

Abstract
Considerable evidence indicates that the basolateral complex of the amygdala (BLA) interacts with efferent brain regions in mediating glucocorticoid effects on memory consolidation. Here, we investigated whether glucocorticoid influences on the consolidation of memory for emotionally arousing training depend on functional interactions between the BLA and the medial prefrontal cortex (mPFC), a brain region involved in higher-order cognitive and affective processing. The glucocorticoid receptor (GR) agonist 11β,17β-dihydroxy-6,21-dimethyl-17α-pregna-4,6-trien-20yn-3-one (RU 28362) administered unilaterally into the left mPFC of male Sprague Dawley rats immediately after inhibitory avoidance training enhanced 48 h retention performance. An ipsilateral, but not contralateral, lesion of the BLA blocked the memory enhancement. In a second experiment, RU 28362 infused into the mPFC after inhibitory avoidance training increased BLA levels of phosphorylated extracellular signal-regulated kinase 1/2 (pErk1/2). Blockade of this pErk1/2 activity in the BLA with the mitogen-activated protein kinase kinase inhibitor PD98059 [2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one] prevented the memory enhancement, suggesting that GR agonist administration into the mPFC enhances memory consolidation via modulation of BLA activity. Conversely, GR agonist infusions administered into the BLA posttraining increased pErk1/2 levels in the mPFC in regulating memory consolidation. Moreover, as assessed with a two-phase inhibitory avoidance procedure designed to separate modulatory influences on memory of context and footshock, posttraining GR agonist infusions into either the BLA or mPFC enhanced memory of the contextual as well as aversive information acquired during inhibitory avoidance training. These findings indicate that glucocorticoid effects on memory consolidation depend on bidirectional interactions between the BLA and mPFC.
Introduction
It is well established that glucocorticoid hormones, released from the adrenal cortex in response to stressful stimuli, strengthen the consolidation of long-term memories of emotionally arousing experiences (de Kloet et al., 1999; Roozendaal, 2000; McGaugh and Roozendaal, 2002; Sandi and Pinelo-Nava, 2007). The basolateral complex of the amygdala (BLA), which has a moderate density of glucocorticoid receptors (GRs), is crucially involved in mediating glucocorticoid effects on memory consolidation. A GR agonist infused into the BLA after training enhances memory consolidation (Roozendaal and McGaugh, 1997b; Roozendaal et al., 2002), whereas lesions or temporary inactivation of the BLA block the memory-enhancing effect of systemically administered glucocorticoids (Roozendaal and McGaugh, 1996; Roozendaal et al., 1996; Quirarte et al., 1997). Glucocorticoid-induced amygdala activation is known to enhance memories of emotionally arousing training experiences via efferent projections that activate many other brain regions, including the hippocampus, dorsal striatum, and neocortical regions (Roozendaal and McGaugh, 1997a; Dolcos et al., 2004; McIntyre et al., 2005; Huff et al., 2006; Tsoory et al., 2008).
A growing body of evidence indicates that BLA neuronal activity is closely regulated by the medial prefrontal cortex (mPFC), a brain region involved in higher-order cognitive and affective processing as well as executive function (Davidson, 2002; Quirk and Gehlert, 2003). The evidence suggests that there are reciprocal connections between the mPFC and BLA (Pérez-Jaranay and Vives, 1991; Rosenkranz and Grace, 2002; Likhtik et al., 2005) that regulate affect and memory (Garcia et al., 1999; Milad and Quirk, 2002; Laviolette et al., 2005; Laviolette and Grace, 2006). Maintenance of mPFC activity is known to constrain BLA activity, whereas stress and glucocorticoids alter mPFC functioning, thereby increasing BLA responses to emotionally arousing stimuli (Lyons et al., 2000; Amat et al., 2005). Like the BLA, mPFC functioning is known to influence the consolidation of memory of emotionally arousing training (Runyan and Dash, 2004; Akirav and Maroun, 2006; Izquierdo et al., 2007). However, previous studies have not investigated whether the mPFC and BLA interact in regulating memory consolidation. Moreover, previous experiments have not investigated whether the mPFC is a locus of glucocorticoid action in regulating memory consolidation.
