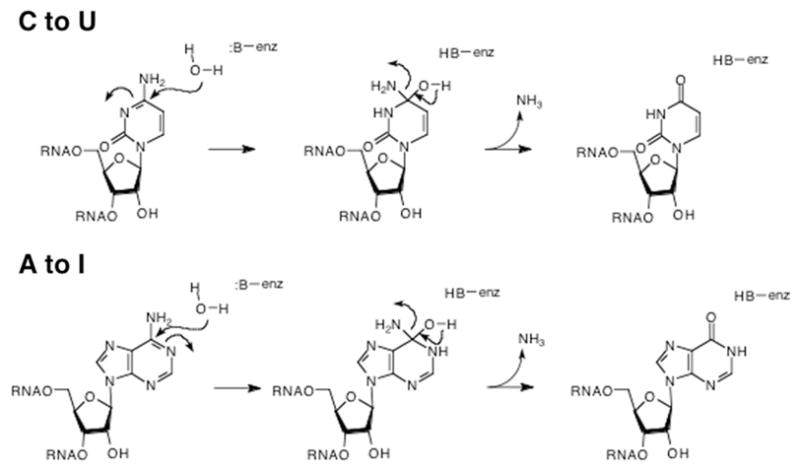
Deaminase mechanisms for C to U and A to I editing. An enzymically activated water molecule attacks the base in an addition–elimination mechanism which ultimately replaces the amine with an oxygen (carbonyl).
In stark contrast to the above RNA editing events, a number of conversions involving pyrimidine to purine transitions have been reported [104–109]. In these cases, it is extremely unlikely that a simple modification of the existing nucleoside could be responsible. In the case of the α-galactosidase mRNA U to A conversion [104], the authors state that “The base change described here cannot be accounted for by deamination reactions such as those implicated in other instances of mammalian nuclear RNA editing. Rather, it must involve some form of base replacement in the RNA through as yet unknown mechanisms.” Interestingly, a G to U editing event has been reported also in the human α-galactosidase mRNA [109]. This conversion occurs in the first position (g + 1) of intron 6 and causes a mis-splicing that deletes exon 6 from the mRNA leading to Fabry disease.
Two mechanisms have been proposed for non-deamination-type base conversions [102]. The first involves nucleoside replacement, reminiscent of the insertion editing process, where the nucleoside is removed prior to the new nucleoside being inserted. Presumably, this would occur through some scission/insertion/ligation process (Fig. 12A). In a number of these cases [105–107], the editing events correct mismatches in the acceptor stem of a tRNA and serve to restore correct base pairing in the stem. Thus, the complementary strand of the stem could serve as the template for this process. Alternatively, a second proposed mechanism is that of transglycosylation (Fig. 12B).
Fig. 12.
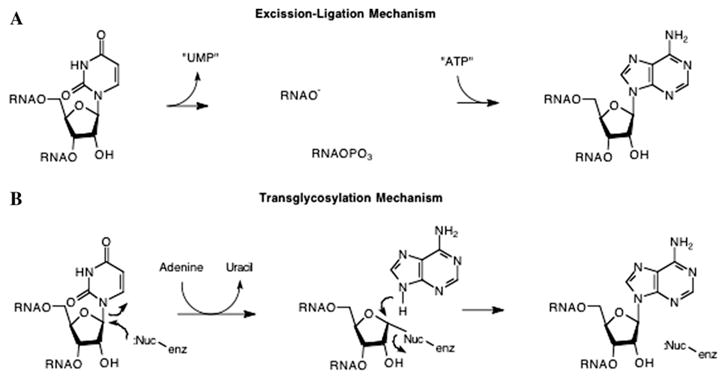
Postulated mechanisms for pyrimidine to purine editing. Mechanisms are shown for the U to A conversion. Similar mechanisms are possible for U to G and C to A conversions. (A) “Excision–Ligation” mechanism, where the elements of UMP are excised from the oligonucleotide followed by the insertion and ligation of the elements of AMP. (B) “Transglycosylation” mechanism, where, similar to tRNA-guanine transglycosylase, an enzymic nucleophile displaces uracil, followed by adenine displacement of the enzymic nucleophile.
