

Abstract
In temporal lobe epilepsy, seizures initiate in or near the hippocampus, which frequently displays loss of neurons, including inhibitory interneurons. It is unclear whether surviving interneurons function normally, are impaired, or develop compensatory mechanisms. We evaluated GABAergic interneurons in the hilus of the dentate gyrus of epileptic pilocarpine-treated GIN mice, specifically a subpopulation of somatostatin interneurons that expresses enhanced green fluorescence protein (GFP). GFP-immunocytochemistry and stereological analyses revealed substantial loss of GFP-positive hilar neurons (GPHNs) but increased GFP-positive axon length per dentate gyrus in epileptic mice. Individual biocytin-labeled GPHNs in hippocampal slices from epileptic mice also had larger somata, more axon in the molecular layer, and longer dendrites than controls. Dual whole-cell patch recording was used to test for monosynaptic connections from hilar GPHNs to granule cells. Unitary IPSCs (uIPSCs) recorded in control and epileptic mice had similar average rise times, amplitudes, charge transfers, and decay times. However, the probability of finding monosynaptically connected pairs and evoking uIPSCs was 2.6 times higher in epileptic mice compared to controls. Together, these findings suggest that surviving hilar somatostatin interneurons enlarge, extend dendrites, sprout axon collaterals in the molecular layer, and form new synapses with granule cells. These epilepsy-related changes in cellular morphology and connectivity may be mechanisms for surviving hilar interneurons to inhibit more granule cells and compensate for the loss of vulnerable interneurons.
Introduction
Hilar somatostatin interneurons are GABAergic (Somogyi et al., 1984; Kosaka et al., 1988; Esclapez and Houser, 1995) and abundant, accounting for 25% of all inhibitory interneurons in the dentate gyrus of rats (Buckmaster and Jongen-Rêlo, 1999) and monkeys (Austin and Buckmaster, 2004). Their axon collaterals concentrate in the middle and outer molecular layer (Bakst et al., 1986), synapse with dendrites of granule cells (Milner and Bacon, 1989; Leranth et al., 1990; Katona et al., 1999), and extend across broad septotemporal spans of the hippocampus (Boyett and Buckmaster, 2001; Buckmaster et al., 2002). Thus, they are ideally positioned to inhibit granule cells at sites of perforant path input, the major afferent of the dentate gyrus.
There is substantial loss of this class of inhibitory cell in patients with temporal lobe epilepsy (de Lanerolle et al., 1989, Mathern et al., 1995; Sundstrom et al., 2001) and rodent models (Sloviter, 1987; Freund et al., 1992; Lowenstein et al., 1992; Schwarzer et al., 1995; Houser and Esclapez, 1996; Sun et al., 2007). Surviving somatostatin cells may be structurally altered in that their somata appear larger (Buckmaster and Dudek, 1997). Other types of hippocampal interneurons in patients with temporal lobe epilepsy appear to develop new dendritic branches (Tóth et al., 2007). The possibility of dendritic growth also is supported by previous studies in normal mice, which reveal that neocortical interneurons, but not pyramidal cells, continue to retract and extend dendritic branches into adulthood (Lee et al., 2006). On the other hand, application of the chemoconvulsant kainic acid to hilar somatostatin interneurons causes acute excitotoxic damage to dendrites (Oliva et al., 2002) that could result in pruning.
The possibility of interneuron axon sprouting is suggested by increased expression of markers of GABAergic axons and synapses in the dentate gyrus of patients with temporal lobe epilepsy (Babb et al., 1989; Mathern et al., 1995, 1999; Patrylo et al., 1999; Wittner et al., 2001; Arellano et al., 2004) and rodent models (Davenport et al., 1990; Schwarzer et al., 1995, 1997; Houser and Esclapez, 1996; Bausch and Chavkin, 1997; Mathern et al., 1997; Esclapez and Houser, 1999; Fritschy et al., 1999; André et al., 2001; Knuesel et al., 2001; Boulland et al., 2007). In addition, GABA levels in hippocampal tissue increase in epileptic rats (Cavalheiro et al., 1994). However, it is difficult to distinguish genuine axon sprouting and synaptogenesis from increased marker expression by pre-existing structures (Bausch, 2005).
In the present study, we used immunocytochemistry, dual whole-cell patch recording, biocytin labeling, and mice in which a subpopulation of hilar somatostatin interneurons expresses enhanced green fluorescent protein (Oliva et al., 2000), to test whether surviving hilar somatostatin interneurons in the pilocarpine model of temporal lobe epilepsy enlarge, extend dendrites, sprout axons, and form new synapses with granule cells.
Materials and Methods
Animals.
All experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Stanford University Institutional Animal Care and Use Committee. GIN mice (FVB-Tg(GadGFP)4570Swn/J, The Jackson Laboratory) (Oliva et al., 2000) were treated with pilocarpine (300 mg/kg, i.p.) 20 min after atropine methylbromide (5 mg/kg, i.p.) at 27–69 d of age. Diazepam (10 mg/kg, i.p.) was administered 2 h after the onset of stage 3 or greater seizures (Racine, 1972) and repeated as needed to suppress convulsions. During recovery, mice received lactated ringers with dextrose subcutaneously. Control mice included animals that were treated identically but did not develop status epilepticus, as well as naive mice. There were no significant differences in results of green fluorescent protein (GFP)-positive hilar neurons (GPHNs) in naive and pilocarpine-treated control mice, so data were combined. Video monitoring, beginning at least 10 d after pilocarpine treatment, verified spontaneous motor convulsions in all epileptic mice. No controls were observed to have spontaneous seizures.
Green fluorescent protein immunocytochemistry.
For GFP-immunocyto chemistry experiments, the control group consisted of 8 mice: half female, half male, half naive, and half pilocarpine-treated controls. The epileptic group consisted of 8 mice (3 female, 5 male). Mice were killed with urethane (2 gm/kg, i.p.) at an average of 88 d old (range 55–219 d old), which was an average of 50 d (range 24–154 d) after pilocarpine-induced status epilepticus. They were perfused through the ascending aorta at 15 ml/min for 2 min with 0.9% sodium chloride and 30 min with 4% paraformaldehyde in 0.1 m phosphate buffer (PB, pH 7.4) at 4°C. Brains were postfixed overnight at 4°C. Left hippocampi were isolated, equilibrated in 30% sucrose in PB, gently straightened, frozen, and sectioned perpendicular to septotemporal axis with a sliding microtome set at 40 μm. Sections were stored at −20°C in preservation solution consisting of 30% ethylene glycol and 25% glycerol in 50 mm PB. Balanced numbers of control and epileptic mice were processed together in each batch. Beginning at random points near the septal pole of the hippocampus and extending through the entire septotemporal length, 1-in-6 series of sections were rinsed in PB and treated with 1% H2O2 for 2 h. After rinses in PB and 0.1 m Tris-buffered saline (TBS, pH 7.4), sections were treated with blocking solution consisting of 3% goat serum (Vector Laboratories), 2% bovine serum albumin (BSA), and 0.3% Triton X-100 in 0.05 m TBS for 1 h. Sections were rinsed in TBS and incubated for 40 h at 4°C in rabbit anti-GFP serum (1:2000, Invitrogen) diluted in 1% goat serum, 0.2% BSA, and 0.3% Triton X-100 in 0.05 m TBS. After rinses in TBS, sections incubated for 2 h in biotinylated goat anti-rabbit serum (1:500, Vector Laboratories) in secondary diluent consisting of 2% BSA, and 0.3% Triton X-100 in 0.05 m TBS. After rinses in TBS, sections incubated for 2 h in avidin-biotin-horseradish peroxidase complex (1:500, Vector Laboratories) in secondary diluent. After rinses in TBS and 0.1 m tris buffer (TB, pH 7.6), sections were placed for 5 min in chromogen solution consisting of 0.02% diaminobenzidine, 0.04% NH4Cl, and 0.015% glucose oxidase in TB and then transferred to fresh chromogen solution with 0.1% β-d-glucose for 13 min. The reaction was stopped in rinses of TB, and sections were mounted and dried on gelatin-coated slides. Sections dehydrated in a series of ethanols and xylenes and were coverslipped with DPX. Somatostatin-immunocytochemical processing was identical, except rabbit anti-somatostatin serum was used (1:4000, Peninsula Laboratories).
The total number of GPHNs per dentate gyrus was estimated using the optical fractionator method (West et al., 1991). Investigators were blind to experimental groups during analysis. Starting at random points near the septal pole of the hippocampus, 1-in-6 series of GFP-immunostained sections were sampled. Using Stereo Investigator (MBF Biosciences) and a microscope equipped with a 10× objective, contours were drawn around the hilus, which was defined by its border with granule cell layer and straight lines drawn from the ends of granule cell layer to the proximal end of the CA3 pyramidal cell layer. The entire hilar area was sampled. Total section thickness was used for dissector height, and only labeled somata not cut at upper surfaces of sections were counted. This modification of the optical fractionator method facilitates analysis of tissue sectioned thinly to enhance staining; however, it increases the probability of underestimating cell numbers. Using a 100× objective, 1741 GFP-positive somata were counted in 246 sections from 16 mice. The total number of GPHNs per dentate gyrus was estimated by multiplying counted neurons by a factor for section sampling (6). To analyze GPHN soma size, one or two sections from the mid-septotemporal level of the hippocampus from each mouse were evaluated. Using Neurolucida (MBF Biosciences) and a microscope equipped with a 100× objective, an investigator blind to experimental groups drew contours around 123 somata in 16 mice and recorded areas and maximum diameters. Adjacent thionin-stained sections were analyzed similarly to measure areas of 657 Nissl-stained hilar somata.