To address these issues, a first experiment examined whether the specific GR agonist 11β,17β-dihydroxy-6,21-dimethyl-17α-pregna-4,6-trien-20yn-3-one (RU 28362) administered into the mPFC immediately after emotionally arousing inhibitory avoidance training enhances memory consolidation and whether such memory enhancement requires an intact BLA. The next experiment investigated whether posttraining GR agonist infusions into the mPFC increase BLA activity and whether such BLA activation is required for mediating the memory-modulatory effects of intra-mPFC GR agonist administration. BLA activity was assessed by determining phosphorylation levels of extracellular signal-regulated kinase 1/2 (pErk1/2), a signaling cascade implicated in memory consolidation (Duvarci et al., 2005; Merino and Maren, 2006). Because of the strong reciprocal interaction between these brain regions, the experiments also examined whether the well known memory enhancement induced by GR agonist infusions into the BLA depends on concurrent changes in pErk1/2 activity levels within the mPFC. A final experiment investigated whether RU 28362 infused into either the mPFC or BLA induces comparable effects on memory of the contextual and aversive information acquired during inhibitory avoidance training.
Materials and Methods
Subjects.
Adult male Sprague Dawley rats (270–320 g at time of surgery) from Charles River Breeding Laboratories were housed individually in a temperature-controlled (22°C) vivarium room and maintained on a standard 12 h light/dark cycle (7:00 A.M. to 7:00 P.M. lights on). Food and water were available ad libitum. Training and testing were performed during the light phase of the cycle between 10:00 A.M. and 3:00 P.M., at the rat nadir of the circadian cycle for corticosterone. All experimental procedures were in compliance with the National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
Surgery.
Animals were adapted to the vivarium for at least 1 week before surgery. They were anesthetized with sodium pentobarbital (50 mg/kg of body weight, i.p.), given atropine sulfate (0.4 mg/kg, i.p.) to maintain respiration, and were subsequently injected with 3 ml of saline to facilitate clearance of these drugs and prevent dehydration. The skull was positioned in a stereotaxic frame (David Kopf Instruments), and either unilateral (left hemisphere) or bilateral stainless-steel guide cannulae (23 gauge) were implanted with the cannula tips 1.5 mm above the prelimbic region of the mPFC [11 mm; coordinates: anteroposterior (AP), +3.7 mm from bregma; mediolateral (ML), ±0.7 mm from midline; dorsoventral (DV), −2.4 mm from skull surface] or 2 mm above the BLA [15 mm; coordinates: AP, −2.8 mm; ML, ±5.0 mm; DV, −6.5 mm (Paxinos and Watson, 2005)]. The cannulae were fixed to the skull with two anchoring screws and dental cement. Stylets (11- or 15-mm-long 00-insect dissection pins) inserted into each cannula to maintain patency were removed only for drug infusions. Other groups of rats received a unilateral neurotoxic lesion of either the ipsilateral or contralateral BLA induced by NMDA (Sigma-Aldrich; 1.25 mg per 100 μl of phosphate buffer, pH 7.4) in addition to a unilateral cannula in the left mPFC. The NMDA solution was backfilled into a 30 gauge needle, which was attached by a polyethylene tube to a 10 μl Hamilton microsyringe driven by a minipump (Sage Instruments). The needle tip was inserted into the BLA (coordinates: AP, −2.8 mm; ML, ±5.0 mm; DV, −8.6 mm), and NMDA (2.5 μg per 0.2 μl of phosphate buffer) was infused over a 25 s period. The injection needle was retained in place for an additional 3 min to optimize diffusion. Sham operations used the same procedure except that the needle was lowered only to the level of the caudate/putamen (coordinates: AP, −2.8 mm; ML, ±5.0 mm; DV, −6.5 mm) and removed after 3 min without infusion. After surgery, the rats were retained in an incubator until recovered from anesthesia and were then returned to their home cages. Rats were allowed to recover from surgery for 7 d before initiation of training.