It is known from studies in our lab and others’ [22,25,27–30,110] that the RNA recognition properties of eubacterial tRNA-guanine transglycosylase are limited to a U-G-U sequence in a stem–loop structure. This sort of recognition motif is likely to occur in many RNAs. While it remains to be shown, it is quite possible that TGT or a similarly evolved transglycosylase could recognize and “modify” other RNA species than tRNAs. One complicating factor in this hypothesis is the fact that TGT exhibits an exquisite recognition of the aminopyrimidone motif that is shared by guanine and queuine. An enzyme that would catalyze a pyrimidine to purine transglycosylation would have to be able to recognize both the “leaving” pyrimidine and the “incoming” purine with high fidelity. Detailed experiments utilizing RNAs radiolabeled in both the ribose and base, such as those described by Niswender [102], could be used to discriminate between these potential mechanisms.
5. Summary
While certainly not novel in enzymology, transglycosylation is a rarely described mechanism for RNA modification. Transglycosylation is an extremely efficient process for incorporating highly modified bases such as queuine and archaeosine into RNA. In these cases, it is difficult to conceive of a series of enzymatic steps that would convert a purine to a pyrrolopyrimidine, while maintaining the integrity of the RNA backbone in an efficient fashion. Transglycosylation is also a requisite process for “isomerizing” an N-nucleoside into a C-nucleoside as is the case for pseudouridine formation. Finally, transglycosylation is an attractive possibility for certain RNA editing events (e.g., pyrimidine to purine conversions) that cannot occur via the known, more straightforward enzymatic reactions (e.g., deaminations).
Acknowledgments
Work in the Garcia lab has been supported by grants from the National Institutes of Health (GM45968, GM065489), the National Science Foundation (MCB-9720139), and the University of Michigan, College of Pharmacy Vahlteich and Up-John Research Funds. J.D.K. was a Pharmaceutical Sciences Training Program Trainee (NIH GM07767) and received support from the American Foundation for Pharmaceutical Education and the University of Michigan, Horace H. Rackham School of Graduate Studies.
References
- 1.Crain PF, McCloskey JA. Nucleic Acids Res. 1996;24:98–99. doi: 10.1093/nar/24.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Söll D. Science (Washington, DC) 1971;173:293–298. doi: 10.1126/science.173.3994.293. [DOI] [PubMed] [Google Scholar]
- 3.Kasai H, Ohashi Z, Harada F, Nishimura S, Oppenheimer NJ, Crain PF, Liehr JG, vonMinden DL, McCloskey JA. Biochemistry. 1975;14:4198. doi: 10.1021/bi00690a008. [DOI] [PubMed] [Google Scholar]
- 4.Gregson JM, Crain PF, Edmonds CG, Gupta R, Hashizume T, Phillipson DW, McCloskey JA. J Biol Chem. 1993;268:10076–10086. [PubMed] [Google Scholar]
