

Abstract
The role of neuropeptide Y Y2 receptor (Y2R) in human diseases such as obesity, mood disorders, and alcoholism could be better resolved by the use of small-molecule chemical probes that are substantially different from the currently available Y2R antagonist, N-[(1S)-4-[(aminoiminomethyl)amino]-1-[[[2-(3,5-dioxo-1,2-diphenyl-1,2,4-triazolidin-4-yl)ethyl]amino]carbonyl]butyl]-1-[2-[4-(6,11-dihydro-6-oxo-5H-dibenz[b,e]azepin-11-yl)-1-piperazinyl]-2-oxoethyl]-cyclopentaneacetamide) (BIIE0246). Presented here are five potent, selective, and publicly available Y2R antagonists identified by a high-throughput screening approach. These compounds belong to four chemical scaffolds that are structurally distinct from the peptidomimetic BIIE0246. In functional assays, IC50 values between 199 and 4400 nM against the Y2R were measured, with no appreciable activity against the related NPY-Y1 receptor (Y1R). Compounds also displaced radiolabeled peptide YY from the Y2R with high affinity (Ki values between 1.55 and 60 nM) while not displacing the same ligand from the Y1R. In contrast to BIIE0246, Schild analysis with NPY suggests that two of the five compounds behave as competitive antagonists. Profiling against a panel of 40 receptors, ion channels, and transporters found in the central nervous system showed that the five Y2R antagonists demonstrate greater selectivity than BIIE0246. Furthermore, the ability of these antagonists to penetrate the blood-brain barrier makes them better suited for pharmacological studies of Y2R function in both the brain and periphery.
Neuropeptide Y (NPY) is one of the most abundant neuropeptides in the mammalian brain (Catapano and Manji, 2007) and was originally identified in 1982 based on its structural similarity to peptide YY (70% homology with PYY) and pancreatic polypeptide (50% homology with PP) (Tatemoto, 1982). Each of these 36-residue peptides is processed from a 94- to 95-residue prohormone (Balasubramaniam, 1997). Four related type 1 G protein-coupled receptor (GPCR) family members (Y1, Y2, Y4, and Y5) mediate biological responses to NPY. These receptors couple to the Gαi signaling pathway and inhibit adenylate cyclase, preventing cAMP production.
Although NPY receptors have mainly been of interest to the pharmaceutical industry as therapeutic targets for obesity and feeding disorders, NPY has numerous roles in the body, including modulation of feeding behavior, circadian rhythm, learning, memory, vascular remodeling, cell proliferation, and angiogenesis (Tatemoto, 1982). Human studies suggest that NPY is involved in mood disorders and in alcoholism (Wahlestedt et al., 1989; Thorsell et al., 2006). In patients with major depression, several studies show that NPY levels in the central nervous system (CNS) are low and correlate inversely to anxiety (Widerlöv et al., 1988; Heilig, 2004; Nikisch et al., 2005). In addition to major depressive disorders, human genetic studies show that polymorphisms in the NPY gene also associate with alcoholism (Wahlestedt et al., 1986; Zhu et al., 2003; Mottagui-Tabar et al., 2005).
The NPY-Y2 receptor (Y2R) is located presynaptically on neurons and in part serves to negatively regulate neurotransmitter release in the brain (King et al., 1999). Antagonism of Y2R would thus be expected to increase NPY levels in the CNS and may prove useful in treating certain psychiatric diseases. Consistent with this, Y2R-null mice demonstrate reduced anxiety-like behavior compared with wild type (Redrobe et al., 2003). In addition, administration of the Y2R antagonist BIIE0246 decreased ethanol consumption in rats and induced antidepressant-like effects in mice (Rimondini et al., 2005).
Few efforts have focused on developing Y2R antagonists, with only two selective Y2R antagonists being reported other than BIIE0246: the peptide ligand T4-[NPY 33-36]4 (Grouzmann et al., 1997); and the small molecule JNJ-5207787 (Bonaventure et al., 2004). Small-molecule diamines have also been reported as Y2R ligands. However, these compounds have not been shown to be functionally active or selective for the Y2R (Andres et al., 2003).
The most widely used Y2R antagonist, BIIE0246, is selective with high receptor affinity and is efficacious both in vitro and in vivo (Doods et al., 1999). However, there are significant drawbacks associated with this compound. For instance, it has been suggested that BIIE0246 behaves as an insurmountable antagonist (Dautzenberg and Neysari, 2005), an undesirable trait for a probe or potential therapeutic agent, because this mode of inhibition might block the Y2R from further activation, which would lead to long-term loss of Y2R function. As a consequence, studies with BIIE0246 are limited to in vitro binding and functional determinations of Y2R activity. Furthermore, this peptidomimetic is big (molecular weight of 896) with a large polar surface area (219 Å2), and both of these factors contribute to poor brain penetration. For example, experiments performed in vivo require direct injection of BIIE0246 to the desired site (Abbott et al., 2005; Rimondini et al., 2005). Similar challenges exist for the studies involving peptides such as T4-[NPY 33-36]4. In contrast, the small molecule JNJ-5207787 has improved properties over T4-[NPY 33-36]4 and BIIE0246. For example, ex vivo receptor autoradiography studies reveal that JNJ-5207787 is able to cross the blood-brain barrier and occupy Y2R receptor binding sites (Bonaventure et al., 2004). However, the lack of commercial availability of JNJ-520778 prevents further investigation into its pharmacology.
As a result of the drawbacks of current antagonists, there is a need to identify brain-penetrant Y2R antagonists to better define the role of the receptor in the CNS. This report presents five novel, selective, brain-penetrant small-molecule Y2R antagonists, discovered from a high-throughput screening (HTS) approach, that seem to have a different mechanism of action from BIIE0246 and are better suited for neuropharmacology studies.
Materials and Methods
Unless otherwise stated, reagents were purchased from commercial sources. The cAMP biosensor assay, ACTOne Membrane Potential assay kit, was purchased from BD Biosciences (Rockville, MD). Puromycin, Ro 20-1724, and isoproterenol were from Sigma-Aldrich (St. Louis, MO). Neuropeptide Y was from American Peptide (Sunnyvale, CA). BIIE0246 and BIBP3226 were from Tocris Bioscience (Ellisville, MO). Human liver microsomes were from Xenotech (Lenexa, KS). Black, clear-bottomed 1536-well tissue culture-treated microtiter plates were from Greiner Bio-One (Longwood, FL). The National Institutes of Health Molecular Libraries Small-Molecule Repository (MLSMR, San Francisco, CA) provided compound collections (dissolved at concentrations up to 10 mM in DMSO) for the primary and follow-up assays. For dose-response assays, compounds were provided by the MLSMR or were ordered as powders and prepared as 10 mM stock solutions in DMSO.
For studies involving powders, the compounds SF-11, SF-21, and SF-22 were obtained from ChemBridge (San Diego, CA). The compounds SF-31 and SF-41 were purchased from Chemical Diversity (San Diego, CA) and Specs (Delft, Holland), respectively. Reference and ordering information for all compounds can be obtained from PubChem (available at http://pubchem.ncbi.nlm.nih.gov/) by conducting queries with the following substance identifiers (SIDs): SF-11, SID 17507305; SF-12, SID 17431723; SF-13, SID 56365809; SF- 14, SID 17433143; SF-15, SID 17505667; SF-16, SID 56365810; SF-21, SID 17413392; SF-22, SID 17413034; SF-23, SID 56365811; SF-24, SID 17412946; SF-31, SID 4242079; SF-32, SID 4255014; SF-33, SID 14736723; SF-34, SID 56365812; SF-35, SID 4241999; SF-36, SID 56365813; SF-37, SID 4246387; SF-38, SID 4244380; SF-39, SID 14731189; SF-41, SID 22413249; and SF-42, SID 22413387.
Cell Culture.
The cAMP biosensor assay cell lines were purchased from BD Biosciences as human embryonic kidney (HEK) 293 cells stably expressing a cyclic nucleotide-gated (CNG) channel and either Y2R or Y1R. Cells were cultured in T-175-cm2 flasks at 37°C and 95% relative humidity. Cells were plated and maintained in growth medium consisting of Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 25 mM HEPES, 5 mM l-glutamine, 250 μg/ml G418 (Geneticin), 1 μg/ml puromycin, and 1% antibiotic mix containing penicillin, streptomycin, and neomycin.
Primary HTS of the MLSMR Compound Collection.