Inhibitory avoidance apparatus and procedures.
For all experiments, the rats were trained in an inhibitory avoidance apparatus consisting of a trough-shaped alley (91 cm long, 15 cm deep, 20 cm wide at the top, and 6.4 cm wide at the bottom) divided into two compartments, separated by a sliding door that opened by retracting into the floor (McGaugh et al., 1988). The starting compartment (31 cm) was made of opaque white plastic and well lit; the shock compartment (60 cm) was made of dark, electrifiable metal plates and was not illuminated. Training and testing were conducted in a sound- and light-attenuated room.
For one-trial inhibitory avoidance training, the rats were placed in the starting compartment of the apparatus, facing away from the door, and were allowed to enter the dark (shock) compartment. After the rat stepped completely into the dark compartment, the sliding door was closed and a single inescapable footshock (0.35 mA; 1 s) was delivered. The rats were removed from the shock compartment 15 s later and, after drug treatment, returned to their home cages. For the modified, two-phase inhibitory avoidance procedure (Malin and McGaugh, 2006), on the first day (context training), the rat was placed into the starting compartment and allowed to freely explore the inhibitory avoidance apparatus for 3 min. On day 2 (shock training), each rat was placed into the dark compartment, facing away from the starting compartment, with the retractable door closed. The rat then received an inescapable footshock and immediately afterward was removed from the training apparatus. Rats of the mPFC experiment received a footshock intensity of 0.7 mA for 1 s. As the vehicle contained 1% ethanol, which is slightly impairing when infused into the BLA, the BLA-cannulated rats received a somewhat higher footshock intensity of 1.0 mA for 2 s to ensure memory in all experimental groups. For both one-trial and two-phase inhibitory avoidance, retention was tested 48 h after training by placing the rat into the starting compartment of the inhibitory avoidance apparatus and measuring the latency to enter the former shock compartment with all four paws (maximum latency of 600 s). Longer latencies were interpreted as indicating better retention. Shock was not administered on the retention test trial.
Drug treatment.
The specific GR agonist RU 28362 (Roussel Uclaf) was administered into either the mPFC (3 or 10 ng in 0.5 μl) or BLA (1 or 3 ng in 0.2 μl). Receptor binding studies have shown that this compound has selective and high affinity for GRs (Teutsch et al., 1981). RU 28362 was first dissolved in 100% ethanol and subsequently diluted in 0.9% saline to reach a final ethanol concentration of 1%. Bilateral infusions of RU 28362 or an equivalent volume of vehicle (1% ethanol in saline) into these brain regions were given immediately after inhibitory avoidance training by using a 30 gauge injection needle connected to a 10 μl Hamilton microsyringe with polyethylene (PE-20) tubing. The use of posttraining drug administration provides direct support for the view that the treatment affects memory consolidation processes and that the retention performance is, thus, not confounded by possible effects on attentional, motivational, or sensory-perceptual mechanisms at the time of training or test (McGaugh, 1966). For mPFC infusions, the injection needle protruded 1.5 mm beyond the tip of the cannula and a 0.5 μl injection volume was infused over a period of 35 s by an automated syringe pump (Sage Instruments). For BLA infusions, the injection needle protruded 2.0 mm beyond the cannula tip and a 0.2 μl injection volume was infused during 25 s. The injection needles were retained within the cannulae for an additional 20 s after drug infusion to maximize diffusion and to prevent backflow of drug into the cannulae. For some experiments, the GR agonist was administered unilaterally into either the mPFC or BLA together with the specific mitogen-activated protein kinase kinase (MEK) inhibitor 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98059) into the other brain region immediately after inhibitory avoidance training. PD98059 acts by binding to the inactivated form of MEK, thereby preventing its phosphorylation by MEK kinase. PD98059 (Tocris Bioscience) was first dissolved in 100% DMSO and subsequently diluted in 0.9% saline to reach a final DMSO concentration of 6%. The vehicle contained 6% DMSO in saline only. A total of 20 ng of PD98059 in 0.2 μl was infused into the left BLA or 50 ng in 0.5 μl into the left mPFC, using procedures as described above. The infusion volume for the BLA was based on extensive previous evidence that this volume of an excitotoxin administered at an identical injection site induces selective lesions of the BLA (Roozendaal and McGaugh, 1996, 1997a; current data). Furthermore, drug infusions of this volume into the BLA, but not into the adjacent central nucleus of the amygdala, modulate memory consolidation (Roozendaal and McGaugh, 1997b; Roozendaal et al., 2007; Campolongo et al., 2009). The infusion volume for the mPFC was based on findings that infusions of this volume administered into the rostral anterior cingulate cortex, located above the mPFC, produce different effects on the two-phase inhibitory avoidance task (Malin and McGaugh, 2006).