- 5.Garcia GA, Goodenough-Lashua DM. In: Modification and Editing of RNA. Grosjean H, Benne R, editors. ASM Press; Washington, DC: 1998. pp. 135–168. [Google Scholar]
- 6.Okada N, Noguchi S, Kasai H, Shindo-Okada N, Ohgi T, Goto T, Nishimura S. J Biol Chem. 1979;254:3067–3073. [PubMed] [Google Scholar]
- 7.Okada N, Nishimura S. J Biol Chem. 1979;254:3061–3066. [PubMed] [Google Scholar]
- 8.Singhal RP. Prog Nucleic Acid Res Mol Biol. 1983;28:75–80. doi: 10.1016/s0079-6603(08)60083-5. [DOI] [PubMed] [Google Scholar]
- 9.Walden TM, Reyniers JP, Hiatt V, Farkas WR. Proc Soc Exp Biol Med. 1982;170:328. doi: 10.3181/00379727-170-41438. [DOI] [PubMed] [Google Scholar]
- 10.Harada F, Nishimura S. Biochemistry. 1972;11:301–308. doi: 10.1021/bi00752a024. [DOI] [PubMed] [Google Scholar]
- 11.Tsang TH, Buck M, Ames BN. Biochim Biophys Acta. 1983;741:180–196. doi: 10.1016/0167-4781(83)90058-1. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe M, Matsuo M, Tanaka S, Akimoto H, Asahi S, Nishimura S, Katze JR, Hashizume T, Crain PF, McCloskey JA, Okada N. J Biol Chem. 1997;272:20146–20151. doi: 10.1074/jbc.272.32.20146. [DOI] [PubMed] [Google Scholar]
- 13.Howes NK, Farkas WR. J Biol Chem. 1978;253:9082–9087. [PubMed] [Google Scholar]
- 14.Slany RK, Mueller SO. Eur J Biochem. 1995;230:221–228. doi: 10.1111/j.1432-1033.1995.0221i.x. [DOI] [PubMed] [Google Scholar]
- 15.Garcia GA, Koch KA, Chong S. J Mol Biol. 1993;231:489–497. doi: 10.1006/jmbi.1993.1296. [DOI] [PubMed] [Google Scholar]
- 16.Reuter K, Ficner R. J Bacteriol. 1995;177:5284–5288. doi: 10.1128/jb.177.18.5284-5288.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie W, Liu XJ, Huang RH. Nat Struct Biol. 2003;10:781–788. doi: 10.1038/nsb976. [DOI] [PubMed] [Google Scholar]
- 18.Romier C, Ficner R, Reuter K, Suck D. Protein: Struct Funct Genet. 1996;24:516–519. doi: 10.1002/(SICI)1097-0134(199604)24:4<516::AID-PROT11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 19.Romier C, Reuter K, Suck D, Ficner R. EMBO J. 1996;15:2850–2857. [PMC free article] [PubMed] [Google Scholar]
- 20.Ishitani R, Nureki O, Kijimoto T, Watanabe M, Kondo H, Nameki N, Okada N, Nishimura S, Yokoyama S. Acta Crystallogr Sect D Biol Crystallogr. 2000;57:1659–1662. doi: 10.1107/s0907444901011994. [DOI] [PubMed] [Google Scholar]
- 21.Ishitani R, Nureki O, Fukai S, Kijimoto T, Nameki N, Watanabe M, Kondo H, Sekine M, Okada N, Nishimura S, Yokoyama S. J Mol Biol. 2002;318:665–677. doi: 10.1016/S0022-2836(02)00090-6. [DOI] [PubMed] [Google Scholar]
- 22.Curnow AW, Garcia GA. Biochimie. 1994;76:1183–1191. doi: 10.1016/0300-9084(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 23.Reuter K, Chong S, Ullrich F, Kersten H, Garcia GA. Biochemistry. 1994;33:7041–7046. doi: 10.1021/bi00189a004. [DOI] [PubMed] [Google Scholar]