As part of the Molecular Libraries Probe Production Network (MLPCN; available at http://mli.nih.gov/mli/) program, the HTS campaign was executed on the automated Kalypsys robotic platform located at the Scripps Research Institute Molecular Screening Center (Jupiter, FL). Screening results for the Y2R (PubChem AID, 793) and Y1R (PubChem AID, 1040) antagonist primary assays, and all subsequent assays within the HTS screening campaign have been deposited into the MLPCN database PubChem. It is important to note that the MLSMR is constantly adding and removing compounds from the MLPCN screening library, and a net of ∼50,000 compounds was added to the library between the Y2R and Y1R primary screens. Both primary assays were screened against the full MLPCN screening library, which consisted of either 140,094 (Y2R assay) or 196,180 compounds (Y1R assay).
Y2R Primary Campaign Protocol.
For Y2R HTS assays, HEK293-CNG cells were diluted in growth medium and dispensed into 1536-well black-walled, clear-bottomed plates (final concentration, 3750 cells/well) and allowed to incubate for 24 h at 37°C. Next, a 4.5× concentrated membrane potential dye, prepared according to manufacturer's instructions, was dispensed into each well. An initial fluorescent measurement (T0) was performed (510–545 nm excitation and 565–625 nm emission), with the use of a FLIPRTETRA fluorescence reader (Molecular Devices, Sunnyvale, CA), after incubating of the cells for 3 h at room temperature. Test compounds (final concentration, 2.7 μM), DMSO alone (final concentration, 0.27%), or BIIE0246 (final concentration, 1 μM) was then added to sample or appropriate control wells, respectively. The cells were then challenged with NPY (final concentration, 100 nM), isoproterenol (freshly prepared; final concentration, 1 μM) and the phosphodiesterase inhibitor Ro 20-1724 (final concentration, 25 μM). The plates were incubated for 30 min at room temperature before the final fluorescence measurement (T30) was taken, as described above.
Y1R Primary Campaign Protocol.
The Y1R HTS screen was performed using a protocol similar to that used for Y2R, with the following exceptions: a total of 3600 cells/well were seeded into assay plates and allowed to grow for 24 h at 37°C. After the T0 measurement, NPY (final concentration, 25 nM) was added to all wells, followed by the addition of test compounds (final concentration, 3.6 μM) or DMSO to respective sample and control wells. Plates were incubated for 1 h at room temperature, followed by the addition of isoproterenol (freshly prepared; final concentration, 1 μM) and Ro 20-1724 addition (final concentration, 25 μM). After 45 min at room temperature, the final fluorescent measurement was performed (T45).
Calculation of Antagonist Activity.
The data from each campaign were normalized with the T0 measurement by dividing the fluorescence at T30 (for the Y2R) or T45 (for the Y1R) by the initial basal fluorescence at T0. The percentage of inhibition for each compound was determined using calculated ratios on a per-plate basis as follows: % Inhibition = 100 · [1 − ((Test Compound − High Control)/(Low Control − High Control))], where Test Compound indicates the ratio of fluorescence of wells containing test compound, NPY, and isoproterenol. Low Control indicates the average ratio of fluorescence of wells containing DMSO, NPY, and isoproterenol. High Control indicates the average ratio of fluorescence of wells containing either Y2R antagonist (BIIE0246), NPY, and isoproterenol for the Y2R assays, or no NPY and isoproterenol for the Y1R assays.
HTS Hit Selection and Activity Cutoff Criteria.
To determine the inhibitory compounds in each primary screen, two values were calculated: 1) the average percentage of inhibition of all compounds tested; and 2) three times their standard deviation (Hodder et al., 2003). The sum of these two values was used as a cutoff parameter; any compound that exhibited greater percentage of inhibition than the cutoff value was considered active. Z′ values were determined to monitor assay quality and were calculated as described previously (Zhang et al., 1999). Assay plates were rejected and rerun if Z′ was less than 0.5.
HTS Hit Confirmation and Counterscreening Assays.
Test compounds active in the primary HTS campaign were subsequently confirmed using the same assay protocol and hit cutoff value as used in the primary campaign, except that compounds were tested in triplicate. All confirmation and counterscreen assay data have been uploaded to the PubChem website (AID 1257, Y2R hit confirmation; AID 1256, Y1R counterscreen).
Dose-Response Assays.
The potency of compounds that passed confirmation and counterscreening was determined using dose-response assays, and IC50 values were calculated from the resultant data. A 10-point dose-response curve with a 1:3 dilution series from 35 μM to 1.8 nM was used, with compounds being tested in triplicate. Compounds exhibiting an IC50 value less than 10 μM were considered active. Dose-response counterscreens were also performed in Y1R cells to confirm that compounds identified as Y2R antagonists were selective (PubChem AID 1272, Y2R dose-response determinations; AID 1279, Y1R dose-response determinations).
Binding Assays.
Whole-cell binding assays were performed as described previously (Brothers et al., 2006). In brief, Y2R HEK293-CNG and Y1R HEK293-CNG cells were cultured and plated in growth medium as described above, except that 105 cells in 0.5 ml of growth medium were seeded in 24-well poly(d-lysine)-coated cell culture plates. After 24 h, cells were washed twice with a wash buffer consisting of 0.5 ml of Dulbecco's modified Eagle's medium/0.1% bovine serum albumin/10 mM HEPES. Next, a range of concentrations of each antagonist was added to the cells, followed by 20 nM 125I-PYY (final concentration) in 0.25 ml in the same buffer. After incubation at room temperature for 60 min (Nikisch et al., 2005), the supernatant was removed, the cells were washed twice and solubilized, and radioactivity of the lysate was determined. Ki values from 125I-PYY displacement were determined using a one-site competition binding model and the Cheng-Prusoff equation (Cheng and Prusoff, 1973). A Kd of 0.27 nM for 125I-PYY as reported previously (Rose et al., 1995) and a concentration of 20 nM for 125I-PYY in the competition binding experiments were used for Ki determination.
Selectivity binding studies were conducted by the National Institute of Mental Health's Psychoactive Drug Screening Program (NIMH PDSP). Methodological details are available at the PDSP web site (http://pdsp.med.unc.edu/), where comprehensive protocols are described that are similar to those reported previously (Armbruster et al., 2007).
Schild Analysis.
A 12-point dose-response curve was performed in quadruplicate with the cAMP biosensor assay in 384-well plates for NPY (1:3 dilution series from 1 μM to 6.6 pM) and in the presence of five different concentrations of BIIE0246 (1, 3, 10, 30, and 50 nM) or the five HTS lead compounds (100, 300, 1000, 3000, and 5000 nM). Identical with HTS assay protocols, agonist (NPY) and antagonists were administered simultaneously to Y2R HEK293-CNG cells. The EC50 values for NPY at varying antagonist concentrations were used for Schild analysis.
Hepatic Microsomal Stability Assays.
Microsome stability was evaluated by incubating 1 μM compound with 2 mg/ml hepatic microsomes from either human, rat, or mouse in 100 mM potassium phosphate buffer, pH 7.4. The reactions were held at 37°C with continuous shaking. The reaction was initiated by adding NADPH (final concentration, 1 mM) and the final incubation volume was 300 μl. Aliquots (40 μl) were removed at 0, 3, 5, 10, 20, and 30 min and added to 160 μl of acetonitrile containing an internal standard (propranolol) to stop the reaction and precipitate the protein. At the end of the assay, the samples were centrifuged through a 0.45-μm filter plate (Solventer low-binding hydrophilic plates; Millipore Corporation, Billerica, MA) and analyzed by LC-MS/MS. The data were log-transformed, and results are reported as half-life.
Plasma Protein Binding Experiments.
An ultracentrifugation method was used for the evaluation of plasma protein binding plasma (Nakai et al., 2004). Plasma sample (1 ml with 1 μM test compound) was prepared, and 900 μl was transferred to a 2-ml polycarbonate ultracentrifuge tube. The sample was centrifuged at 400,000g for 2 h using an Optima Max ultracentrifuge (130,000 rpm maximum; Beckman Coulter, Fullerton, CA) with a TLA 120.2 rotor held at 25°C. The middle layer (2–3 mm below the surface using the described conditions) was used to determine the amount of unbound drug. The fraction of compound unbound to plasma proteins was determined using LC-MS/MS in comparison with the total amount of drug in the uncentrifuged sample.
Hepatic Clearance Calculations.
In vitro intrinsic hepatic clearance was calculated from the microsomal incubation data using methods similar to those described previously (Lipscomb and Poet, 2008). Specifically, the plasma protein binding data were combined with the results from the microsome stability experiments to predict the theoretical hepatic clearance and hepatic extraction ratio.
Brain Penetration Assays.