Histology.
The rats were deeply anesthetized with an overdose of sodium pentobarbital and perfused transcardially with 0.9% saline followed by 4% formaldehyde solution (w/v). After decapitation, the brains were removed and immersed in fresh 4% formaldehyde. At least 24 h before sectioning, the brains were transferred to a 20% sucrose (w/v) solution for cryoprotection. Coronal sections of 50 μm were cut on a freezing microtome, mounted on gelatin-coated slides, and stained with cresyl violet. The sections were examined under a light microscope and the location of injection needle tips in the mPFC and BLA or lesions of the BLA were determined according to the standardized atlas plates of Paxinos and Watson (2005) by an observer blind to drug treatment condition. For all experiments, only rats with needle tips within the boundaries of the mPFC or BLA were included in the data analysis. Approximately 14% of the animals were excluded from analysis because of either cannula misplacement or damage to the targeted tissue.
Western blotting for p-Erk1/2 and mitogen-activated protein kinase.
Fifteen minutes after training and immediate posttraining drug treatment, rats were deeply anesthetized with sodium pentobarbital and brains were rapidly removed and flash frozen by submersion for 2 min in a beaker of 2-methylbutane sitting in a dry ice–ethanol bath. Tissue punches (1 mm in diameter) were taken from either the BLA or mPFC, bilaterally, with a glass pipette. Tissue punches were sonicated in 0.1 m phosphate buffer, pH 7.4 [containing 2% SDS, 10% glycerol, 20 μm leupeptin, 0.1 mm TLCK (N-α-p-tosyl-l-lysine chloromethyl ketone), NS 1 mm PMSF]. Protein concentrations were determined with a microplate reader and Pierce protein assay kit. Approximately 15 μg of protein from each sample was heated in sample buffer with reducing agent (Bio-Rad), loaded, and run on 12% Tris-HCl gels (Bio-Rad). Samples from left and right BLA or mPFC of an individual animal were loaded in adjacent wells. Gels were then electroblotted to nitrocellulose membranes. Membranes were treated as previously described (Sharma and Carew, 2002). Membranes were first processed to detect phosphorylated p44/42 mitogen-activated protein (MAP) kinase (pErk1/2) immunoreactivity and then stripped and reprobed with antibody to total MAP kinase (1:1000 and 1:2000, respectively; Cell Signaling Technology). Bio-Rad markers were run on all gels to determine the relative mobility of the immunoreactive bands. For quantification of immunoblot results, films were scanned and converted into TIF files for analysis using Scion Image software. Phosphorylated Erk1/2 immunoreactivity levels were normalized to the total MAP kinase immunoreactivity value for each sample (Patterson et al., 2001; Sharma et al., 2003).
Immunohistochemistry.