- 24.Goodenough-Lashua DM, Garcia GA. Bioorg Chem. 2003;31:331–344. doi: 10.1016/S0045-2068(03)00069-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curnow AW, Kung FL, Koch KA, Garcia GA. Biochemistry. 1993;32:5239–5246. doi: 10.1021/bi00070a036. [DOI] [PubMed] [Google Scholar]
- 26.Curnow AW, Garcia GA. J Biol Chem. 1995;270:17264–17267. doi: 10.1074/jbc.270.29.17264. [DOI] [PubMed] [Google Scholar]
- 27.Kung FL, Garcia GA. FEBS Lett. 1998;431:427–432. doi: 10.1016/s0014-5793(98)00801-1. [DOI] [PubMed] [Google Scholar]
- 28.Kung FL, Nonekowski S, Garcia GA. RNA. 2000;6:233–244. doi: 10.1017/s135583820099191x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nonekowski ST, Garcia GA. RNA. 2001;7:1432–1441. [PMC free article] [PubMed] [Google Scholar]
- 30.Nonekowski ST, Kung FL, Garcia GA. J Biol Chem. 2002;277:7178–7182. doi: 10.1074/jbc.M111077200. [DOI] [PubMed] [Google Scholar]
- 31.Grosjean H, Edqvist J, Straby K. Arch Int Physiol Biochim Biophys. 1993;101:B13. [Google Scholar]
- 32.Grosjean H, Edqvist J, Straby KB, Giege R. J Mol Biol. 1996;255:67–85. doi: 10.1006/jmbi.1996.0007. [DOI] [PubMed] [Google Scholar]
- 33.Haumont E, Droogmans L, Grosjean H. Eur J Biochem. 1987;168:219. doi: 10.1111/j.1432-1033.1987.tb13408.x. [DOI] [PubMed] [Google Scholar]
- 34.Hoops GC, Townsend LB, Garcia GA. Biochemistry. 1995;34:15381–15387. doi: 10.1021/bi00046a047. [DOI] [PubMed] [Google Scholar]
- 35.Hoops GC, Townsend LB, Garcia GA. Biochemistry. 1995;34:15539–15544. doi: 10.1021/bi00047a020. [DOI] [PubMed] [Google Scholar]
- 36.Hoops GC, Park J, Garcia GA, Townsend LB. J Heterocyclic Chem. 1996;33:767–781. [Google Scholar]
- 37.Chong S, Curnow AW, Huston TJ, Garcia GA. Biochemistry. 1995;34:3694–3701. doi: 10.1021/bi00011a026. [DOI] [PubMed] [Google Scholar]
- 38.Garcia GA, Chong SR. J Protein Chem. 1997;16:11–17. doi: 10.1023/a:1026334726357. [DOI] [PubMed] [Google Scholar]
- 39.Noguchi S, Nishimura Y, Hirota Y, Nishimura S. J Biol Chem. 1982;257:6544–6550. [PubMed] [Google Scholar]
- 40.Grädler U, Ficner R, Garcia GA, Stubbs MT, Klebe G, Reuter K. FEBS Lett. 1999;454:142–146. doi: 10.1016/s0014-5793(99)00793-0. [DOI] [PubMed] [Google Scholar]
- 41.Romier C, Meyer JEW, Suck D. FEBS Lett. 1997;416:93–98. doi: 10.1016/s0014-5793(97)01175-7. [DOI] [PubMed] [Google Scholar]
- 42.Todorov KW. Medicinal Chemistry. University of Michigan; Ann Arbor: 2003. [Google Scholar]
- 43.Romier C, Reuter K, Suck D, Ficner R. Biochemistry. 1996;35:15734–15739. doi: 10.1021/bi962003n. [DOI] [PubMed] [Google Scholar]
- 44.Porter DJT, Merrill BM, Short SA. J Biol Chem. 1995;270:15551–15556. doi: 10.1074/jbc.270.26.15551. [DOI] [PubMed] [Google Scholar]
- 45.Porter DJT, Short SA. J Biol Chem. 1995;270:15557–15562. doi: 10.1074/jbc.270.26.15557. [DOI] [PubMed] [Google Scholar]
- 46.Danzin C, Cardinaud R. Eur J Biochem. 1974;48:255–262. doi: 10.1111/j.1432-1033.1974.tb03763.x. [DOI] [PubMed] [Google Scholar]