The total length of GFP-positive axon in the granule cell layer plus molecular layer was estimated by investigators blind to experimental groups. Starting at random points near the septal pole of the hippocampus, 1-in-12 series of GFP-immunostained sections were sampled. Contours were drawn around the granule cell layer plus molecular layer in each section, and areas were recorded. Using Stereo Investigator (MBF Biosciences), sample points were determined randomly and systematically. Counting grids were 350 × 350 μm, and counting frames were 25 × 25 μm. All GFP-positive axons within counting frames and throughout the entire depth of sections were reconstructed using a 100× objective, and cumulative length at each sample site was recorded. A total of 164,767 μm axon was measured at 341 sample sites in 124 sections from 16 mice. Total axon length per dentate gyrus (granule cell layer plus molecular layer) was estimated by multiplying measured axon length by factors for section sampling (12) and area sampling, which was the total area of the granule cell layer plus molecular layer divided by the analyzed area (number of counting frames per section times counting frame area).
Numbers of GFP-positive axon crossings of the granule cell layer were measured in a section from the mid-septotemporal level of the hippocampus from each mouse. Using Neurolucida (MBF Bioscience) and a microscope equipped with a 100× objective, a line was drawn along the middle of the granule cell layer starting at the tip of the superior blade, to the apex, and then to the tip of the inferior blade. All GFP-positive axon crossings were counted. Number of crossings per length of the granule cell layer was calculated. A total of 3201 crossings were counted in 16 sections from 16 mice.
Slice preparation.
Male and female mice were used for hippocampal slice experiments. There were no significant gender differences in any of the parameters analyzed in the present study. The control group consisted of 13 naive and 13 pilocarpine-treated controls. The epileptic group consisted of 31 mice, which were used for slice experiments when they were 92 ± 4 d old, an average of 48 d (range 12–82 d) after pilocarpine-induced status epilepticus. Mice were deeply anesthetized with urethane (1.5 g/kg, i.p.) and decapitated. Tissue blocks including the dentate gyrus were removed rapidly and stored for 3 min in ice-cold modified artificial CSF (mACSF) containing (in mm): 230 sucrose, 2.5 KCl, 10 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, 2.5 CaCl2, and 10 d-glucose. Horizontal slices (300 μm) were prepared with a microslicer (Leica VT1000S). All slices were from the same septotemporal level of the hippocampus, which corresponded to 3.6–4.4 mm below the dorsal surface of the brain (4.08 ± 0.04 mm and 4.10 ± 0.04 mm in control and epileptic mice, respectively, p = 0.74, t test) in the atlas of Franklin and Paxinos (2007). Slices were incubated at 32°C for 30 min in a submersion-type holding chamber that contained 50% mACSF and 50% normal ACSF, which consisted of (in mm): 126 NaCl, 3 KCl, 2 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, and 10 d-glucose. After that, slices were transferred to normal ACSF at 32°C for 1 h. ACSF was aerated continuously with a mixture of 95% O2 and 5% CO2. Slices were maintained at room temperature until used for recording.
Recording.
Cells were visualized with Nomarski optics (40×, Nikon) and an infrared-sensitive video camera (Hamamatsu Photonics). Fluorescence was used to identify GFP-positive neurons in the hilus. Whole-cell patch-clamp recordings were obtained at 32 ± 1°C. Interneurons were recorded in current-clamp mode (Axopatch 1D, Molecular Devices). Interneuron pipette solution contained (in mm): 100 potassium gluconate, 40 HEPES, 20 biocytin, 10 EGTA, 5 MgCl2, 2 disodium ATP, and 0.3 sodium GTP. Granule cells were recorded in voltage-clamp mode (Axopatch 200B, Molecular Devices). Granule cell pipette solution contained (in mm): 120 cesium methanesulfonate, 20 biocytin, 10 HEPES, 5 NaCl, 5 QX-314, 2 magnesium ATP, 0.3 sodium GTP, and 0.1 BAPTA. The measured liquid junction potential was 7 mV, and all membrane potentials were corrected accordingly. Patch electrodes were pulled from borosilicate glass (1.5 mm outer diameter, 0.75 mm inner diameter, 3–4 MΩ). Seal resistance was >5 GΩ, and only data obtained from electrodes with access resistance <20 MΩ and <20% change during recordings were included. Series resistance was 80% compensated, and compensation was readjusted during experiments, when necessary. Data were acquired (pCLAMP, Molecular Devices) and stored on computer for off-line analysis. Membrane currents and potentials were low-pass filtered at 2 kHz and digitized at 10 kHz.
Input resistance was determined in interneurons by measuring peak voltage responses to current steps ≤±10 pA and calculating slopes of regression lines in current-voltage plots. Unitary IPSCs (uIPSCs) were recorded in granule cells. IPSCs were enhanced and EPSCs minimized by holding granule cells at 0 mV. Action potentials were evoked in GFP-positive hilar neurons by brief (1.2 ms) depolarizing (1.5 nA) current injection, while uIPSCs were recorded in granule cells. Trains of 20 action potentials at 50 Hz were evoked at 0.1 Hz in interneurons. IPSCs were analyzed using Mini Analysis (Synaptosoft). The threshold for event detection was 3 times root mean square noise level. Latencies of uIPSCs were measured from the peak of presynaptic action potentials to 5% of uIPSC amplitude. Rise time was measured as the interval between points corresponding to 10 and 90% of peak amplitude during the rising phase. Amplitude was measured as the difference between peak amplitude and baseline. Decay time (τ) was measured as the interval between points corresponding to 100% and 37% of peak amplitude during the falling phase. Charge transfer was measured as the area under individual uIPSCs. For uIPSC analysis, parameters were measured from traces averaged after uIPSC failures were excluded. For paired-pulse and multipulse analysis, averaged traces included failures.
Biocytin-labeling.
After recording, slices were placed in 4°C 4% paraformaldehyde in PB at least overnight and then stored at −20°C in preservation solution. Slices were rinsed in 0.5% Triton X-100 and 0.1 m glycine in PB and then placed in blocking solution containing 0.5% Triton X-100, 2% goat serum (Vector Laboratories), and 2% BSA in PB for 4 h. Slices incubated with mouse antibody to NeuN (1:1000, MAB377; Millipore Bioscience Research Reagents) in blocking solution overnight. After rinsing, slices incubated with Alexa Fluor 594 streptavidin (5 μg/ml) and Alexa Fluor 488 goat anti-mouse serum (10 μg/ml; Invitrogen) in blocking solution overnight. Slices were rinsed, mounted on slides, and coverslipped with Vectashield (Vector Laboratories).
Interneuron reconstruction.
For dendritic reconstruction, biocytin-labeled neurons were scanned with a confocal microscope (LSM 5 Pascal; Zeiss) at a magnification just large enough to include the entire dendritic arbor. Stack height was adjusted to include all dendritic processes. Optical section interval was 3 μm. For high-magnification reconstruction of axon-dendrite appositions, a 100× objective and 0.5 μm optical section intervals were used. For axon reconstruction, a 20× objective was used to collect image stacks at 1 μm optical section intervals through the entire slice depth containing axon. Montages were collected to sample the entire area of the dentate gyrus molecular layer containing axon. Somata, dendrites, and axons were reconstructed and measured using Neurolucida (MBF Biosciences).
All chemicals and drugs were obtained from Sigma unless specified otherwise. Results are reported as mean ± SEM. Statistics were performed using Excel (Microsoft) and Sigma Stat (Systat) with p < 0.05 considered significant.
Results
Green fluorescent protein immunoreactivity
In mice, unlike other species examined, axons in the dentate gyrus molecular layer are only weakly immunoreactive for somatostatin (Buckmaster et al., 1994). However, in GIN mice a subset of somatostatin interneuron axons in the molecular layer can be visualized readily by GFP-immunoreactivity (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Therefore, we used GFP-immunocytochemistry in GIN mice to test whether surviving hilar somatostatin-immunoreactive interneurons enlarge and sprout axons after pilocarpine-induced status epilepticus.
Total numbers of GPHNs per dentate gyrus were estimated. Control mice had 827 ± 97 GPHNs per dentate gyrus (Fig. 1A,D). Epileptic mice had only 479 ± 74, which was 58% of controls (p = 0.01, t test). GPHNs were more numerous in sections from the temporal pole of the hippocampus (Fig. 1G). In epileptic mice, average numbers of GPHNs per section were reduced along the septotemporal axis of the hippocampus but most severely at the temporal pole. These findings reveal GPHN distributions and patterns of loss similar to that of somatostatin-immunoreactive hilar neurons in a rat model of temporal lobe epilepsy (Buckmaster and Dudek, 1997).