Fifteen minutes after training and immediate posttraining drug treatment, rats were deeply anesthetized with pentobarbital and fixed by transcardial perfusion of 4% paraformaldehyde. Brains were postfixed for 24 h at room temperature and then kept in 0.1 m Tris-buffered saline (TBS) at 4°C. Twenty-five micrometer sections were cut from the relevant brain area using a cryostat and transferred into TBS with 0.2% Triton X-100. Free-floating sections were incubated for 2 h in a blocking solution containing 10% of the appropriate normal serum in TBS with phosphatase inhibitors (20 mm sodium fluoride and 2 mm sodium orthovanadate), after which they were incubated overnight at 4°C with rabbit anti-phospho-p44/42 MAP kinase (pErk1/2) as primary antibody overnight (1:1000; Cell Signaling). The sections were then incubated for 2 h with biotinylated goat anti-rabbit IgG (1:400; Jackson ImmunoResearch Laboratories) and visualized using ABC kits (Vector Laboratories) according to the manufacturer's instructions.
Statistics.
Inhibitory avoidance training and retention latencies were analyzed with one- or two-way ANOVAs. The source of the detected significances was determined by Fisher's post hoc tests. To determine whether learning had occurred, paired t tests were used to compare the training and retention latencies of the vehicle groups. Western blot data were expressed as ratio of normalized pErk1/2 across hemispheres (ipsilateral to infusion/contralateral to infusion), and ratios were compared across groups with one-way ANOVAs. Data are expressed as mean ± SEM. Values of p < 0.05 were considered statistically significant. The number of rats per group is indicated in the figure legends.
Results
Ipsilateral, but not contralateral, lesions of the BLA block inhibitory avoidance retention enhancement induced by posttraining intra-mPFC infusions of a GR agonist
This experiment examined whether immediate posttraining infusions of a GR agonist administered into the mPFC would enhance 48 h retention performance of inhibitory avoidance training and whether this retention enhancement depends on an intact BLA. To address this issue, unilateral infusions of the GR agonist RU 28362 (3 or 10 ng) or vehicle were administered into the left mPFC immediately after inhibitory avoidance training to rats with an NMDA-induced lesion of either the ipsilateral or contralateral BLA.
Average step-through latencies for all groups during training (i.e., before footshock or drug treatment) were 12.6 + 0.6 s (mean + SEM). In rats with lesions or sham lesions of the left BLA, a two-way ANOVA for entrance latencies during training showed no BLA lesion effect (F(1,62) = 0.15; p = 0.70). Also, rats in different posttraining drug groups did not differ in their latencies during training (F(2,62) = 0.88; p = 0.42). In rats with lesions or sham lesions of the right BLA, there were also no significant lesion (F(1,59) = 0.04; p = 0.84) or drug group effects (F(2,59) = 0.77; p = 0.49) (data not shown).
Lesions of the ipsilateral BLA blocked the memory-enhancing effect of posttraining intra-mPFC infusions of RU 28362. Figure 1A shows 48 h retention latencies of rats with sham- or NMDA-induced lesions of the left BLA given posttraining infusions of RU 28362 into the left mPFC. Retention latencies of sham-lesioned rats treated with vehicle were significantly longer than their entrance latencies during training (paired t test, p < 0.001), indicating memory for the training experience. Two-way ANOVA for retention latencies showed no significant RU 28362 effect (F(2,62) = 1.78; p = 0.17) but did show a significant lesion (F(1,62) = 14.01; p < 0.0005) and interaction effect between both factors (F(2,62) = 3.27; p < 0.05). Additional analysis with Fisher's post hoc tests showed that, in sham-lesioned rats, posttraining infusions of the higher dose of RU 28362 (10 ng) enhanced retention (p < 0.05, compared with vehicle), whereas retention latencies of animals given the lower dose (3 ng) approached, but failed to reach, significance (p = 0.06). A lesion of the BLA made ipsilaterally to the mPFC infusion side alone did not affect retention (p = 0.71), but blocked the retention-enhancing effect of RU 28362 infused into the mPFC. Retention latencies of BLA-lesioned rats given either dose of RU 28362 were significantly shorter than those of corresponding sham-lesioned rats (both, p < 0.05).
Figure 1.