Plasma and brain levels of the compounds were assessed in C57BL6 mice 30 min after dosing 10 mg/kg i.p. Samples were formulated at 2 mg/ml in 10:10:80 DMSO/Tween/water. Blood was collected into EDTA-containing tubes at 30 min, and plasma was generated using standard centrifugation techniques. Brain samples were frozen upon collection, and all samples were stored at −80°C until analyzed. Brain tissue was not perfused before freezing to prevent diffusion of the compound out of the tissue during the process. Plasma samples were analyzed by treating 25 μl of plasma with 125 μl of acetonitrile containing an internal standard (propranolol) and filtering through a Millipore Multiscreen Solvinter 0.45-μm low-binding polytetrafluoroethylene hydrophilic filter. The filtrate was analyzed by LC-MS/MS using an API Sciex 4000 (Applied Biosystems, Foster City, CA). Multiple reaction-monitoring methods were developed in positive-ion mode, and concentrations were determined using a standard curve between 2 and 2000 ng/ml. Samples with concentrations outside of the curve were diluted with blank plasma and reanalyzed. Similar conditions were used to determine brain levels, except the samples were weighed, and acetonitrile was added (10×, weight by volume). The samples were sonicated to extract the compound from the brain matrix and then filtered as described above. A density of 1 g/ml was used to convert compound per milligram of tissue into molar equivalents.
Cell Viability Assays.
Cytotoxicity assays were performed as described previously (Madoux et al., 2008). Y2R HEK293-CNG cells were seeded at 500 cells/well in 1536-well plates in 5 μl of growth medium. Compounds (in DMSO) prepared as 10-point, 1:3 serial dilutions and added to cells (highest final concentration, 99 μM). Plates were then incubated for 72 h at 37°C. After incubation, 5 μl of CellTiter-Glo (Promega, Madison, WI) was added to each well, and the plates were allowed to incubate for 15 min at room temperature. Luminescence was then measured (ViewLux plate reader; PerkinElmer Life and Analytical Sciences, Turku, Finland). Viability was calculated as a percentage relative to control cells treated with either DMSO alone (0% cytotoxicity) or 100 μM doxorubicin (100% cytotoxicity).
Data Analysis and Statistics.
Data were analyzed using MDL Assay Explorer (version 3.1; Symyx Software, Santa Clara, CA) or Prism (version 5.01; GraphPad Software Inc., San Diego, CA). Curve-fitting and IC50 determinations were performed using the variable slope sigmoidal dose-response analysis tool in Prism. Ki values were determined using a one-site competition binding model. Replicates of at least three data points for each treatment group within an experiment were analyzed by unpaired, two-tailed t test or using one-way analysis of variance followed by Tukey's test. Schild nonlinear regression analysis was performed in Prism using the Gaddum/Schild EC50 shift analysis tool. Statistical significance was ascertained by F test comparing the calculated Schild slope with a hypothetical Schild analysis with slope of unity.
Results
High-Throughput Screening Assay for NPY Receptor Antagonists.
GPCRs that couple to the Gαi signaling pathway, such as Y1R and Y2R, modulate intracellular cAMP concentrations via adenylate cyclase. Several HTS-compatible assays may be used to measure Gαi-coupled receptor activity, including use of GPCR-fluorescent fusion proteins (Milligan et al., 2004), reporter genes (Doucette et al., 2009), or measurement of cellular cAMP concentrations directly (Eglen, 2005). For the research presented here, an HTS-compatible cell-based cAMP assay was used (Visegrády et al., 2007). In this assay format, measurement of Gαi protein-coupled receptor antagonism in mammalian cells is facilitated by the presence of a “cAMP biosensor” (i.e., a modified CNG channel). Activation of the CNG channel by cAMP results in membrane depolarization. Depolarization is detected using a fluorescent membrane potential dye.
In the NPY receptor cAMP biosensor assay protocols (Fig. 1), isoproterenol, a β-adrenergic receptor agonist, was used to induce intracellular cAMP production and therefore membrane depolarization via the activated CNG channel. The addition of NPY receptor agonist, through inhibition of adenylate cyclase, inhibits the accumulation of cAMP induced by isoproterenol. As designed, an antagonist would be expected to block NPY-agonized receptor signaling, leading to increased cAMP levels, decreased membrane polarization, and consequently increased dye fluorescence.
Fig. 1.

Principle of the cAMP biosensor assay used for the HTS campaign. A, addition of NPY receptor agonist stimulates the receptor, activates Gαi protein, and decreases cAMP production via inhibition of adenylate cyclase (AC). Antagonism of the receptor increases production of cAMP by the same pathway, which serves to activate and open the CNG ion channel. The relocation of extracellular Ca2+ and Na+ from the extracellular space to the cytosol causes membrane depolarization. B, a membrane potential fluorescent indicator (D) is used to measure the cell membrane polarization state. Before cellular depolarization, indicator dye molecules are mainly extracellular with minimal fluorescence. Upon depolarization, dye redistributes into the cytoplasm, becoming fluorescent.
Y2R Antagonist HTS Campaign.
To identify selective Y2R antagonists via an HTS approach, Y2R and Y1R cAMP biosensor assays were developed in 1536-well plates in a total assay volume of 9 μl/well. The assays used HEK cells expressing either the Y2R or the Y1R and the CNG channel (necessary to affect a membrane potential change). Experimental conditions were optimized to give the best balance between assay performance (determined by Z′ factor), reagent consumption, and suitability of the protocol for robotic-based screening. Several parameters were optimized independently for the Y2R and Y1R antagonist assays such as cell number, NPY concentration, and assay incubation times (see Materials and Methods section for final conditions).
Parallel Counterscreen Outcomes and Hit Confirmation.
As part of a MLPCN research effort (Lazo et al., 2007), the Y2R cAMP biosensor assay was screened against a collection of 140,094 compounds from the MLSMR. All MLMSR compounds were tested for antagonism at a nominal concentration of 2.7 μM. Compounds that antagonized Y2R activity greater than the 17.08% inhibition cutoff (determined from the mean response plus 3 standard deviations for the entire compound data set) were considered active (“hits”). A total of 1384 compounds met this cutoff for the Y2R antagonist campaign.
The Y1R cAMP biosensor assay was used as a parallel counterscreen during the Y2R screening campaign with the goal to streamline Y2R hit validation (Fig. 2). Specifically, all compounds tested in the Y2R antagonist HTS assay were also tested in the Y1R antagonist HTS assay to remove cAMP biosensor assay-specific artifact and nonselective antagonists from further consideration. Of the 1384 compounds found active in the Y2R screen, 624 were also active in the Y1R screen. Removal of compounds found active in both assays left 760 potentially Y2R-selective antagonists. From this subset of compounds active in the Y2R assay, 707 compounds were available for further testing from the MLSMR. Fresh aliquots of these 707 compounds were retested at a single concentration (2.7 μM) in triplicate using the same assay conditions as the original screen. A total of 228 of these compounds confirmed activity (i.e., inhibited Y2R activity greater than 17.08%). From these 228 confirmed hits, 123 were found to have significant activity when tested in triplicate at a single concentration (3.6 μM) in the Y1R assay; these 123 compounds were not considered further.
Fig. 2.

Correlation plot of inhibition results from the Y2R and Y1R primary HTS assays. Gray circles represent the response, expressed as the percentage of inhibition, for each compound tested in the Y1R (y-axis) and Y2R (x-axis) antagonism format cAMP biosensor assays. A total of 140,094 compounds were tested for inhibition in both the Y2R and Y1R primary HTS assays (details in text). The five HTS-derived Y2R antagonists presented in this manuscript are distributed in the lower quadrant of the plot, represented by the following symbols: ■, SF-11; +, SF-21; ♦, SF-22; ●, SF-31; and ▲, SF-41. The vertical and horizontal dashed lines represent the hit cutoff values of the Y2R (17.08%) and Y1R (20.11%) assays.
As a final step in the Y2R screening campaign, 119 of 125 Y2R-selective compounds (with six unavailable from the MLSMR) were freshly prepared as 10-point titrations (1:3 serial dilutions from 35 μM to 1.8 nM). Antagonism was evaluated over this concentration range in both the Y2R and the Y1R cAMP biosensor assays. From this set, 72 compounds antagonized the Y2R with an IC50 value less than 10 μM, whereas 74 compounds antagonized the Y1R with an IC50 value less than 10 μM (Fig. 3).
Fig. 3.

Correlation plot of results from the Y2R and Y1R dose-response assays. The IC50 values of 119 compounds are plotted for both the Y2R (x-axis) and Y1R (y-axis) cAMP biosensor assays. The five selective Y2R antagonists can be found in the upper part of the plot, represented by ■, SF-11; +, SF-21; ♦, SF-22; ●, SF-31; and ▲, SF-41. The five corresponding structures of antagonists, as well as previously described NPY receptor antagonists BIIE0246 and JNJ-5207787 (Y2R) and BIBP3226 (Y1R), are illustrated.
Identification of Selective Y2R Antagonists.