Figure 1.
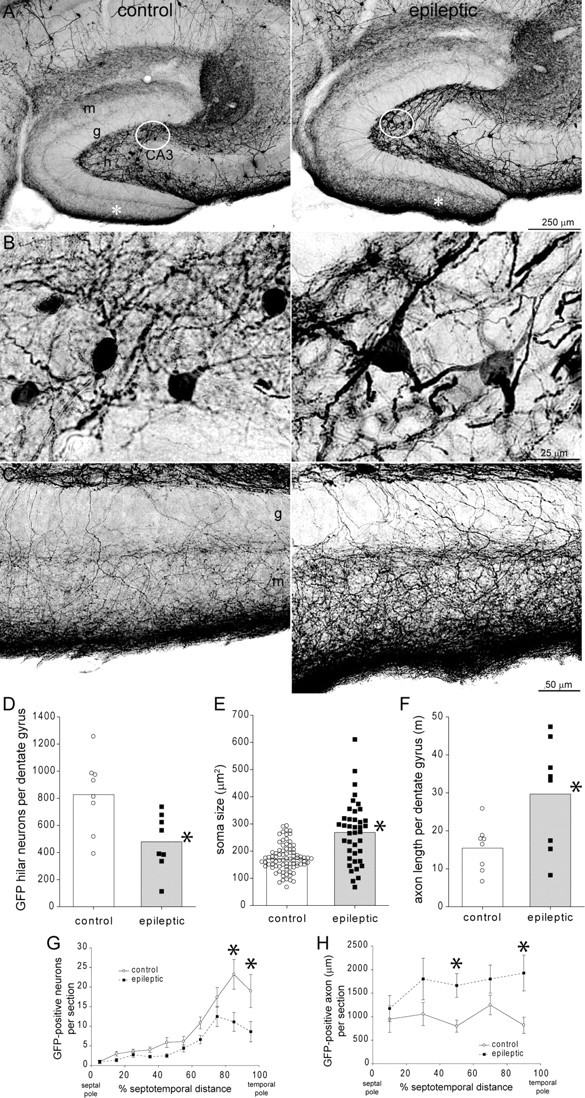
GFP-immunoreactivity in the dentate gyrus of a control (left) and epileptic pilocarpine-treated GIN mouse (right). A, Within the dentate gyrus, more GFP-positive neurons are evident in the hilus (h) of the control compared to the epileptic mouse. g, Granule cell layer; m, molecular layer; CA3, CA3 pyramidal cell layer. B, High-magnification views of circled regions in A reveal larger GFP-positive somata in the epileptic mouse. C, High-magnification views of regions indicated by asterisks in A reveal more intensely labeled GFP-positive axons extending through the granule cell layer and ramifying within the molecular layer, especially the outer molecular layer, in the epileptic mouse. D, The average number of GFP-positive hilar neurons per dentate gyrus (bars) was reduced in epileptic mice (*p = 0.01, t test, n = 8 controls, 8 epileptic mice). E, The average soma area (bars) was increased in epileptic mice (*p = 0.001, t test). Symbols indicate values of each measured soma. Many somata in epileptic mice exceeded the maximum of control mice. F, The average cumulative length of GFP-positive axon in the granule cell layer plus molecular layer per dentate gyrus (bars) was increased in epileptic mice (*p = 0.02, t test). G, GFP-positive hilar neurons were more numerous in sections from the temporal hippocampus. Epileptic mice had fewer neurons/section at all septotemporal levels, but especially in the temporal hippocampus. Values indicate mean ± SEM, *p < 0.001, repeated-measures ANOVA, Holm–Sidak method. H, Despite neuron loss, GFP-positive axon length per section was greater at all septotemporal levels of the hippocampus in epileptic mice. *p < 0.05, repeated-measures ANOVA, Holm–Sidak method.
Surviving neurons in epileptic mice appeared larger than GPHNs in controls (Fig. 1B). Average soma area in epileptic mice was 269 ± 21 μm2, which was 158% of controls (170 ± 10 μm2, p = 0.001, t test) (Fig. 1E). Average maximum diameter of somata in epileptic mice (26.1 ± 1.0 μm) was 134% of controls (19.5 ± 0.5 μm, p < 0.0001, t test). Average soma area of all surviving Nissl-stained hilar neurons in epileptic mice was 216 ± 21 μm2, which was 126% of controls (172 ± 7 μm2, p = 0.058, t test). Average soma size could increase in epileptic mice because cells enlarge or because small cells die. To help distinguish between these possibilities, the soma area of each measured GPHN was plotted (Fig. 1E). Many cells in epileptic mice had larger soma areas than the maximum in controls. These findings suggest surviving hilar somatostatin interneurons enlarge in epileptic mice.
The entire dentate gyrus appeared to be larger in epileptic mice (Fig. 1A). The Cavalieri method (West et al., 1991) was used to calculate volumes of the granule cell layer plus molecular layer in GFP-immunoprocessed sections. In epileptic mice the volume of the granule cell layer plus molecular layer was 1.34 ± 0.15 mm3, which was 161% of controls (0.83 ± 0.04 mm3, p = 0.004, t test). To test whether the larger volume might be attributable, at least in part, to more granule cells, the number of granule cells per dentate gyrus was estimated using the optical fractionator method in adjacent Nissl-stained sections. Control mice had 476,000 ± 24,000 granule cells per dentate gyrus, which was in the range of previous studies (Kempermann et al., 1997; Abusaad et al., 1999; Bonthius et al., 2004). Epileptic mice had 586,000 ± 41,000, which was 123% of controls (p = 0.03, t test). These findings reveal that the dentate gyrus of epileptic mice is larger and contains more granule cells than that of controls.
Hilar somatostatin interneurons project axon collaterals to the middle and outer molecular layer (Bakst et al., 1986; Buckmaster et al., 2002). Similar to previous somatostatin-immunocytochemistry studies of tissue from patients (Mathern et al., 1995) and rat models of temporal lobe epilepsy (Buckmaster and Dudek, 1997), the GFP-positive axon plexus in the molecular layer appeared to be more intensely labeled and developed in epileptic mice (Fig. 1C). Average numbers of GFP-positive axon crossings of the granule cell layer in sections from the mid-septotemporal level were similar in control (0.14 ± 0.02 crossings per 1 μm-length of granule cell layer) and epileptic mice (0.13 ± 0.02 crossings, p = 0.73, t test), despite fewer GPHNs in epileptic mice. Stereological techniques were used to estimate the total length of GFP-immunoreactive axon per dentate gyrus (molecular layer plus granule cell). Control mice had an average length of 15.5 ± 2.2 m (Fig. 1F). In epileptic mice, average GFP-positive axon length per dentate gyrus was increased to 29.7 ± 5.1 m, which was 192% of control levels (p = 0.02, t test) (Fig. 1D). To test whether increased axon length in epileptic mice was concentrated at a specific septotemporal level of the hippocampus, average length of GFP-positive axon per section was plotted against section position along the septotemporal axis (Fig. 1H). Average values of epileptic mice were greater than those of controls at all septotemporal levels. Together, these findings suggest that surviving somatostatin interneurons sprout axon collaterals along the entire septotemporal axis of the hippocampus. However, as with any immunoreactivity-based axon analysis, it is unclear to what extent increased axon length is attributable to genuine sprouting versus upregulated antigen expression in pre-existing axons. Furthermore, from immunocytochemical data alone it is difficult or impossible to identify sources of putative sprouted axon collaterals, because GFP-positive neurons throughout the CNS could potentially project into the dentate gyrus.
Biocytin-labeling
To more directly evaluate structural changes of surviving hilar somatostatin interneurons, patch-clamp recording was used in hippocampal slices to label individual GPHNs (Fig. 2A). Average soma size of biocytin-labeled GPHNs in epileptic mice was 225 ± 13 μm2, which was 160% of controls (141 ± 6 μm2, p < 0.0001, t test) (Fig. 2D), similar to results based on GFP-immunoreactivity (Fig. 1B,E). Consistent with increased cell size, average input resistance of GPHNs in epileptic mice was 296 ± 21 MΩ, which was 55% of controls (535 ± 38 MΩ, p < 0.0001, t test).
Figure 2.