- 47.Miao S, Ziser L, Aebersold R, Withers SG. Biochemistry. 1994;33:7029–7032. doi: 10.1021/bi00189a002. [DOI] [PubMed] [Google Scholar]
- 48.Vocadlo DJ, MacKenzie LF, He S, Zeikus GJ, Withers SG. Biochem J. 1998;335:449–455. doi: 10.1042/bj3350449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong AW, He S, Grubb JH, Sly WS, Withers SG. J Biol Chem. 1998;273:34057–34062. doi: 10.1074/jbc.273.51.34057. [DOI] [PubMed] [Google Scholar]
- 50.Zechel DL, He S, Dupont C, Withers SG. Biochem J. 1998;336:139–145. doi: 10.1042/bj3360139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vocadlo DJ, Mayer C, He SM, Withers SG. Biochemistry. 2000;39:117–126. doi: 10.1021/bi991958d. [DOI] [PubMed] [Google Scholar]
- 52.McCarter JD, Withers SG. J Biol Chem. 1996;271:6889–6894. doi: 10.1074/jbc.271.12.6889. [DOI] [PubMed] [Google Scholar]
- 53.Horenstein BA, Parkin DW, Estupinan B, Schramm VL. Biochemistry. 1991;30:10788–10795. doi: 10.1021/bi00108a026. [DOI] [PubMed] [Google Scholar]
- 54.Werner RM, Stivers JT. Biochemistry. 2000;39:14054–14064. doi: 10.1021/bi0018178. [DOI] [PubMed] [Google Scholar]
- 55.Oppenheimer NJ. Mol Cell Biochem. 1994;138:245–251. doi: 10.1007/BF00928468. [DOI] [PubMed] [Google Scholar]
- 56.Kittendorf JD, Barcomb LM, Nonekowski ST, Garcia GA. Biochemistry. 2001;40:14123–14133. doi: 10.1021/bi0110589. [DOI] [PubMed] [Google Scholar]
- 57.Gopaul DN, Meyer SL, Degano M, Sacchettini JC, Schramm VL. Biochemistry. 1996;35:5963–5970. doi: 10.1021/bi952998u. [DOI] [PubMed] [Google Scholar]
- 58.Degano M, Almo SC, Sacchettini JC, Schramm VL. Biochemistry. 1998;37:6277–6285. doi: 10.1021/bi973012e. [DOI] [PubMed] [Google Scholar]
- 59.Xu Y, Grubmeyer C. Biochemistry. 1998;37:4114–4124. doi: 10.1021/bi972519m. [DOI] [PubMed] [Google Scholar]
- 60.Zechel DL, Withers SG. Acc Chem Res. 2000;33:11–18. doi: 10.1021/ar970172+. [DOI] [PubMed] [Google Scholar]
- 61.MacLeod AM, Lindhorst T, Withers SG, Warren RA. Biochemistry. 1994;33:6371–6376. doi: 10.1021/bi00186a042. [DOI] [PubMed] [Google Scholar]
- 62.Wang Q, Trimbur D, Graham R, Warren RA, Withers SG. Biochemistry. 1995;34:14554–14562. doi: 10.1021/bi00044a034. [DOI] [PubMed] [Google Scholar]
- 63.Kittendorf JD, Sgraja T, Reuter K, Klebe G, Garcia GA. J Biol Chem. 2003;278:42369–42376. doi: 10.1074/jbc.M304323200. [DOI] [PubMed] [Google Scholar]
- 64.Singer CE, Smith GR, Cortese R, Ames BN. Nat New Biol. 1972;238:72–74. doi: 10.1038/newbio238072a0. [DOI] [PubMed] [Google Scholar]
- 65.Erdmann VA, Huysmans E, Vandenbergh A, DeWachter R. Nucleic Acids Res. 1983;11:r105–r133. [PMC free article] [PubMed] [Google Scholar]
- 66.Shibita H, Ro-Choi TS, Reddy P, Choi YC, Henning D, Busch H. J Biol Chem. 1975;250:3909–3920. [PubMed] [Google Scholar]
- 67.Limbach PA, Crain PF, McCloskey JA. Nucleic Acids Res. 1994;22:2183–2196. doi: 10.1093/nar/22.12.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kammen HO, Marvel CC, Hardy L, Penhoet EE. J Biol Chem. 1988;263:2255–2263. [PubMed] [Google Scholar]