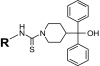
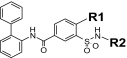
Any compound chosen for further follow-up studies was required to exhibit an IC50 value of less than 5 μM at Y2R and greater than 35 μM at Y1R. Five compounds met or exceeded these criteria: SF-11, SF-21, SF-22, SF-31, and SF-41(Fig. 3). They fall into four chemotypes, classified as SF-1 through SF-4, representing piperidinecarbothioamide, arylsulfamoylbenzamide, aryl-1,2,4-oxadiazole, and arylsulfonylmethylisoxazole scaffolds, and are not structurally similar to the known Y2R antagonists BIIE0246 and JNJ-5207787. Molecular weight, partition coefficient (as logP), and polar surface area (PSA) were calculated for the five HTS-derived compounds as well as BIIE0246 (Table 1). The molecular weight of all five HTS-derived antagonists was less than 500; taken together, the logP and PSA values suggest that they are lipophilic and are likely to be cell membrane-permeable.
TABLE 1.
Comparison of calculated physical properties for different Y2R antagonists
| ID | Molecular Weight | logP | Polar Surface Area |
|---|---|---|---|
| SF-11 | 447 | 5.1 | 44.7 |
| SF-21 | 471 | 6.5 | 75.3 |
| SF-22 | 491 | 6.9 | 75.3 |
| SF-31 | 415 | 4.7 | 86.5 |
| SF-41 | 341 | 3.9 | 60.2 |
| BIIE0246 | 896 | 4 | 219 |
Fresh powders of all compounds were characterized by LC-MS and retested at the Y2R and the Y1R. All compounds confirmed antagonism in the Y2R assay with IC50 values of less than 5 μM, and in the case of SF-11, SF-21, and SF-22, the IC50 values were less than 1 μM (Fig. 4A). For all compounds tested, the maximum inhibition observed was within 20% of that observed for the positive control (BIIE0246). Furthermore, four compounds (SF-11, SF-22, SF-31, and SF-41) were inactive in the Y1R assay even at the highest concentration tested (35 μM; Fig. 4B), whereas SF-21 exhibited minor but significant inhibition (10% at 35 μM).
Fig. 4.

A, antagonist dose-response curves from the Y2R cAMP biosensor assay. Y2R antagonists SF-11 (■, IC50 = 199 ± 10 nM), SF-21 (+, IC50 = 440 ± 30 nM), SF-22 (♦, IC50 = 750 ± 30 nM), SF-31 (●, IC50 = 1200 ± 100 nM), and SF-41 (▲, IC50 = 4400 ± 40 nM) are graphed. For comparison, the IC50 for BIIE0246 is 0.58 ± 0.56 nM. B, antagonist dose-response curves from the Y1R cAMP biosensor assay. Symbols for the different compounds are the same as those described in A. For comparison, BIBP3226 (BIBP), a Y1R antagonist, is shown (x, IC50 = 3046 ± 982 nM). C, results of 125I-PYY displacement with Y2R-expressing cells. Percentage of binding symbols for the different compounds are the same as those described in A. SF-11 (Ki = 1.55 ± 0.93 nM), SF-21 (Ki = 1.93 ± 0.86 nM), SF-22 (Ki = 2.25 ± 0.49 nM), SF-31 (Ki = 6.0 ± 2.8 nM), and SF-41 (Ki = 60.3 ± 36.2 nM) versus concentration is graphed. Comparison Ki = 0.02 ± 0.01 nM for BIIE0246. D, results of 125I-PYY displacement with Y1R-expressing cells. Displacement was measured at a single high concentration (50 μM) of Y1R-selective antagonist (BIBP3226) or HTS-derived antagonist. Error bars on all graphs represent the standard deviation of at least three separate experiments.
Y2R Antagonist Structure-Activity Relationship.
As a first step in evaluating structure-activity relationships for the newly identified Y2R antagonists, 16 close analogs of the five antagonists were procured. All were then tested for IC50 values in both the Y2R and Y1R cAMP biosensor assays (Table 2). Although all 16 analogs demonstrated activity at the Y2R, none of the newly obtained compounds demonstrated improved potency compared with the antagonists derived from HTS efforts. In addition, four analogs belonging to the aryl-1,2,4-oxadiazole scaffold (SF-33, SF-34, SF-35, and SF-36) exhibited greater than 30% inhibition in the Y1R assay, with SF-34 being equipotent in both assays.
TABLE 2.
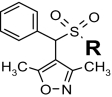
Structure-activity relationships for Y2R-selective antagonists SF numbers of compounds from HTS are in boldface type.
| ID | R | IC50(Maximum
Inhibition) |
CC50(Maximum Toxicity) | ID | R1 | R2 | IC50(Maximum
Inhibition) |
CC50(Maximum Toxicity) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Y2R | Y1R | Y2R | Y1R | |||||||
|
|
|||||||||
| μM | μM | μM | μM | |||||||
| SF-11 |
|
0.199 ± 0.01 | >35.4 (inactive) | >33 | SF-21 | Cl |
|
0.44 ± 0.03 | >35.4 (10 ± 2%) | 19.7 ± 7.2 |
| SF-12 |
|
1.1 ± 0.1 | >35.4 (5 ± 4%) | >99 (37 ± 8%) | SF-22 | CH3 |
|
0.75 ± 0.03 | >35.4 (inactive) | 61 ± 30 |
| SF-13 |
|
1.3 ± 0.1 | >35.4 (inactive) | >33 | SF-23 | CH3 |
|
2.1 ± 0.2 | >35.4 (inactive) | >33 |
| SF-14 |
|
2.99 ± 0.1 | >35.4 (inactive) | >99 (49 ± 6%) | SF-24 | Cl |
|
3.96 ± 0.3 | >35.4 (inactive) | >33 |
| SF-15 |
|
4.9 ± 0.57 | >35.4 (4 ± 3%) | >99 (41 ± 6%) |
|
|||||
| SF-16 |
|
>35.4 (43 ± 5%) | >35.4 (inactive) | >33 | SF-31 |
|
1.2 ± 0.1 | >35.4 (inactive) | >99 (43 ± 7%) | |
|
SF-32 |
|
4.9 ± 0.1 | >35.4 (inactive) | >99 (27 ± 11%) | |||||
| SF-41 |
|
4.4 ± 0.04 | >35.4 (inactive) | >99 (35 ± 9%) | SF-33 |
|
5.5 ± 0.8 | >35.4 (34 ± 4%) | 30.8 ± 2.7 | |
| SF-42 |
|
8.6 ± 0.5 | >35.4 (inactive) | >99 (9 ± 8%) | SF-34 |
|
17.3 ± 6.7 | 17.3 ± 3.4 | >33 | |
| SF-35 |
|
>11.8 | >11.8 | 17.2 ± 0.4 | ||||||
| SF-36 |
|
>35.4 (46 ± 4%) | >35.4 (36 ± 11%) | >11 | ||||||
| SF-37 |
|
>35.4 (45 ± 7%) | >35.4 (inactive) | >33 | ||||||
| SF-38 |
|
>35.4 (43 ± 4%) | >35.4 (6 ± 3%) | >33 | ||||||
| SF-39 |
|
>35.4 (4 ± 2%) | >35.4 (inactive) | 22.9 ± 2.5 | ||||||
Assessment of Compound Cytotoxicity.
All five HTS-derived Y2R antagonists and their 16 structural analogs were tested as 10-point titrations (in the range 5 nM to 99 μM) for their effect on the viability of Y2R-expressing HEK cells (Table 2). After a 72-h incubation, the five compounds identified by HTS (SF-11, SF-21, SF-22, SF-31, and SF-41) were toxic [in terms of their 50% cytotoxic concentration (CC50) values] at concentrations that were 10- to 100-fold higher than their respective IC50 values for the Y2R. In general, the 16 analogs were also toxic at compound concentrations significantly higher than their respective Y2R IC50 values. However, nine analogs (SF-16, SF-24, SF-33, SF-34, SF-35, SF-36, SF-37, SF-38, and SF-39) demonstrated less than a 10-fold difference between the Y2R IC50 values and their corresponding CC50 values.
Binding of 125I-PYY to Cells Treated with Y2R-Selective Antagonists.
Radioligand displacement studies were used to measure the ability of SF-11, SF-21, SF-22, SF-31, or SF-41 to block agonist binding. These five compounds were titrated for eight-point dose-response curves (1:6 serial dilutions from 50 μM to 1.8 pM), and radioligand displacement was evaluated over this concentration range in HEK cells expressing the Y2R or the Y1R. Calculations of Ki values were performed using the Cheng-Prusoff model (Cheng and Prusoff, 1973). All five compounds competed 125I-PYY binding in Y2R-expressing cells with Ki values of 1.55 ± 0.93 nM for SF-11, 1.93 ± 0.86 nM for SF-21, 2.25 ± 0.49 nM for SF-22, 6.0 ± 2.8 nM for SF-31, and 60.3 ± 36.2 nM for SF-41, respectively (Fig. 4C). In contrast, none of the tested compounds significantly blocked 125I-PYY binding in Y1R-expressing cells (Fig. 4D).