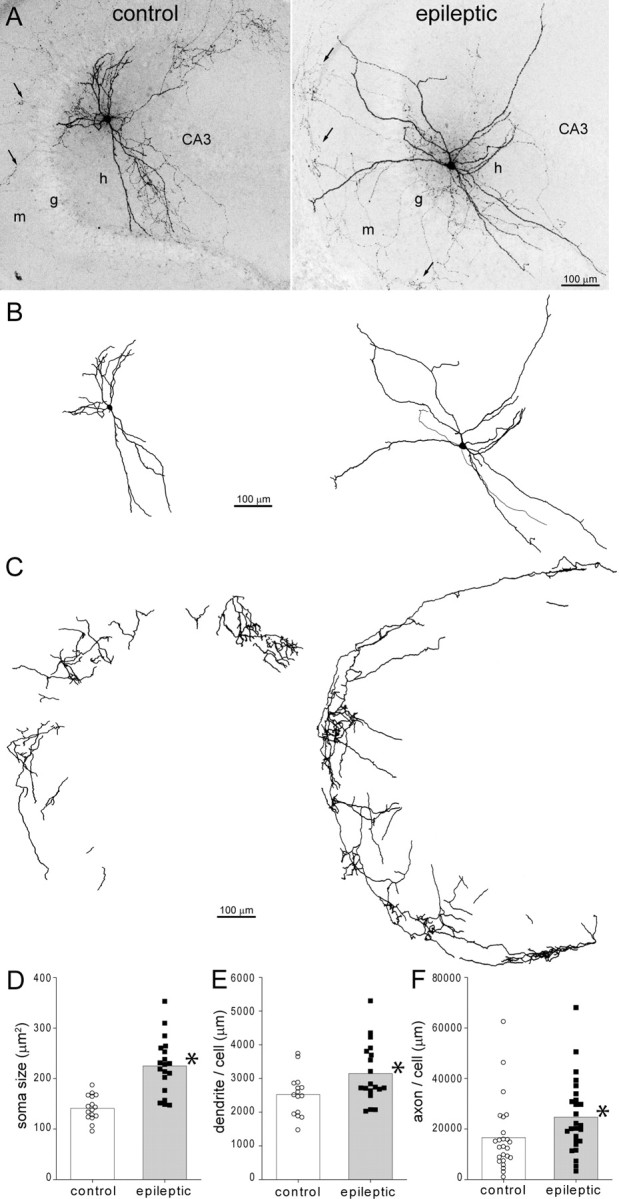
Biocytin-labeled GFP-positive hilar somatostatin interneurons in a control (left) and epileptic pilocarpine-treated GIN mouse (right). A, Biocytin-labeled GFP-positive neurons in the hilus (h) of 300 μm thick slices. Arrows indicate axon in the molecular layer (m). g, Granule cell layer; CA3, CA3 pyramidal cell layer. B, Soma and dendrite reconstructions of cells shown in A. C, Axon reconstructions in the molecular layer of cells shown in A. Only axon in the molecular layer, not hilus or granule cell layer, was reconstructed. D, Average soma area (bars) was increased in epileptic mice (*p < 0.0001, t test, n = 16 controls, 19 epileptic mice). E, Average dendritic length per cell (bars) was increased in epileptic mice (*p = 0.02, t test). F, Average axon length in the dentate gyrus molecular layer per cell (bars) was increased in epileptic mice (*p < 0.05, t test, n = 25 control, 26 epileptic mice).
As in previous reports of hilar somatostatin neurons in other rodent species (Milner and Bacon, 1989; Leranth et al., 1990; Katona et al., 1999; Buckmaster et al., 2002), in mice the dendrites of GPHNs ramified primarily in the hilus with occasional projections through the granule cell layer and into the molecular layer. The average number of dendritic extensions into the molecular layer per cell was similar in control (1.1 ± 0.5) and epileptic mice (1.2 ± 0.3). As previously reported for putative somatostatin interneurons in the hilus of monkeys (Buckmaster and Amaral, 2001), filamentous, undulating dendritic branches containing long, thin spines were a common feature of GPHNs in control and epileptic GIN mice. To reduce amputation of dendrites by slice preparation, only cells deep in slices were targeted for biocytin-labeling. Nevertheless, some amputation was unavoidable. There were no significant differences between control and epileptic mice in the average number of amputated dendrites per cell for primary (0.25 vs 0.35 in control and epileptic mice, respectively), secondary (0.55 vs 0.35), tertiary (0.75 vs 0.60), or total dendrites (3.0 vs 2.6). The average number of primary dendrites was similar in control (4.7 ± 0.3) and epileptic mice (5.0 ± 0.3). However, epileptic mice had an average of 3147 ± 202 μm total dendritic length/cell, which was 125% of controls (2523 ± 154 μm, p = 0.01, t test) (Fig. 2E). GPHNs in epileptic mice had 122 ± 23 dendritic endings compared to 90 ± 26 in controls, but the difference was not significant (p = 0.36, t test). These findings suggest that surviving hilar somatostatin interneurons elongate dendritic branches in epileptic mice.
In slices containing biocytin-labeled GPHNs, axons could be distinguished from dendrites by their smaller diameter, presence of boutons, and lack of spines. In both control and epileptic mice, biocytin-labeled axons extended through the hilus and granule cell layer and ramified within the molecular layer, especially, but not exclusively, in the middle and outer molecular layer (Fig. 2A). In both experimental groups, some cells displayed axon projections into stratum lacunosum-moleculare of CA3, a site of a dense GFP-immunoreactive axon plexus (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Also, in both experimental groups, some cells extended a large-diameter axon toward the fimbria, suggesting a possible contralateral projection, which occurs in other species (Bakst et al., 1986). Evaluating biocytin-labeled axons avoided potential confounding effects of antigen expression-level changes in the GFP-immunoreactive axon analysis described above. Axon collaterals of hilar somatostatin interneurons project far along the septotemporal axis of the hippocampus (Boyett and Buckmaster, 2001; Buckmaster et al., 2002), but our hippocampal slices were only 300 μm thick to optimize slice health and visualization of cells for recording. Therefore, slice preparation amputated substantial amounts of axon, likely adding variability to measured axon lengths. Well-labeled axons with low background staining and at least one projection into the molecular layer were included for analysis. These criteria were applied without regard to experimental group. And, as with all other anatomical analyses in the present study, investigators were blind to experimental group during data analysis. The total length of biocytin-labeled axon in the molecular layer per interneuron was measured in 48 slices containing a single biocytin-labeled interneuron and 3 slices containing a biocytin-labeled interneuron plus 1–2 biocytin-labeled granule cells, whose axons were clearly distinguishable from those of the interneuron. In epileptic mice, average total axon length/cell within the molecular layer of slices was 24,726 ± 2882 μm, which was 149% of controls (16,637 ± 2794 μm, p < 0.05, t test) (Fig. 2C,F). Figure 2C illustrates axon collaterals in the molecular layer, not the hilus or granule cell layer. These findings suggest that in epileptic mice, axons of surviving hilar somatostatin interneurons elongate in the molecular layer, where granule cell dendrites are located.
Paired recording
GFP-immunocytochemistry and biocytin-labeling data suggested somatostatin interneurons that survived pilocarpine-induced status epilepticus sprouted axon collaterals. We used paired recordings from GPHNs and granule cells to test whether such sprouting resulted in functional synaptic connections. The probability of detecting synaptically coupled pairs was measured in hippocampal slices from control and epileptic pilocarpine-treated mice. Action potentials were evoked in presynaptic GPHNs, and the incidence of finding synaptically coupled granule cells, as indicated by uIPSCs, was determined. In 17 of 101 pairs from control and 36 of 90 pairs from epileptic mice, uIPSCs were recorded at consistent and short latencies (2.9 ± 0.1 ms), indicating monosynaptic somatostatin interneuron-to-granule cell synapses (Fig. 3C). The probability of synaptic coupling was significantly higher in epileptic mice (257% of controls, p < 0.005, χ2 test). Biocytin-labeling revealed axons from GPHNs projecting into the molecular layer near dendritic arbors of recorded granule cells (Fig. 3A). Appositions between GPHN axons and granule cell dendrites were found consistently (Fig. 3B). In a subset of synaptically coupled pairs (n = 17 and 29 in control and epileptic mice, respectively), 20–70 responses in granule cells were recorded to 50 Hz trains, delivered at 0.1 Hz, of 20 action potentials in GPHNs. The response to the first interneuron action potential of each train was averaged (failures excluded) to evaluate uIPSC characteristics. Average amplitude (36 ± 6 pA vs 37 ± 5 pA), charge transfer (293 ± 62 fC vs 329 ± 57 fC), rise-time (1.8 ± 0.2 ms vs 1.8 ± 0.1 ms), and decay time (8.7 ± 0.8 ms vs 9.7 ± 0.6 ms) were similar in control and epileptic mice, respectively. Average failure rate of uIPSCs in epileptic mice (0.22 ± 0.03) was 183% of controls (0.12 ± 0.04), but the difference was not significant (p = 0.06, t test). These findings suggest that hilar somatostatin interneurons in slices from epileptic mice synapse with more granule cells than in controls and uIPSC characteristics are similar in control and epileptic mice.
Figure 3.