Ipsilateral, but not contralateral, lesions of the BLA block inhibitory avoidance retention enhancement induced by posttraining intra-mPFC infusions of a GR agonist. Step-through latencies (mean + SEM) in seconds on the 48 h inhibitory avoidance retention test of rats with unilateral sham- or NMDA-induced lesions of the BLA given the GR agonist RU 28362 (3 or 10 ng in 0.5 μl) into the left mPFC immediately after training. A, Ipsilateral BLA lesion. B, Contralateral BLA lesion. ✷p < 0.05; ✷✷p < 0.01 compared with the corresponding vehicle group; ♦p < 0.05 compared with the corresponding sham lesion–RU 28362 group (n = 8–14 per group). C, Injection needle tips in the mPFC of 20 randomly selected sham-lesioned (black circles) and 20 BLA-lesioned rats (black diamonds) included in the experiment. D, Smallest (black area) and largest (gray area) ipsilateral and contralateral BLA lesions from rats used in the analysis. Adapted from Paxinos and Watson (2005).
In contrast, a lesion of the contralateral BLA did not block the retention enhancement induced by immediate posttraining intra-mPFC administration of RU 28362 (Fig. 1B). Two-way ANOVA for 48 h retention latencies showed a significant RU 28362 effect (F(2,59) = 7.85; p < 0.001), but no lesion effect (F(1,59) = 0.10; p = 0.75) or interaction effect between both factors (F(2,59) = 0.01; p = 0.99). Administration of the higher dose of RU 28362 (10 ng) into the mPFC enhanced retention in both sham-lesioned (p < 0.05) and BLA-lesioned rats (p < 0.01).
Cannula placement in the mPFC is shown in Figure 1C. All injection needle tips of rats included in the analysis were localized in the prelimbic region of the mPFC. A total of 10 rats with improper cannula placement or extensive tissue damage at the injection needle tip site was excluded from additional analysis. Histological examination of the lesioned BLA indicated that the lesioned area was characterized by pyknosis and loss of neurons, accompanied by extensive gliosis. In several animals, the BLA lesion was not complete, leaving most of the anterior part intact, but typically 50% or more of the nucleus was damaged. Thirteen rats with improper lesions of either the ipsilateral or contralateral BLA were excluded from analysis. In some animals, whose data were included in the analyses, minor damage was seen in the cortex adjacent to the BLA but the central nucleus of the amygdala was intact in all animals. Figure 1D shows the extent of minimum and maximum BLA lesions of rats included in the experiment.
GR agonist infusions into the mPFC after inhibitory avoidance training increase pErk1/2 levels in the BLA
As indicated above, manipulation of mPFC activity can alter BLA neuronal activity (Rosenkranz and Grace, 2002; Likhtik et al., 2005). And, as the BLA is critically involved in modulating memory consolidation, it seems possible that manipulation of GR activity in the mPFC after inhibitory avoidance training enhanced memory consolidation via influencing BLA activity. To examine whether posttraining infusions of RU 28362 into the mPFC modulate BLA activity, pErk1/2 levels in the BLA were assessed in rats given vehicle or RU 28362 (3 or 10 ng in 0.5 μl) into the left mPFC immediately after inhibitory avoidance training. As the findings of the previous experiment indicate that the GR agonist infused into the mPFC was only blocked by an ipsilateral, and not contralateral BLA lesion, we compared pErk1/2 levels, normalized to total MAP kinase levels (Patterson et al., 2001; Sharma et al., 2003), in the BLA of the stimulated side with that of the nonstimulated control side. Figure 2A shows the ratio of pErk1/2 expression in the BLA across the two hemispheres. One-way ANOVA revealed a significant difference across groups (F(2,24) = 5.02; p < 0.05). For rats given infusions of RU 28362 into the mPFC after inhibitory avoidance training, the ratio of pErk1/2 protein levels in the BLA between the ipsilateral and contralateral side was significantly greater than that seen in rats receiving vehicle infusions (3 ng, p < 0.05; 10 ng, p < 0.01). Importantly, pErk1/2 levels in the BLA did not increase after infusions of RU 28362 into the mPFC of control rats that were not trained on the inhibitory avoidance task (F(2,14) = 1.59; p = 0.24). Figure 2B shows a representative example of an immunoblot illustrating pErk1/2 and total MAP kinase levels in the ipsilateral and contralateral BLA of the different experimental groups.