- 69.Nurse K, Wrzesinski J, Bakin A, Lane BG, Ofengand J. RNA. 1995;1:102–112. [PMC free article] [PubMed] [Google Scholar]
- 70.Wrzesinski J, Nurse K, Bakin A, Lane BG, Ofengand J. RNA. 1995;1:437–448. [PMC free article] [PubMed] [Google Scholar]
- 71.Wrzesinski J, Nurse K, Bakin A, Lane BG, Ofengand J. Biochemistry. 1995;34:8904–8913. doi: 10.1021/bi00027a043. [DOI] [PubMed] [Google Scholar]
- 72.Kaya Y, Ofengand J. RNA. 2003;9:711–721. doi: 10.1261/rna.5230603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koonin EV. Nucleic Acids Res. 1996;24:2411–2415. doi: 10.1093/nar/24.12.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Massenet S, Motorin Y, Lafontaine DLJ, Hurt EC, Grosjean H, Branlant C. Mol Cell Biol. 1999;19:2142–2154. doi: 10.1128/mcb.19.3.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Becker HF, Motorin Y, Planta RJ, Grosjean H. Nucleic Acids Res. 1997;25:4493–4499. doi: 10.1093/nar/25.22.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ganot P, Bortolin ML, Kiss T. Cell. 1997;89:799–809. doi: 10.1016/s0092-8674(00)80263-9. [DOI] [PubMed] [Google Scholar]
- 77.Ni J, Tien AL, Fournier MJ. Cell. 1997;89:565–573. doi: 10.1016/s0092-8674(00)80238-x. [DOI] [PubMed] [Google Scholar]
- 78.Charette M, Gray MW. IUBMB Life. 2000;49:341–351. doi: 10.1080/152165400410182. [DOI] [PubMed] [Google Scholar]
- 79.Mullenbach GT, Kammen HO, Penhoet EE. J Biol Chem. 1976;251:4570–4578. [PubMed] [Google Scholar]
- 80.Frendewey DA, Kladianos DM, Moore VG, Kaiser II. Biochem Biophys Acta. 1982;697:31–40. doi: 10.1016/0167-4781(82)90042-2. [DOI] [PubMed] [Google Scholar]
- 81.Pogolotti AL, Ivanetich KM, Somner H, Santi DV. Biochem Biophys Res Commun. 1986;70:972–978. doi: 10.1016/0006-291x(76)90687-2. [DOI] [PubMed] [Google Scholar]
- 82.Santi DV, Hardy LW. Biochemistry. 1987;26:8599–8606. doi: 10.1021/bi00400a016. [DOI] [PubMed] [Google Scholar]
- 83.Prior JJ, Santi DV. J Biol Chem. 1984;259:2429–2434. [PubMed] [Google Scholar]
- 84.Zhao XM, Horne DA. J Biol Chem. 1997;272:1950–1955. doi: 10.1074/jbc.272.3.1950. [DOI] [PubMed] [Google Scholar]
- 85.Ramamurthy V, Swann SL, Spedaliere CJ, Mueller EG. Biochemistry. 1999;38:13106–13111. doi: 10.1021/bi9913911. [DOI] [PubMed] [Google Scholar]
- 86.Giorgianni F, Beranova S, Wesdimiotis C, Viola RE. Biochemistry. 1995;34:3529–3535. doi: 10.1021/bi00011a006. [DOI] [PubMed] [Google Scholar]
- 87.Huang LX, Pookanjanatavip M, Gu XG, Santi DV. Biochemistry. 1998;37:344–351. doi: 10.1021/bi971874+. [DOI] [PubMed] [Google Scholar]
- 88.Mueller EG. Nat Struct Biol. 2002;9:320–322. doi: 10.1038/nsb0502-320. [DOI] [PubMed] [Google Scholar]
- 89.Ramamurthy V, Swann SL, Paulson JL, Spedaliere CJ, Mueller EG. J Biol Chem. 1999;274:22225–22230. doi: 10.1074/jbc.274.32.22225. [DOI] [PubMed] [Google Scholar]