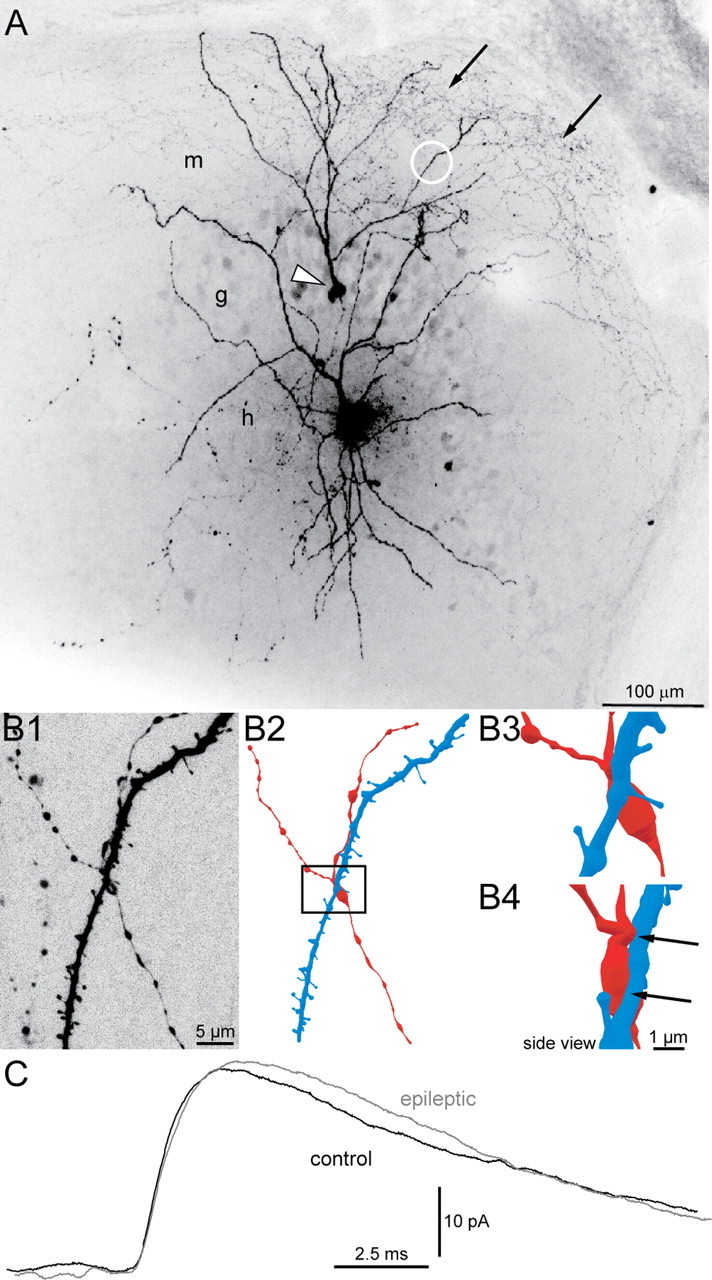
uIPSCs recorded in granule cells and generated by green fluorescent protein-positive hilar somatostatin interneurons in slices from control and epileptic pilocarpine-treated GIN mice. A, A biocytin-labeled interneuron in the hilus (h) and granule cell (arrowhead) in a slice from an epileptic mouse. The interneuron's axon (arrows) is concentrated in the molecular layer (m). g, Granule cell layer. B, An apposition of the interneuron's axon and the granule cell's dendrite. B1, High-magnification view of the region indicated by the white circle in A showing the spiny granule cell dendrite and interneuron axon with varicosities. B2, Reconstruction of the granule cell dendrite (blue) and interneuron axon (red). B3, High-magnification view of the area in the rectangle in B2. B4, Side view with axon-dendrite appositions indicated by arrows. C, Average uIPSCs from a control (black) and epileptic mouse (gray).
To further evaluate uIPSCs, responses to trains of interneuron action potentials were evaluated. Traces (42 ± 3 and 44 ± 3 per pair in control and epileptic mice, respectively) were averaged (uIPSC failures included). Average amplitudes of the first, second, and last (20th) uIPSC of each trace were measured (Fig. 4A). Paired-pulse (second/first) and multipulse (20th/first) ratios were calculated and averaged (Fig. 4B). The average paired-pulse ratio of uIPSCs in epileptic mice (66 ± 6%) was larger than that of controls (37 ± 3%, p = 0.002, t test). Similarly, the average multipulse ratio in epileptic mice (41 ± 5%) was larger than in controls (21 ± 4%, p = 0.008, t test).
Figure 4.
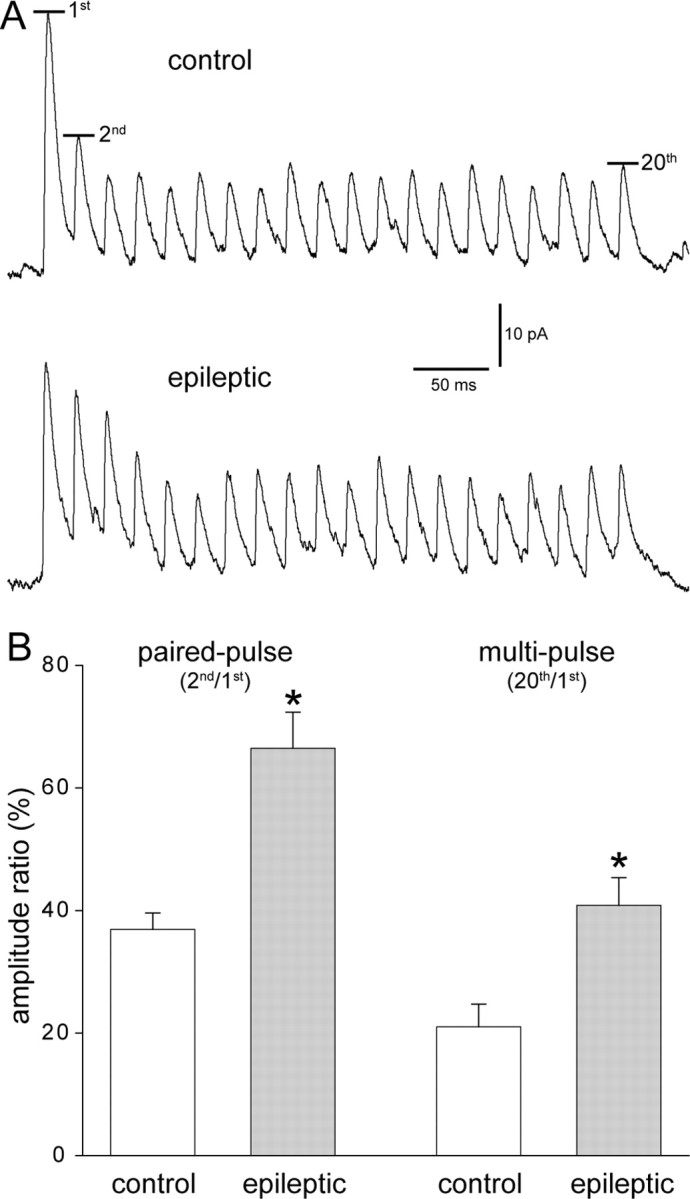
Paired- and multipulse depression was reduced at interneuron-to-granule cell synapses in epileptic mice. A, Trains of uIPSCs recorded in granule cells and generated by green fluorescent protein-positive hilar somatostatin interneurons in slices from control and epileptic pilocarpine-treated GIN mice. Action potentials (20) were evoked in interneurons at 50 Hz every 10 s. At least 20 traces were recorded and averaged. Average amplitudes of the first, second, and 20th uIPSCs were measured. B, Paired- and multipulse amplitude ratios were larger in epileptic mice (*p < 0.009, t test, n = 8 control, 15 epileptic mice).
Discussion
The principal findings of this study are that surviving GFP-positive hilar somatostatin interneurons in epileptic pilocarpine-treated GIN mice have larger somata, longer dendrites, more axon in the molecular layer, and are more likely to synapse with granule cells than in controls.
Surviving hilar somatostatin interneurons enlarge and increase their connectivity
Soma size and dendritic length of GPHNs increased in epileptic mice. Many cells were killed, presumably during pilocarpine-induced status epilepticus, suggesting the possibility of selective loss of smaller cells in epileptic animals. This seems unlikely, however, because ranges of soma area and dendritic length values in epileptic mice extended higher than ranges in control animals, suggesting that at least some cells in epileptic mice grew after pilocarpine treatment. Apparently increased dendritic and axonal lengths might be attributable to epilepsy-related upregulation of antigen and subsequent superior immunocytochemical labeling of pre-existing structures in epileptic mice. GFP expression in GIN mice is driven by an upstream regulatory sequence from the murine Gad1 gene (Oliva et al., 2000), and glutamic acid decarboxylase (GAD) is upregulated in the dentate gyrus of rat models of temporal lobe epilepsy (Feldblum et al., 1990; Esclapez and Houser, 1999). Thus, superior immunolabeling of axons might account for the larger proportional increase in average GFP-labeled axon length in epileptic animals (1.9-fold) compared to the relatively smaller increase in biocytin-labeled axon length per cell (1.5-fold). Another possibility is that in epileptic mice more GFP-positive axons project into the molecular layer from outside the dentate gyrus.