Schild Analysis of BIIE0246- and Y2R-Selective Compounds.
To investigate the molecular pharmacology of the Y2R antagonists, Schild analysis was performed on BIIE0246 and the five HTS leads using the Y2R cAMP biosensor assay. NPY was used as the challenge. From these experiments, the Schild slopes were 2.79 ± 0.09 for BIIE0246, 0.79 ± 0.03 for SF-11, 0.98 ± 0.05 for SF-21, 1.3 ± 0.1 for SF-22, 1.55 ± 0.12 for SF-31, and 1.23 ± 0.08 for SF-41 (see Supplementary Fig. 1).
Metabolic Stability Experiments.
The metabolic stability of SF-11, SF-21, SF-22, SF-31, and SF-41 along with BIIE0246 were evaluated by incubation with human, mouse, and rat liver microsomes (Table 3). The concentration of microsomal protein was normalized to 2 mg/ml from each species, and exogenous NADPH was added to initiate the reactions. BIIE0246 demonstrated the longest half-life of all compounds tested. Among the five compounds identified by HTS, SF-31 had the longest half-life in all species. The shortest half-life in rodent and human microsomes was observed with SF-21 and SF-22 with half-lives of approximately 1 min.
TABLE 3.
Results of microsome stability studies (expressed as T1/2), protein binding experiments [expressed as fraction unbound (fu)], calculated hepatic clearance (Clhepatic), and compound extracted per pass for five Y2R antagonists and BIIE0246
| ID and Species | T 1/2 | fu | Clhepatic | Extracted per Pass |
|---|---|---|---|---|
| min | % | |||
| SF-11 | ||||
| Mouse | 4 | 0.03 | 5.7 | 6.3 |
| Rat | 3 | 0.02 | 6.1 | 11 |
| Human | 10 | 0.01 | 0.3 | 1.4 |
| SF-21 | ||||
| Mouse | 1 | 0.82 | 90 | 100 |
| Rat | 1 | 0.13 | 50 | 90 |
| Human | 8 | 0.23 | 7 | 34 |
| SF-22 | ||||
| Mouse | 1 | 1 | 90 | 100 |
| Rat | 1 | 0.19 | 53 | 96 |
| Human | 6 | 0.28 | 10 | 49 |
| SF-31 | ||||
| Mouse | 14 | 0.12 | 6.4 | 7.2 |
| Rat | 6 | 0.07 | 10 | 18 |
| Human | 13 | 0.07 | 1.5 | 7.4 |
| SF-41 | ||||
| Mouse | 4 | 0.39 | 51 | 57 |
| Rat | 1 | 0.19 | 53 | 96 |
| Human | 3 | 0.19 | 12 | 60 |
| BIIE0246 | ||||
| Mouse | 166 | 0.079 | 0.4 | 0.4 |
| Rat | 27 | 0.022 | 0.8 | 1.4 |
| Human | 23 | 0.015 | 0.2 | 0.9 |
Plasma Protein Binding Experiments and Hepatic Clearance Calculations.
An ultracentrifugation method was used for the evaluation of plasma protein binding plasma (Nakai et al., 2004); the fraction of compound unbound to plasma proteins was determined using LC-MS/MS. As noted in Table 3, the fraction of compound unbound to plasma proteins was high except for SF-11. In addition, in vitro intrinsic hepatic clearance (Clhepatic) was calculated using methods similar to those described previously (Lipscomb and Poet, 2008). Overall, BIIE0246 had the lowest hepatic clearance; the hepatic clearance of the five HTS-derived antagonists was high except for SF-11 and SF-31, in which it was less than 10 (Table 3). The hepatic extraction ratios (calculated as the percentage extracted per pass) for the five HTS-derived compounds were also higher than those calculated for BIIE0246.
Blood-Brain Barrier Penetration Studies.
To determine whether the five antagonists identified by HTS were brain-penetrant, each compound was injected intraperitoneally into adult mice at 10 mg/kg, and levels in the brain tissue and plasma were measured after 30 min. All five newly identified antagonists were highly brain-penetrant, with brain-to-plasma concentration ratios ranging between 36% (SF-21) and 115% (SF-41) (Table 4). At 30 min, SF-11, SF-21, SF-22, and SF-31 had brain concentrations greater than their respective cell-based IC50 values. High concentrations of BIIE0246 were detected in the plasma after intraperitoneal dosing, but the achieved brain levels were only 2% of plasma levels. All compounds seemed to be well tolerated, with the exception of SF-11, which caused significant lethargy in the mice.
TABLE 4.
Results of blood-brain barrier penetration studies for five Y2R antagonists and BIIE0246
| ID | Brain Penetration
Studies |
||
|---|---|---|---|
| Plasma Concentration | Brain Concentration | Ratio [B]/[P] | |
| μM | % | ||
| SF-11 | 8.5 ± 3.3 | 3.7 ± 1.7 | 44 |
| SF-21 | 1.1 ± 0.1 | 0.4 ± 0.03 | 36 |
| SF-22 | 1.6 ± 0.6 | 1.7 ± 0.6 | 106 |
| SF-31 | 9.0 ± 0.7 | 4.5 ± 0.3 | 50 |
| SF-41 | 1.3 ± 0.3 | 1.5 ± 0.2 | 115 |
| BIIE0246 | 12 ± 2 | 0.2 ± 0.1 | 2 |
Selectivity Profiling Using the NIMH Psychoactive Drug Screening Program.
The selectivity of SF-11, SF-21, SF-22, SF-31, and SF-41 was further assessed by the NIMH PDSP, in which the compounds were screened for binding to receptors (n = 35), ion channels (n = 2), and transporters (n = 3) typically found in the CNS (Fig. 5). Initial screens were conducted in quadruplicate at a final concentration of 10 μM as detailed previously (Armbruster et al., 2007). For comparison, BIIE0246 was also tested; JNJ-5207787 could not be tested because of the lack of availability of this compound. In the initial screen, BIIE0246 had significant activity at 15 targets in the screening panel, whereas the Y2R antagonists discovered in this HTS effort were active at fewer targets. That is, significant radioligand displacement was observed with two targets for SF-11 (the 5-HT2B serotonin receptor and the dopamine transporter), two targets for SF-21 (the 5-HT2B receptor and the dopamine transporter), three targets for SF-22 (the 5-HT2B and 5-HT6 serotonin receptors, as well as the dopamine transporter), and three targets for SF-41 (5-HT2C, 5-HT6, and the κ opioid receptor).
Fig. 5.

Radioligand displacement screening assay results for 40 different receptors, ion channels, and transporters found in the CNS. Measured percentage of activity versus target name is graphed. All compounds were tested at 10 μM. Inhibition greater than the 50% cutoff (dotted line) is considered significant. Negative binding values, obtained when nonspecific binding was higher than specific binding, are reported as 0. Abbreviations are given for serotonin receptor subtypes (5HT1A-7); adrenergic receptor subtypes (α1A-β3); rat brain GABA1 and GABA3 sites (BZP sites); dopamine receptor subtypes: (D2, D4) the dopamine (DAT), norepinephrine (NET), and serotonin (SERT) transporters; prostaglandin receptor subtypes (EP3, EP4); histamine receptor subtypes (H1, H2, H4); muscarinic receptor subtypes (M1–M5); opioid receptor subtypes (MOR, DOR, KOR), and the σ1 receptor.
Dose-response curves were generated where significant displacement was observed, and Ki values were determined (Table 5). BIIE0246 was found to bind three members of the panel with high affinity (defined as Ki < 1 μM). In contrast, only one of the HTS-derived compounds, SF-22, demonstrated comparable potency, binding the 5-HT2B receptor with a Ki value of 250 nM. SF-31 did not significantly displace radioligand from any of the 40 members in the screening panel.
TABLE 5.