- 90.Gu XR, Liu Y, Santi DV. Proc Natl Acad Sci USA. 1999;96:14270–14275. doi: 10.1073/pnas.96.25.14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hoang C, Ferre-D’Amare AR. Cell. 2001;107:929–939. doi: 10.1016/s0092-8674(01)00618-3. [DOI] [PubMed] [Google Scholar]
- 92.Spedaliere CJ, Mueller EG. RNA. 2004;10:192–199. doi: 10.1261/rna.5100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Spedaliere CJ, Ginter JM, Johnston MV, Mueller EG. J Am Chem Soc. 2004;126:12758–12759. doi: 10.1021/ja046375s. [DOI] [PubMed] [Google Scholar]
- 94.Arluison V, Hountondji C, Robert B, Grosjean H. Biochemistry. 1998;37:7268–7276. doi: 10.1021/bi972671o. [DOI] [PubMed] [Google Scholar]
- 95.Garcia GA, Tierney DL, Chong S, Clark K, Penner-Hahn JE. Biochemistry. 1996;35:3133–3139. doi: 10.1021/bi952403v. [DOI] [PubMed] [Google Scholar]
- 96.Benne R, Van den Burg J, Brakenhoff JP, Sloff P, Van Boom JH, Tromp MC. Cell. 1986;46:819–826. doi: 10.1016/0092-8674(86)90063-2. [DOI] [PubMed] [Google Scholar]
- 97.Benne R. RNA Editing: The Alteration of Protein Coding Sequences of RNA. Ellis Horwood; New York: 1993. [Google Scholar]
- 98.Bass B. In: The RNA World. Gesteland RF, Atkins JF, editors. Cold Spring Harbor Press; Cold Spring Harbor, NY: 1993. pp. 383–419. [Google Scholar]
- 99.Chan L, Chang BHJ, Nakamuta M, Li WH, Smith LC. Biochim Biophys Acta. 1997;1345:11–26. doi: 10.1016/s0005-2760(96)00156-7. [DOI] [PubMed] [Google Scholar]
- 100.Maas S, Rich A. Bioessays. 2000;22:790–802. doi: 10.1002/1521-1878(200009)22:9<790::AID-BIES4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 101.Horton TL, Landweber LF. Curr Opin Microbiol. 2002;5:620–626. doi: 10.1016/s1369-5274(02)00379-x. [DOI] [PubMed] [Google Scholar]
- 102.Niswender CM. Cell Mol Life Sci. 1998;54:946–964. doi: 10.1007/s000180050225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gray MW. IUBMB Life. 2003;55:227–233. doi: 10.1080/1521654031000119425. [DOI] [PubMed] [Google Scholar]
- 104.Novo FJ, Kruszewski A, MacDermot KD, Goldspink G, Gorecki DC. Nucleic Acids Res. 1995;23:2636–2640. doi: 10.1093/nar/23.14.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yokobori S, Paabo S. Proc Natl Acad Sci USA. 1995;92:10432–10435. doi: 10.1073/pnas.92.22.10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Laforest MJ, Roewer I, Lang BF. Nucleic Acids Res. 1997;25:626–632. doi: 10.1093/nar/25.3.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lonergan KM, Gray MW. Science (Washington, DC) 1993;259:812–816. doi: 10.1126/science.8430334. [DOI] [PubMed] [Google Scholar]
- 108.Yokobori SI, Paabo S. Nature (London) 1995;377:490. doi: 10.1038/377490a0. [DOI] [PubMed] [Google Scholar]
- 109.Sakuraba H, Eng CM, Desnick RJ, Bishop DF. Genomics. 1992;12:643–650. doi: 10.1016/0888-7543(92)90288-4. [DOI] [PubMed] [Google Scholar]
- 110.Nakanishi S, Ueda T, Hori H, Yamazaki N, Okada N, Watanabe K. J Biol Chem. 1994;269:32221–32225. [PubMed] [Google Scholar]