Although upregulation of GFP expression may contribute, increased immuno-positive and especially biocytin-labeled axon lengths in epileptic mice are likely attributable to axon sprouting by surviving GPHNs. Previous studies suggest that under other pathophysiological conditions GABAergic interneurons in the dentate gyrus might sprout axons and form new synapses. For example, weeks after lesioning perforant path input to the dentate gyrus, GAD activity (Nadler et al., 1974) and immunoreactivity (Goldowitz et al., 1982) increase in the outer molecular layer, gephyrin punctae numbers increase in the molecular layer (Simbürger et al., 2000), and GABAergic axons labeled with anterograde tracer sprout in the outer molecular layer (Deller et al., 1995). In the dentate gyrus of Alzheimer's disease mouse models, immunoreactivity of GABAergic axons, including somatostatin-immunoreactivity, increases (Palop et al., 2007). These findings suggest somatostatin interneurons might respond to a variety of conditions by sprouting axons.
However, it is unclear whether all surviving hilar somatostatin interneurons respond similarly. In GIN mice only 16% of hilar somatostatin interneurons express GFP (Oliva et al., 2000), so a substantial proportion was not evaluated in the present study. Furthermore, the dentate gyrus contains many different classes of interneurons (Freund and Buzsáki, 1996). In patients with temporal lobe epilepsy, calbindin-immunoreactive interneurons enlarge and extend dendrites (Maglóczky et al., 2000), as they do in a rat freeze microgyrus model of developmental epileptogenesis (Kharazia et al., 2003). On the other hand, parvalbumin-immunoreactive interneurons appear to have shrunken somata and shorter dendrites in patients (Zhu et al., 1997). Biocytin-labeling revealed no significant differences in soma or dendritic morphology of dentate basket cells in control and epileptic pilocarpine-treated rats (Zhang and Buckmaster, 2009). Therefore, each class of interneurons may respond differently in epilepsy; furthermore, changes may be specific in different types and models of epilepsy.
Characteristics of circuit changes
Increased axon length appears to enable surviving hilar somatostatin interneurons to synapse with more granule cells, as suggested by the increased probability of finding synaptically coupled GPHNs and granule cells in epileptic mice. If individual interneurons developed more synapses onto single granule cells, one would expect increased amplitude of uIPSCs, assuming no reductions in quantal size or release probability. However, average uIPSC amplitude was similar in control and epileptic mice, suggesting that most new synapses were with granule cells that were new targets. The present study cannot determine whether axon sprouting in epileptic mice extends septotemporal spans of surviving hilar somatostatin interneurons beyond their control levels. However, results of previous experiments that used a retrograde tracer in combination with somatostatin-immunocytochemistry suggest surviving interneurons sprout new axon collaterals within their normal, pre-epileptic axonal span but not farther along the septotemporal axis of the hippocampus (Boyett and Buckmaster, 2001).
Hilar somatostatin interneurons appear to hypertrophy and expand their connectivity, but many questions persist. One possibility is that GPHNs with low connectivity were selectively vulnerable to epileptogenic injury. Another possibility is that new synapses were not formed. Instead, connectivity only appeared to increase, because synapses that were too weak to be detected in controls became stronger in epileptic mice. However, evidence for somatic and dendritic hypertrophy suggests that axon sprouting is likely, and synaptogenesis may be the most parsimonious explanation for the findings of the present study.
The mechanisms underlying these morphological changes are unclear, but neurotrophins are possible contributors. For example, brain-derived neurotrophic factor promotes neuron growth and GABAergic synaptogenesis (Vicario-Abejón et al., 1998) and is upregulated by seizures (Isackson et al., 1991). Previous studies in other species and other systems suggest a correlation between neuronal cell body size and the number of axonal targets (McPhedran et al., 1965; Easter, 1979). Perhaps GFP-positive hilar somata enlarge in response to signals that indicate additional cellular machinery is needed to support their longer dendrites and axons and new synapses.
Functional implications
The functional consequences of the morphological changes in GPHNs are unclear. One possibility is that interneurons sprout axons to adapt to a larger target volume. Pilocarpine-induced status epilepticus stimulates neurogenesis in the dentate gyrus (Parent et al., 1997), which persists in mouse models of temporal lobe epilepsy (Jessberger et al., 2005). Epileptic mice had larger dentate gyrus volumes and more granule cells than controls, which would provide more synaptic targets for interneuron axons.
Another possibility is that interneurons sprout axons and form new synapses to compensate for interneurons and synapses lost during the epileptogenic injury. Shortly after epileptogenic injuries, before GABAergic axon sprouting has had sufficient time to develop, miniature IPSC frequency, which depends primarily on GABAergic synapse number and synaptic release probability, is substantially reduced in rat models of temporal lobe epilepsy (Kobayashi and Buckmaster, 2003; Shao and Dudek, 2005; Sun et al., 2007) (but see Leroy et al., 2004). These findings are consistent with the loss of interneurons, including hilar somatostatin interneurons. After axon sprouting by interneurons, mIPSC frequencies would be expected to return toward control levels, but they do not (Kobayashi and Buckmaster, 2003; Shao and Dudek, 2005; Sun et al., 2007). The discrepancy between mIPSC frequency and number of GABAergic synapses with granule cells might be attributable to different pools of synaptic vesicles for miniature and action potential dependent release of GABA (Mathew et al., 2008) or to reduced probability of GABA release in epileptic animals (Zhang and Buckmaster, 2009). Consistent with the latter hypothesis, paired-pulse ratios of uIPSCs in epileptic mice were larger than those of controls. Together, these findings suggest surviving hilar somatostatin interneurons sprout axon collaterals and form new synapses to partially compensate for the loss of other interneurons; however, their synapses with granule cells might be less reliable. Nevertheless, during high-frequency trains of action potentials, GPHNs generated uIPSCs that persisted and did not fade. Previous studies that have evaluated inhibition of granule cells in epilepsy report mixed results. Some studies found normal or enhanced inhibitory synaptic responses in granule cells (Haas et al., 1996; Isokawa, 1996; Bausch and Chavkin, 1997; Buckmaster and Dudek, 1999; Okazaki et al., 1999); whereas, others reported reduced granule cell inhibition (Williamson et al., 1999; Kobayashi and Buckmaster, 2003; Sayin et al., 2003). Findings of the present study suggest a possible mechanism (axon sprouting and synaptogenesis) that might contribute to recovered evoked inhibitory responses in granule cells, depending on stimulation parameters.
Although increased connectivity of hilar somatostatin cells might restore inhibition that is lost when vulnerable interneurons die, paradoxical proconvulsive effects also are possible. Excessive GABAergic synaptic input can depolarize neurons, especially in dendrites (Staley et al., 1995). GABAergic axon sprouting and synaptogenesis might exacerbate depolarizing GABA responses. The reversal potential for GABAA receptor-mediated events (EGABA) becomes more depolarized in some hippocampal neurons of patients with temporal lobe epilepsy (Cohen et al., 2002; Huberfeld et al., 2007). Similarly, EGABA depolarizes in rat granule cells after pilocarpine-induced status epilepticus but returns to normal in chronically epileptic animals (Pathak et al., 2007). Regardless of EGABA, GABAergic axon sprouting in epileptic tissue has been proposed to be potentially hypersynchronizing (Babb et al., 1989). Neurons with enhanced incoming and outgoing connectivity (“hubs”) can promote hyperexcitability (Morgan and Soltesz, 2008). The present study reveals increased outgoing connectivity by GFP-positive hilar interneurons. Their larger dendritic arbors suggest they may receive more synaptic input, which is supported by preliminary results (Halabisky et al., 2007). In conclusion, hypertrophy and hyper-connectivity of hilar somatostatin interneurons appears to have a partial compensatory effect, however, there are other possibilities that require further study.
Footnotes
This work was supported by the Epilepsy Foundation and by National Institute of Neurological Disorders and Stroke and National Center for Research Resources of National Institutes of Health. Thanks to Dr. Khushdev Thind for assistance with soma area analysis.