Radioligand displacement assay Ki values for BIIE0246 and five Y2R antagonists
Target names correspond to those in Fig. 5.
| Target |
Ki
from PDSP |
|||||
|---|---|---|---|---|---|---|
| BIIE0246 | SF-11 | SF-21 | SF-22 | SF-31 | SF-41 | |
| nM | ||||||
| 5-HT2B | 7093 ± 869 | 1212 ± 161 | 255 ± 26 | |||
| 5-HT2C | 9943 ± 1696 | |||||
| 5-HT5A | 1361 ± 206 | |||||
| 5-HT6 | 2853 ± 419 | 2606 ± 359 | ||||
| α1A | 360 ± 31 | |||||
| α1D | 1294 ± 163 | |||||
| β3 | 1645 ± 160 | |||||
| D2 | 7294 ± 2509 | |||||
| D4 | 3151 ± 256 | |||||
| DAT | 2222 ± 430 | 4989 ± 1047 | 5525 ± 987 | 4005 ± 676 | ||
| H2 | 2979 ± 2979 | |||||
| M2 | 2452 ± 391 | |||||
| M3 | 1573 ± 280 | |||||
| M4 | 2232 ± 358 | |||||
| M5 | 1899 ± 231 | |||||
| MOR | 323 ± 31 | |||||
| DOR | 1132 ± 181 | |||||
| KOR | 948 ± 183 | 1044 ± 57 | ||||
Discussion
In the Y2R screening campaign, all compounds tested in the Y2R cAMP biosensor primary assay were also counterscreened in the Y1R cAMP biosensor primary assay (Fig. 2). We have found this approach useful for rapidly prioritizing selective and potent chemical probes (Madoux et al., 2008). Because both the Y2R and Y1R assays used the same detection technology, the counterscreen was effective at removing nonspecific GPCR agonists and compounds that nonspecifically modulate GPCR signaling. For example, the Gs-coupled adenosine receptor agonist 5′-N-ethylcarboxamidoadenosine (Baraldi et al., 2006) (PubChem SID 7975554) was found active in both the Y1R and Y2R primary screening campaigns. Likewise, the laxative bisacodyl (SID 855868), a known activator of adenylate cyclase (Ratnaike and Jones, 1998), was removed from further consideration.
The elimination of compounds found active at the Y1R during each successive screening stage of the hit-to-lead process resulted in the rapid identification of five selective Y2R antagonists (SF-11, SF-21, SF-22, SF-31, and SF-41), with the most potent, SF-11, antagonizing the Y2R with an IC50 value of 199 nM (Fig. 4A). Although the five HTS antagonists were found to be Y2R-selective in the cAMP biosensor dose-response assays, it was necessary to determine whether their efficacy was a result of displacement of agonist from the NPY receptor. Through the use of competitive binding studies, all five antagonists and BIIE0246 were shown to effectively block 125I-PYY binding to the Y2R. Their rank-order potency was comparable with that found in the Y2R cAMP biosensor functional assay (Fig. 4, A and C). It is interesting that BIIE0246 blocked 125I-PYY binding with a Ki of 0.02 nM, lending further support to the notion that it behaves as an insurmountable antagonist (Dautzenberg and Neysari, 2005). Conversely, all five novel antagonists failed to block 125I-PYY binding to the Y1R as high as 50 μM, confirming functional assay results (Fig. 4D).
Mechanism of action for the five antagonists and for BIIE0246 was investigated via Schild analysis (Arunlakshana and Schild, 1959) (Supplementary Fig. 1). It has been reported that BIIE0246 effectively competes with agonist and functionally inhibits the Y2R (Dautzenberg and Neysari, 2005; Ziemek et al., 2006). However, Schild analysis has not been reported for BIIE0246, and when performed in this study, it resulted in a steep slope (2.79 ± 0.09). Because BIIE0246 is known to be an insurmountable antagonist when administered before Y2R agonists, this high slope suggests a more sophisticated mechanism of action for BIIE0246, such as a binding site for the antagonist that is different from the orthosteric (NPY) site. In contrast, Schild slopes for the five HTS compounds were close to unity. One compound, SF-21, resulted in a slope that is not statistically different from unity (0.98 ± 0.05, P = 0.62), suggesting that it behaves as a true competitive antagonist and binds to the orthosteric site. Although the slope of compound SF-22 is significantly different from unity (1.3 ± 0.1, P < 0.0001), SF-21 and SF-22 are closely related, making it unlikely that they exhibit substantially different mechanisms of action. The three other antagonists exhibited Schild slopes that are close to unity but nonetheless are statistically different (namely 0.79 ± 0.03 and P < 0.0001 for SF-11, 1.55 ± 0.12 and P < 0.0001 for SF-31, and 1.23 ± 0.08 and P = 0.0001 for SF-41).
When assessed in silico, all five HTS antagonists exhibit several physical properties desirable for CNS probes. Compared with drugs that act in the periphery, brain-penetrant drugs tend to be more lipophilic and rigid, have fewer hydrogen bonds, fewer formal charges, and lower polar surface area (van de Waterbeemd et al., 1998; Mahar Doan et al., 2002). Optimal molecular properties for brain penetration have been proposed (van de Waterbeemd et al., 1998), namely that desirable compounds exhibit a logP between 1 and 4, molecular weight less than 450, and a polar surface area of less than 90 Å2. All five Y2R antagonists display molecular properties close to these optimums, with the exception of a higher computed logP value for all but SF-41 (Table 1).
Microsome stability studies were conducted for all five HTS antagonists as a first step toward evaluating the suitability of these compounds for CNS applications in vivo. To estimate the ability of the liver to remove compounds from the blood in the absence of confounding factors, in vitro intrinsic hepatic clearances were calculated from the microsome and protein plasma binding studies (Lipscomb and Poet, 2008). Studies in mouse, rat, and human microsomes (Table 3) showed that across all species, the antagonists exhibited moderate to low stability (in terms of T1/2) compared with BIIE0246. Among the identified Y2R antagonists, SF-21, SF-22, and SF-41 were rapidly metabolized in microsomal incubations, and the predicted in vivo hepatic clearance is high. In contrast, SF-11 was rapidly metabolized by rat and mouse hepatic microsomes but was highly bound to plasma protein (when 1 μM SF-11 was added to mouse plasma, only 30 nM compound was unbound to plasma proteins). High plasma protein binding would be expected to decrease compound diffusion into hepatocytes and thus decrease the rate of elimination. SF-31 was the most stable of the identified antagonists in mouse microsomal incubations and had moderate plasma protein binding. Both SF-11 and SF-31 had predicted hepatic clearance values lower than 10 and favorable hepatic extraction ratios.
Because the Y2R is implicated in neurological and behavioral disorders, mouse blood-brain barrier penetration experiments were conducted on all five HTS compounds and BIIE0246. In consideration of the predicted high hepatic extraction ratio of three of the antagonists, all compounds were dosed by intraperitoneal injection at 10 mg/kg, and compound concentrations were evaluated in plasma and brain at 30 min. In general, intraperitoneal dosing allows the compound to be absorbed over time, and drug levels can be maintained over a short time period because metabolized compound is replenished by additional drug being absorbed from the intraperitoneal space. As the intraperitoneal reservoir is depleted and additional compound cannot be absorbed, drug concentrations can rapidly decrease. Therefore, the 30-min time point was chosen because of the rapid hepatic metabolism predicted from the microsomal studies; continued absorption of compound from the intraperitoneal space maintained a higher plasma concentration and facilitated the assessment of brain exposure.
Brain penetration for all five HTS antagonists was high, signifying their potential for in vivo neurological studies. Brain concentrations determined for all five compounds (0.4–4.5 μM) are comparable with levels achieved by the small molecule JNJ-5207787 (Cmax of 2.6 μM at 30 min) (Bonaventure et al., 2004). Because of the potential for rapid hepatic metabolism, SF-21, SF-22, and SF-41 may not be suitable for rodent in vivo efficacy studies. Although a formal pharmacokinetic study was not conducted for the five compounds, plasma levels measured in the brain penetration experiment after intraperitoneal dosing in mice were consistent with the microsomal stability data.
The five antagonists identified by HTS were evaluated for their ability to displace radioligands from a panel of GPCRs (35), ion channels (2), and transporters (3) found in the CNS. Among these targets were dopamine, serotonin, muscarinic, histamine, adrenergic and opioid receptors (including subtypes), as well as dopamine, serotonin, and norepinephrine transporters, and rat (whole brain) GABA ion channels. BIIE0246, used as a control, displaced radioligand from 15 members of this panel at a test concentration of 10 μM, whereas each of the other compounds tested were active at three or fewer panel members (Fig. 4). In dose-response studies (Table 5), BIIE0246 displaced radioligand from three receptors with submicromolar potency: the α1A adrenergic receptor (Ki = 360 ± 31 nM) and the μ and κ opioid receptors (Ki = 323 ± 31 and 948 ± 183 nM, respectively). In contrast, SF-22 was the only HTS compound that displaced radioligand with appreciable potency (Ki = 255 ± 26 nM for the serotonin 5-HT2B receptor). In summary, the results from selectivity profiling against this CNS panel provide further evidence for these newly discovered compounds as more selective Y2R antagonists than the commonly used Y2R antagonist, BIIE0246.