References
- Abusaad I, MacKay D, Zhao J, Stanford P, Collier DA, Everall IP. Stereological estimates of the total number of neurons in the murine hippocampus using the optical dissector. J Comp Neurol. 1999;408:560–566. doi: 10.1002/(sici)1096-9861(19990614)408:4<560::aid-cne9>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- André V, Marescaux C, Nehlig A, Fritschy JM. Alterations of hippocampal GABA system contribute to development of spontaneous recurrent seizures in the rat lithiumpilocarpine model of temporal lobe epilepsy. Hippocampus. 2001;11:452–468. doi: 10.1002/hipo.1060. [DOI] [PubMed] [Google Scholar]
- Arellano JI, Muñoz A, Ballesteros-Yáñez I, Sola RG, DeFelipe J. Histopathology and reorganization of chandelier cells in the human epileptic sclerotic hippocampus. Brain. 2004;127:45–64. doi: 10.1093/brain/awh004. [DOI] [PubMed] [Google Scholar]
- Austin JE, Buckmaster PS. Recurrent excitation of granule cells with basal dendrites and low interneuron density and inhibitory postsynaptic current frequency in the dentate gyrus of macaque monkeys. J Comp Neurol. 2004;476:205–218. doi: 10.1002/cne.20182. [DOI] [PubMed] [Google Scholar]
- Babb TL, Pretorius JK, Kupfer WR, Crandall PH. Glutamate decarboxylase-immunoreactive neurons are preserved in human epileptic hippocampus. J Neurosci. 1989;9:2562–2574. doi: 10.1523/JNEUROSCI.09-07-02562.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakst I, Avendano C, Morrison JH, Amaral DG. An experimental analysis of the origins of somatostatin-like immunoreactivity in the dentate gyrus of the rat. J Neurosci. 1986;6:1452–1462. doi: 10.1523/JNEUROSCI.06-05-01452.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch SB. Axonal sprouting of GABAergic interneurons in temporal lobe epilepsy. Epilepsy Behav. 2005;7:390–400. doi: 10.1016/j.yebeh.2005.07.019. [DOI] [PubMed] [Google Scholar]
- Bausch SB, Chavkin C. Changes in hippocampal circuitry after pilocarpine-induced seizures as revealed by opioid receptor distribution and activation. J Neurosci. 1997;17:477–492. doi: 10.1523/JNEUROSCI.17-01-00477.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonthius DJ, McKim R, Koele L, Harb H, Karacay B, Mahoney J, Pantazis NJ. Use of frozen sections to determine neuronal number in the murine hippocampus and neocortex using the optical dissector and optical fractionator. Brain Res Protocols. 2004;14:45–57. doi: 10.1016/j.brainresprot.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Boulland JL, Ferhat L, Tallak Solbu T, Ferrand N, Chaudhry FA, Storm-Mathisen J, Esclapez M. Changes in vesicular transporters for γ-aminobutyric acid and glutamate reveal vulnerability and reorganization of hippocampal neurons following pilocarpine-induced seizures. J Comp Neurol. 2007;503:466–485. doi: 10.1002/cne.21384. [DOI] [PubMed] [Google Scholar]
- Boyett JM, Buckmaster PS. Somatostatin-immunoreactive interneurons contribute to lateral inhibitory circuits in the dentate gyrus of control and epileptic rats. Hippocampus. 2001;11:418–422. doi: 10.1002/hipo.1056. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Amaral DG. Intracellular recording and labeling of mossy cells and proximal CA3 pyramidal cells in macaque monkeys. J Comp Neurol. 2001;430:264–281. doi: 10.1002/1096-9861(20010205)430:2<264::aid-cne1030>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. Neuron loss, granule cell axon reorganization, and functional changes in the dentate gyrus of epileptic kainate-treated rats. J Comp Neurol. 1997;385:385–404. [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. In vivo intracellular analysis of granule cell axon reorganization in epileptic rats. J Neurophysiol. 1999;81:712–721. doi: 10.1152/jn.1999.81.2.712. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Jongen-Rêlo AL. Highly specific neuron loss preserves lateral inhibitory circuits in the dentate gyrus of kainate-induced epileptic rats. J Neurosci. 1999;19:9519–9529. doi: 10.1523/JNEUROSCI.19-21-09519.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Kunkel DD, Robbins RJ, Schwartzkroin PA. Somatostatin-immunoreactivity in the hippocampus of mouse, rat, guinea pig, and rabbit. Hippocampus. 1994;4:167–180. doi: 10.1002/hipo.450040207. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Yamawaki R, Zhang GF. Axon arbors and synaptic connections of a vulnerable population of interneurons in the dentate gyrus in vivo. J Comp Neurol. 2002;445:360–373. doi: 10.1002/cne.10183. [DOI] [PubMed] [Google Scholar]
- Cavalheiro EA, Fernandes MJ, Turski L, Naffah-Mazzacoratti MG. Spontaneous recurrent seizures in rats: amino acid and monoamine determination in the hippocampus. Epilepsia. 1994;35:1–11. doi: 10.1111/j.1528-1157.1994.tb02905.x. [DOI] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Davenport CJ, Brown WJ, Babb TL. Sprouting of GABAergic and mossy fiber axons in dentate gyrus following intrahippocampal kainate in the rat. Exp Neurol. 1990;109:180–190. doi: 10.1016/0014-4886(90)90072-z. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Robbins RJ, Spencer DD. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989;495:387–395. doi: 10.1016/0006-8993(89)90234-5. [DOI] [PubMed] [Google Scholar]
- Deller T, Frotscher M, Nitsch R. Morphological evidence for the sprouting of inhibitory commissural fibers in response to the lesion of the excitatory entorhinal input to the rat dentate gyrus. J Neurosci. 1995;15:6868–6878. doi: 10.1523/JNEUROSCI.15-10-06868.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easter SS., Jr The growth and development of the superior oblique muscle and trochlear nerve in juvenile and adult goldfish. Anat Rec. 1979;195:683–697. doi: 10.1002/ar.1091950408. [DOI] [PubMed] [Google Scholar]
- Esclapez M, Houser CR. Somatostatin neurons are a subpopulation of GABA neurons in the rat dentate gyrus: evidence from colocalization of pre-prosomatostatin and glutamate decarboxylase messenger RNAs. Neuroscience. 1995;64:339–355. doi: 10.1016/0306-4522(94)00406-u. [DOI] [PubMed] [Google Scholar]
- Esclapez M, Houser CR. Up-regulation of GAD65 and GAD67 in remaining hippocampal GABA neurons in a model of temporal lobe epilepsy. J Comp Neurol. 1999;412:488–505. [PubMed] [Google Scholar]
- Feldblum S, Ackermann RF, Tobin AJ. Long-term increase of glutamate decarboxylase mRNA in a rat model of temporal lobe epilepsy. Neuron. 1990;5:361–371. doi: 10.1016/0896-6273(90)90172-c. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. Ed 3. New York: Elsevier; 2007. The mouse brain in stereological coordinates. [Google Scholar]
- Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Freund TF, Ylinen A, Miettinen R, Pitkänen A, Lahtinen H, Baimbridge KG, Riekkinen PJ. Pattern of neuronal death in the rat hippocampus after status epilepticus. Relationship to calcium binding protein content and ischemic vulnerability. Brain Res Bull. 1992;28:27–38. doi: 10.1016/0361-9230(92)90227-o. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Kiener T, Bouilleret V, Loup F. GABAergic neurons and GABAA-receptors in temporal lobe epilepsy. Neurochem Int. 1999;34:435–445. doi: 10.1016/s0197-0186(99)00040-6. [DOI] [PubMed] [Google Scholar]
- Goldowitz D, Vincent SR, Wu JY, Hökfelt T. Immunohistochemical demonstration of plasticity in GABA neurons of the adult rat dentate gyrus. Brain Res. 1982;238:413–420. doi: 10.1016/0006-8993(82)90116-0. [DOI] [PubMed] [Google Scholar]
- Haas KZ, Sperber EF, Moshé SL, Stanton PK. Kainic acid-induced seizures enhance dentate gyrus inhibition by downregulation of GABAB receptors. J Neurosci. 1996;16:4250–4260. doi: 10.1523/JNEUROSCI.16-13-04250.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halabisky BE, Buckmaster PS, Prince DA. Increased excitatory input onto hilar interneurons in a mouse model of pilocarpine-induced epilepsy. Soc Neurosci Abstr. 2007;33:333–1. [Google Scholar]
- Houser CR, Esclapez M. Vulnerability and plasticity of the GABA system in the pilocarpine model of spontaneous recurrent seizures. Epilepsy Res. 1996;26:207–218. doi: 10.1016/s0920-1211(96)00054-x. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- Isokawa M. Decrement of GABAA receptor-mediated inhibitory postsynaptic currents in dentate granule cells in epileptic hippocampus. J Neurophysiol. 1996;75:1901–1908. doi: 10.1152/jn.1996.75.5.1901. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Römer B, Babu H, Kempermann G. Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp Neurol. 2005;196:342–351. doi: 10.1016/j.expneurol.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Katona I, Acsády L, Freund TF. Postsynaptic targets of somatostatin-immunoreactive interneurons in the rat hippocampus. Neuroscience. 1999;88:37–55. doi: 10.1016/s0306-4522(98)00302-9. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH. Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proc Natl Acad Sci U S A. 1997;94:10409–10414. doi: 10.1073/pnas.94.19.10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharazia VN, Jacobs KM, Prince DA. GluR1 and calbindin expression are altered in interneurons of neocortical microgyral malformations. Neuroscience. 2003;120:207–218. doi: 10.1016/s0306-4522(03)00282-3. [DOI] [PubMed] [Google Scholar]
- Knuesel I, Zuellig RA, Schaub MC, Fritschy JM. Alterations in dystrophin and utrophin expression parallel the reorganization of GABAergic synapses in a mouse model of temporal lobe epilepsy. Eur J Neurosci. 2001;13:1113–1124. doi: 10.1046/j.0953-816x.2001.01476.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci. 2003;23:2440–2452. doi: 10.1523/JNEUROSCI.23-06-02440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka T, Wu JY, Benoit R. GABAergic neurons containing somatostatin-like immunoreactivity in the rat hippocampus and dentate gyrus. Exp Brain Res. 1988;71:388–398. doi: 10.1007/BF00247498. [DOI] [PubMed] [Google Scholar]
- Lee WC, Huang H, Feng G, Sanes JR, Brown EN, So PT, Nedivi E. Dynamic remodeling of dendritic arbors in GABAergic interneurons of adult visual cortex. PLoS Biology. 2006;4:e29. doi: 10.1371/journal.pbio.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leranth C, Malcolm AJ, Frotscher M. Afferent and efferent synaptic connections of somatostatin-immunoreactive neurons in the rat fascia dentata. J Comp Neurol. 1990;295:111–122. doi: 10.1002/cne.902950110. [DOI] [PubMed] [Google Scholar]
- Leroy C, Poisbeau P, Keller AF, Nehlig A. Pharmacological plasticity of GABAA at dentate gyrus synapses in a rat model of temporal lobe epilepsy. J Physiol. 2004;557:473–487. doi: 10.1113/jphysiol.2003.059246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglóczky Z, Wittner L, Borhegyi Z, Halász P, Vajda J, Czirják S, Freund TF. Changes in the distribution and connectivity of interneurons in the epileptic human dentate gyrus. Neuroscience. 2000;96:7–25. doi: 10.1016/s0306-4522(99)00474-1. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Pretorius JK, Leite JP. Reactive synaptogenesis and neuron densities for neuropeptide Y, somatostatin, and glutamate decarboxylase immunoreactivity in the epileptogenic human fascia dentata. J Neurosci. 1995;15:3990–4004. doi: 10.1523/JNEUROSCI.15-05-03990.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathern GW, Bertram EH, 3rd, Babb TL, Pretorius JK, Kuhlman PA, Spradlin S, Mendoza D. In contrast to kindled seizures, the frequency of spontaneous epilepsy in the limbic status model correlates with greater aberrant fascia dentata excitatory and inhibitory axon sprouting, and increased staining for N-methyl-D-aspartate, AMPA, and GABAA receptors. Neuroscience. 1997;77:1003–1019. doi: 10.1016/s0306-4522(96)00516-7. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Mendoza D, Lozada A, Pretorius JK, Dehnes Y, Danbolt NC, Nelson N, Leite JP, Chimelli L, Born DE, Sakamoto AC, Assirati JA, Fried I, Peacock WJ, Ojemann GA, Adelson PD. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology. 1999;52:453–472. doi: 10.1212/wnl.52.3.453. [DOI] [PubMed] [Google Scholar]
- Mathew SS, Pozzo-Miller L, Hablitz JJ. Kainate modulates presynaptic GABA release from two vesicle pools. J Neurosci. 2008;28:725–731. doi: 10.1523/JNEUROSCI.3625-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhedran AM, Wuerker RB, Henneman E. Properties of motor units in a homogeneous red muscle (soleus) of the cat. J Neurophysiol. 1965;28:71–84. doi: 10.1152/jn.1965.28.1.71. [DOI] [PubMed] [Google Scholar]
- Milner TA, Bacon CE. Ultrastructural localization of somatostatin-like immunoreactivity in the rat dentate gyrus. J Comp Neurol. 1989;290:544–560. doi: 10.1002/cne.902900409. [DOI] [PubMed] [Google Scholar]
- Morgan RJ, Soltesz I. Nonrandom connectivity of the epileptic dentate gyrus predicts a major role for neuronal hubs in seizures. Proc Natl Acad Sci U S A. 2008;105:6179–6184. doi: 10.1073/pnas.0801372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler JV, Cotman CW, Lynch GS. Biochemical plasticity of short-axon interneurons: increased glutamate decarboxylase activity in the denervated area of rat dentate gyrus following entorhinal lesion. Exp Neurol. 1974;45:403–413. doi: 10.1016/0014-4886(74)90128-9. [DOI] [PubMed] [Google Scholar]
- Okazaki MM, Molnár P, Nadler JV. Recurrent mossy fiber pathway in rat dentate gyrus: synaptic currents evoked in presence and absence of seizure-induced growth. J Neurophysiol. 1999;81:1645–1660. doi: 10.1152/jn.1999.81.4.1645. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr, Jiang M, Lam T, Smith KL, Swann JW. Novel hippocampal interneuronal subtypes identified using transgenic mice that express green fluorescent protein in GABAergic interneurons. J Neurosci. 2000;20:3354–3368. doi: 10.1523/JNEUROSCI.20-09-03354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva AA, Jr, Lam T, Swann JW. Distally directed dendrotoxicity induced by kainic acid in hippocampal interneurons of green fluorescent protein-expressing transgenic mice. J Neurosci. 2002;22:8052–8062. doi: 10.1523/JNEUROSCI.22-18-08052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, Coulter DA. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrylo PR, van den Pol AN, Spencer DD, Williamson A. NPY inhibits glutamatergic excitation in the epileptic human dentate gyrus. J Neurophysiol. 1999;82:478–483. doi: 10.1152/jn.1999.82.1.478. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Sayin U, Osting S, Hagen J, Rutecki P, Sutula T. Spontaneous seizures and loss of axo-axonic and axo-somatic inhibition induced by repeated brief seizures in kindled rats. J Neurosci. 2003;23:2759–2768. doi: 10.1523/JNEUROSCI.23-07-02759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer C, Williamson JM, Lothman EW, Vezzani A, Sperk G. Somatostatin, neuropeptide Y, neurokinin B and cholecystokinin immunoreactivity in two chronic models of temporal lobe epilepsy. Neuroscience. 1995;69:831–845. doi: 10.1016/0306-4522(95)00268-n. [DOI] [PubMed] [Google Scholar]
- Schwarzer C, Tsunashima K, Wanzenböck C, Fuchs K, Sieghart W, Sperk G. GABAA receptor subunits in the rat hippocampus II: Altered distribution in kainic acid-induced temporal lobe epilepsy. Neuroscience. 1997;80:1001–1017. doi: 10.1016/s0306-4522(97)00145-0. [DOI] [PubMed] [Google Scholar]
- Shao LR, Dudek FE. Changes in mIPSCs and sIPSCs after kainate treatment: evidence for loss of inhibitory input to dentate granule cells and possible compensatory responses. J Neurophysiol. 2005;94:952–960. doi: 10.1152/jn.01342.2004. [DOI] [PubMed] [Google Scholar]
- Simbürger E, Plaschke M, Kirsch J, Nitsch R. Distribution of the receptor-anchoring protein gephyrin in the rat dentate gyrus and changes following entorhinal cortex lesion. Cereb Cortex. 2000;10:422–432. doi: 10.1093/cercor/10.4.422. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–76. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Hodgson AJ, Smith AD, Nunzi MG, Gorio A, Wu JY. Different populations of GABAergic neurons in the visual cortex and hippocampus of cat contain somatostatin- or cholecystokinin-immunoreactive material. J Neurosci. 1984;4:2590–2603. doi: 10.1523/JNEUROSCI.04-10-02590.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Sun C, Mtchedlishvili Z, Bertram EH, Erisir A, Kapur J. Selective loss of dentate hilar interneurons contributes to reduced synaptic inhibition of granule cells in an electrical stimulation-based animal model of temporal lobe epilepsy. J Comp Neurol. 2007;500:876–893. doi: 10.1002/cne.21207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundstrom LE, Brana C, Gatherer M, Mepham J, Rougier A. Somatostatin- and neuropeptide Y-synthesizing neurons in the fascia dentata of humans with temporal lobe epilepsy. Brain. 2001;124:688–697. doi: 10.1093/brain/124.4.688. [DOI] [PubMed] [Google Scholar]
- Tóth K, Wittner L, Urbán Z, Doyle WK, Buzsáki G, Shigemoto R, Freund TF, Maglóczky Z. Morphology and synaptic input of substance P receptor-immunoreactive interneurons in control and epileptic human hippocampus. Neuroscience. 2007;144:495–508. doi: 10.1016/j.neuroscience.2006.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario-Abejón C, Collin C, McKay RD, Segal M. Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci. 1998;18:7256–7271. doi: 10.1523/JNEUROSCI.18-18-07256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Williamson A, Patrylo PR, Spencer DD. Decrease in inhibition in dentate granule cells from patients with medial temporal lobe epilepsy. Ann Neurol. 1999;45:92–99. doi: 10.1002/1531-8249(199901)45:1<92::aid-art15>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Wittner L, Maglóczky Z, Borhegyi Z, Halász P, Tóth S, Erőss L, Szabó Z, Freund TF. Preservation of perisomatic inhibitory input of granule cells in the epileptic human dentate gyrus. Neuroscience. 2001;108:587–600. doi: 10.1016/s0306-4522(01)00446-8. [DOI] [PubMed] [Google Scholar]
- Zhang W, Buckmaster PS. Dysfunction of the dentate basket cell circuit in a rat model of temporal lobe epilepsy. J Neurosci. 2009;29:7846–7856. doi: 10.1523/JNEUROSCI.6199-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu ZQ, Armstrong DL, Hamilton WJ, Grossman RG. Disproportionate loss of CA4 parvalbumin-immunoreactive interneurons in patients with Ammon's horn sclerosis. J Neuropathol Exp Neurol. 1997;56:988–998. doi: 10.1097/00005072-199709000-00004. [DOI] [PubMed] [Google Scholar]