In an effort to better understand the structural requirements for antagonism and to ascertain whether the four identified chemotypes may be amenable to optimization by medicinal chemistry, 16 close structural analogs were tested for dose-dependent antagonism of Y2R or Y1R. Although none of the analogs antagonized the Y2R as well as the five original HTS compounds, all analogs did exhibit some significant Y2R antagonism (Table 2), demonstrating that structural modification of the different chemotypes is possible without total loss of activity at the Y2R. Off-target antagonism of the closely related Y1R was either insignificant or low. In terms of CC50 values, all 21 compounds exhibited relatively low cytotoxicity after 72 h.
In summary, this report describes the use of cell-based HTS to discover selective Y2R antagonists from a screening library available through the auspices of the National Institutes of Health. The HTS compounds SF-11, SF-21, SF-22, SF-31, and SF-41 are small molecules that penetrate the brain and offer promising chemical probes for the studies of Y2R antagonism both in vitro and in vivo. These compounds are structurally distinct from all previously reported NPY receptor ligands, and Schild analysis reveals a marked difference in the mechanism of action between the peptidomimetic BIIE0246 and two of the five compounds. Furthermore, these compounds have improved brain permeability over BIIE0246 and are comparable with JNJ-5207787. The antagonists display an improved selectivity profile compared with BIIE0246 when tested against a panel of receptors, ion channels, and transporters found in the CNS. For these reasons, SF-11, SF-21, SF-22, SF-31, and SF-41 may prove to be valuable small-molecule probes for further studies into the biology of the Y2R in human diseases such as obesity, alcoholism, and psychiatric disorders. A continued medicinal chemistry effort is currently in progress to further optimize these compounds for CNS studies.
Supplementary Material
Acknowledgments
We thank Pierre Baillargeon and Lina DeLuca (Lead Identification Division, Scripps Research Institute Molecular Screening Center, Scripps Florida) for their assistance with compound management. We thank Dr. Louis Scampavia (Lead Identification Division, Scripps Research Institute Molecular Screening Center, Scripps Florida) for LC-MS analysis of the presented compounds. The helpful discussions with Bryan Roth (University of North Carolina at Chapel Hill) on the interpretation of PDSP binding data are appreciated. The support of Dr. Hugh Rosen (Scripps Research Institute, San Diego, CA) is acknowledged.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant R21-NS056950-01S]; and the National Institutes of Health National Institute of Mental Health [Grant U54-MH084512]. Selectivity binding studies were conducted by the National Institute of Mental Health's Psychoactive Drug Screening Program [Contract N01-MH32004].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.058677.
- NPY
- neuropeptide Y
- Y2R
- neuropeptide Y Y2 receptor
- Y1R
- neuropeptide Y Y1 receptor
- 125I-PPY
- radiolabeled peptide YY
- MLPCN
- Molecular Probe Production Center Network
- MLSMR
- Molecular Libraries Small Molecule Repository
- CNS
- central nervous system
- HTS
- high-throughput screening
- GPCR
- G protein-coupled receptor
- Z′
- Z-factor
- DMSO
- dimethyl sulfoxide
- HEK
- human embryonic kidney
- AID
- bioassay identification number
- PSA
- polar surface area
- 5-HT
- 5-hydroxytryptamine
- NIMH PDSP
- National Institute of Mental Health Psychoactive Drug Screening Program
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- CNG
- cyclic nucleotide-gated
- SID
- substance identification number
- BIIE0246
- N-[(1S)-4-[(aminoiminomethyl)amino]-1-[[[2-(3,5-dioxo-1,2-diphenyl-1,2,4-triazolidin-4-yl)ethyl]amino]carbonyl]butyl]-1-[2-[4-(6,11-dihydro-6-oxo-5H-dibenz[b,e]azepin-11-yl)-1-piperazinyl]-2-oxoethyl]-cyclopentaneacetamide)
- BIBP3226
- (2R)-5-(diaminomethylideneamino)-2-[(2,2-diphenylacetyl)amino]-N-[(4-hydroxyphenyl)methyl]pentanamide
- SF-11
- N-(4-ethoxyphenyl)-4-[hydroxy(diphenyl)methyl]piperidine-1-carbothioamide
- SF-12
- 4-[hydroxy(diphenyl)methyl]-N-(4-methoxyphenyl)piperidine-1-carbothioamide
- SF-13
- N-(4-chlorophenyl)-4-[hydroxy(diphenyl)methyl]piperidine-1-carbothioamide
- SF-14
- N-(3, 5-dimethoxyphenyl)-4-[hydroxy(diphenyl)methyl]piperidine-1-carbothioamide
- SF-15
- 5-dimethoxyphenyl)-4-[hydroxy(diphenyl)methyl]piperidine-1-carbothioamide
- SF-16
- N-(4-fluorophenyl)-4-[hydroxy(diphenyl)methyl]piperidine-1-carbothioamide
- SF-21
- 4-chloro-3-[(2,5-dimethylphenyl)sulfamoyl]-N-(2-phenylphenyl)benzamide
- SF-22
- N-2-biphenylyl-3-{[(2,5-dimethylphenyl)amino]sulfonyl}-4-methylbenzamide
- SF-23
- 4-methyl-N-(2-phenylphenyl)-3-(phenylsulfamoyl)benzamide
- SF-24
- 4-chloro-3-[(2-methylphenyl)sulfamoyl]-N-(2-phenylphenyl)benzamide
- SF-31
- 2-(2-methoxyphenyl)-N-[4-[5-(3-methoxyphenyl)-1,2,4-oxadiazol-3-yl]phenyl]acetamide
- SF-32
- N-[4-[5-(2-ethoxyphenyl)-1,2,4-oxadiazol-3-yl]phenyl]-2-(2-methoxyphenyl)acetamide
- SF-33
- 2-(4-methoxyphenyl)-N-[4-[5-(2-methoxyphenyl)-1,2,4-oxadiazol-3-yl]phenyl]acetamide
- SF-34
- N-[4-[5-(4-fluorophenyl)-1,2,4-oxadiazol-3-yl]phenyl]-2-(2-methoxyphenyl)acetamide
- SF-35
- N-[4-[5-(2-ethoxyphenyl)-1,2,4-oxadiazol-3-yl]phenyl]-2-(4-methoxyphenyl)acetamide
- SF-36
- N-[4-[5-(4-chlorophenyl)-1,2,4-oxadiazol-3-yl]phenyl]-2-(2-methoxyphenyl)acetamide
- SF-37
- 2-(4-methoxyphenyl)-N-[4-[5-(3,4,5-trimethoxyphenyl)-1,2,4-oxadiazol-3-yl]phenyl]acetamide
- SF-38
- 2-(4-methoxyphenyl)-N-[4-[5-(3-methoxyphenyl)-1,2,4-oxadiazol-3-yl]phenyl]acetamide
- SF-39
- N-[4-[5-(4-chlorophenyl)-1,2,4-oxadiazol-3-yl]phenyl]-2-(4-methoxyphenyl)acetamide
- SF-41
- 3,5-dimethyl-4-[(4-methylphenyl)sulfonyl-phenylmethyl]-1,2-oxazole
- SF-42
- 4-[benzenesulfonyl(phenyl)methyl]-3,5-dimethyl-1,2-oxazole
- Ro 20-1724
- 4-[(3-butoxy-4-methoxyphenyl)-methyl]-2-imidazolidinone
- JNJ-5207787
- (N-(1-acetyl-2,3-dihydro-1H-indol-6-yl)-3-(3-cyano-phenyl)-N-[1-(2-cyclopentyl-ethyl)-piperidin-4yl]-acrylamide).
References
- Abbott CR, Small CJ, Kennedy AR, Neary NM, Sajedi A, Ghatei MA, Bloom SR. (2005) Blockade of the neuropeptide Y Y2 receptor with the specific antagonist BIIE0246 attenuates the effect of endogenous and exogenous peptide YY(3-36) on food intake. Brain Res 1043: 139–144 [DOI] [PubMed] [Google Scholar]
- Andres CJ, Antal Zimanyi I, Deshpande MS, Iben LG, Grant-Young K, Mattson GK, Zhai W. (2003) Differentially functionalized diamines as novel ligands for the NPY2 receptor. Bioorg Med Chem Lett 13: 2883–2885 [DOI] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL.(2007)Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A 104: 5163–5168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO. (1959) Some quantitative uses of drug antagonists. Br J Pharmacol Chemother 14: 48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniam AA. (1997) Neuropeptide Y family of hormones: receptor subtypes and antagonists. Peptides 18: 445–457 [DOI] [PubMed] [Google Scholar]
- Baraldi PG, Romagnoli R, Preti D, Fruttarolo F, Carrion MD, Tabrizi MA. (2006) Ligands for A2B adenosine receptor subtype. Curr Med Chem 13: 3467–3482 [DOI] [PubMed] [Google Scholar]
- Bonaventure P, Nepomuceno D, Mazur C, Lord B, Rudolph DA, Jablonowski JA, Carruthers NI, Lovenberg TW. (2004) Characterization of N-(1-acetyl-2,3-dihydro-1H-indol-6-yl)-3-(3-cyano-phenyl)-N-[1-(2-cyclopentyl-ethyl)-piperidin-4yl]acrylamide (JNJ-5207787), a small molecule antagonist of the neuropeptide Y Y2 receptor. J Pharmacol Exp Ther 308: 1130–1137 [DOI] [PubMed] [Google Scholar]
- Brothers SP, Janovick JA, Conn PM. (2006) Calnexin regulated gonadotropin-releasing hormone receptor plasma membrane expression. J Mol Endocrinol 37: 479–488 [DOI] [PubMed] [Google Scholar]
- Catapano LA, Manji HK. (2007) G protein-coupled receptors in major psychiatric disorders. Biochim Biophys Acta 1768: 976–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22: 3099–3108 [DOI] [PubMed] [Google Scholar]
- Dautzenberg FM, Neysari S. (2005) Irreversible binding kinetics of neuropeptide Y ligands to Y2 but not to Y1 and Y5 receptors. Pharmacology 75: 21–29 [DOI] [PubMed] [Google Scholar]
- Doods H, Gaida W, Wieland HA, Dollinger H, Schnorrenberg G, Esser F, Engel W, Eberlein W, Rudolf K. (1999) BIIE0246: a selective and high affinity neuropeptide Y Y2 receptor antagonist. Eur J Pharmacol 384: R3–5 [DOI] [PubMed] [Google Scholar]
- Doucette C, Vedvik K, Koepnick E, Bergsma A, Thomson B, Turek-Etienne TC. (2009) κ opioid receptor screen with the Tango β-arrestin recruitment technology and characterization of hits with second messenger assays. J Biomol Screen 14: 381–394 [DOI] [PubMed] [Google Scholar]
- Eglen RM. (2005) Functional G protein-coupled receptor assays for primary and secondary screening. Comb Chem High Throughput Screen 8: 311–318 [DOI] [PubMed] [Google Scholar]
- Grouzmann E, Buclin T, Martire M, Cannizzaro C, Dörner B, Razaname A, Mutter M. (1997) Characterization of a selective antagonist of neuropeptide Y at the Y2 receptor. Synthesis and pharmacological evaluation of a Y2 antagonist. J Biol Chem 272: 7699–7706 [DOI] [PubMed] [Google Scholar]
- Heilig M. (2004) The NPY system in stress, anxiety and depression. Neuropeptides 38: 213–224 [DOI] [PubMed] [Google Scholar]
- Hodder P, Cassaday J, Peltier R, Berry K, Inglese J, Feuston B, Culberson C, Bleicher L, Cosford ND, Bayly C, et al. (2003) Identification of metabotropic glutamate receptor antagonists using an automated high-throughput screening system. Anal Biochem 313: 246–254 [DOI] [PubMed] [Google Scholar]
- King PJ, Widdowson PS, Doods HN, Williams G. (1999) Regulation of neuropeptide Y release by neuropeptide Y receptor ligands and calcium channel antagonists in hypothalamic slices. J Neurochem 73: 641–646 [DOI] [PubMed] [Google Scholar]
- Lazo JS, Brady LS, Dingledine R. (2007) Building a pharmacological lexicon: small molecule discovery in academia. Mol Pharmacol 72: 1–7 [DOI] [PubMed] [Google Scholar]
- Lipscomb JC, Poet TS. (2008) In vitro measurements of metabolism for application in pharmacokinetic modeling. Pharmacol Ther 118: 82–103 [DOI] [PubMed] [Google Scholar]
- Madoux F, Li X, Chase P, Zastrow G, Cameron MD, Conkright JJ, Griffin PR, Thacher S, Hodder P. (2008) Potent, selective and cell penetrant inhibitors of SF-1 by functional ultra-high-throughput screening. Mol Pharmacol 73: 1776–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahar Doan KM, Humphreys JE, Webster LO, Wring SA, Shampine LJ, Serabjit-Singh CJ, Adkison KK, Polli JW. (2002) Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther 303: 1029–1037 [DOI] [PubMed] [Google Scholar]
- Milligan G, Feng GJ, Ward RJ, Sartania N, Ramsay D, McLean AJ, Carrillo JJ. (2004) G protein-coupled receptor fusion proteins in drug discovery. Curr Pharm Des 10: 1989–2001 [DOI] [PubMed] [Google Scholar]
- Mottagui-Tabar S, Prince JA, Wahlestedt C, Zhu G, Goldman D, Heilig M. (2005) A novel single nucleotide polymorphism of the neuropeptide Y (NPY) gene associated with alcohol dependence. Alcohol Clin Exp Res 29: 702–707 [DOI] [PubMed] [Google Scholar]
- Nakai D, Kumamoto K, Sakikawa C, Kosaka T, Tokui T. (2004) Evaluation of the protein binding ratio of drugs by a micro-scale ultracentrifugation method. J Pharm Sci 93: 847–854 [DOI] [PubMed] [Google Scholar]
- Nikisch G, Agren H, Eap CB, Czernik A, Baumann P, Mathé AA. (2005) Neuropeptide Y and corticotropin-releasing hormone in CSF mark response to antidepressive treatment with citalopram. Int J Neuropsychopharmacol 8: 403–410 [DOI] [PubMed] [Google Scholar]
- Ratnaike RN, Jones TE. (1998) Mechanisms of drug-induced diarrhoea in the elderly. Drugs Aging 13: 245–253 [DOI] [PubMed] [Google Scholar]
- Redrobe JP, Dumont Y, Herzog H, Quirion R. (2003) Neuropeptide Y (NPY) Y2 receptors mediate behaviour in two animal models of anxiety: evidence from Y2 receptor knockout mice. Behav Brain Res 141: 251–255 [DOI] [PubMed] [Google Scholar]
- Rimondini R, Thorsell A, Heilig M. (2005) Suppression of ethanol self-administration by the neuropeptide Y (NPY) Y2 receptor antagonist BIIE0246: evidence for sensitization in rats with a history of dependence. Neurosci Lett 375: 129–133 [DOI] [PubMed] [Google Scholar]
- Rose PM, Fernandes P, Lynch JS, Frazier ST, Fisher SM, Kodukula K, Kienzle B, Seethala R. (1995) Cloning and functional expression of a cDNA encoding a human type 2 neuropeptide Y receptor. J Biol Chem 270: 22661–22664 [DOI] [PubMed] [Google Scholar]
- Tatemoto K.(1982)Neuropeptide Y: complete amino acid sequence of the brain peptide. Proc Natl Acad Sci U S A 79: 5485–5489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsell A, Slawecki CJ, El Khoury A, Mathe AA, Ehlers CL. (2006) The effects of social isolation on neuropeptide Y levels, exploratory and anxiety-related behaviors in rats. Pharmacol Biochem Behav 83: 28–34 [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd H, Camenisch G, Folkers G, Chretien JR, Raevsky OA. (1998) Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J Drug Target 6: 151–165 [DOI] [PubMed] [Google Scholar]
- Visegrády A, Boros A, Némethy Z, Kiss B, Keseru GM. (2007) Application of the BD ACTOne technology for the high-throughput screening of Gs-coupled receptor antagonists. J Biomol Screen 12: 1068–1073 [DOI] [PubMed] [Google Scholar]
- Wahlestedt C, Ekman R, Widerlöv E. (1989) Neuropeptide Y (NPY) and the central nervous system: distribution effects and possible relationship to neurological and psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry 13: 31–54 [DOI] [PubMed] [Google Scholar]
- Wahlestedt C, Yanaihara N, Håkanson R. (1986) Evidence for different pre- and post-junctional receptors for neuropeptide Y and related peptides. Regul Pept 13: 307–318 [DOI] [PubMed] [Google Scholar]
- Widerlöv E, Lindström LH, Wahlestedt C, Ekman R. (1988) Neuropeptide Y and peptide YY as possible cerebrospinal fluid markers for major depression and schizophrenia, respectively. J Psychiatr Res 22: 69–79 [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4: 67–73 [DOI] [PubMed] [Google Scholar]
- Zhu G, Pollak L, Mottagui-Tabar S, Wahlestedt C, Taubman J, Virkkunen M, Goldman D, Heilig M. (2003) NPY Leu7Pro and alcohol dependence in Finnish and Swedish populations. Alcohol Clin Exp Res 27: 19–24 [DOI] [PubMed] [Google Scholar]
- Ziemek R, Brennauer A, Schneider E, Cabrele C, Beck-Sickinger AG, Bernhardt G, Buschauer A. (2006) Fluorescence- and luminescence-based methods for the determination of affinity and activity of neuropeptide Y2 receptor ligands. Eur J Pharmacol 551: 10–18 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}