Figure 2.

GR agonist RU 28362 (3 or 10 ng in 0.5 μl) infusions into the mPFC after inhibitory avoidance training increase pErk1/2 levels in the BLA. A, Mean ratio (+SEM) of pErk1/2 in the BLA ipsilateral to mPFC infusions versus pErk1/2 measured in the contralateral BLA is significantly greater in rats administered RU 28362 than the ratio of the comparison across two hemispheres of vehicle-treated rats. ✷p < 0.05; ✷✷p < 0.01 (n = 6–11 per group). No significant differences were observed across hemispheres in rats that were not trained on the inhibitory avoidance task (n = 5–6 per group). B, Density of pErk1/2 bands was normalized to density of total MAP kinase bands on the same blot. Left band is ipsilateral (i) to mPFC infusion, and right is contralateral (c). C, Immunohistochemical demonstration of location of pErk1/2 within the amygdala. Positive cells were present in the BLA as well as in the central part of the lateral division of the central nucleus (CEA).
Some animals were processed for immunocytochemistry to qualitatively confirm the Western blot data and to determine the location of increased pErk1/2 activity within the BLA. As is shown in Figure 2C, posttraining infusions of RU 28362 (10 ng) into the mPFC increased the density of pErk1/2-positive neurons scattered throughout the BLA. The BLA of the vehicle-infused control group was almost devoid of pErk1/2-positive cells. Furthermore, and even more pronounced, more pErk1/2-immunoreactive cells were present in the central part of the lateral division of the central nucleus. The pErk1/2-positive cells represent the multipolar principal cells of the amygdalar subnuclei. Increased density of pErk1/2 cells was also found in the medial amygdalar nucleus, whereas the other subnuclei of the amygdala revealed pErk1/2-stained cells only occasionally. The intercalated mass was characterized by moderate levels of pErk1/2-positive neurons (data not shown). Other brain regions did not show such an increase in pErk1/2 immunoreactivity, illustrating the brain region-specific nature of the GR agonist-induced pErk1/2 changes.
A MEK inhibitor infused into the BLA blocks retention enhancement induced by posttraining infusion of a GR agonist into the mPFC
If the increased pErk1/2 expression in the BLA plays a role in mediating the memory-enhancing effect of RU 28362 infused into the mPFC, then a blockade of pErk1/2 activity in the BLA should prevent the retention enhancement induced by immediate posttraining intra-mPFC administration of the GR agonist. To investigate this issue, rats were given unilateral infusions of the specific MEK inhibitor PD98059 (20 ng in 0.2 μl) or vehicle into the left BLA together with the GR agonist RU 28362 (3 or 10 ng) or vehicle into the ipsilateral mPFC immediately after inhibitory avoidance training. Two-way ANOVA for entrance latencies on the training trial, before footshock and drug treatment, did not show significant differences between groups (p > 0.08 for all comparisons) (data not shown). As shown in Figure 3A, two-way ANOVA for retention latencies revealed a significant RU 28362 effect (F(2,62) = 3.53; p < 0.05), a significant PD98059 effect (F(1,62) = 7.62; p < 0.01), as well as a significant interaction between both factors (F(2,62) = 5.07; p < 0.01). Posttraining infusions of the higher dose of RU 28362 (10 ng) into the mPFC enhanced retention of rats given vehicle into the BLA, compared with the retention of corresponding vehicle-treated rats (p < 0.01). Infusion of a low dose of the MEK inhibitor into the ipsilateral BLA did not impair retention latencies when administered alone (p = 0.34) but blocked the retention-enhancing effect of RU 28362 administered into the mPFC. Retention latencies of RU 28362-treated rats given PD98059 into the BLA were significantly shorter than those of RU 28362-treated rats given vehicle into the BLA (10 ng, p < 0.01).
Figure 3.
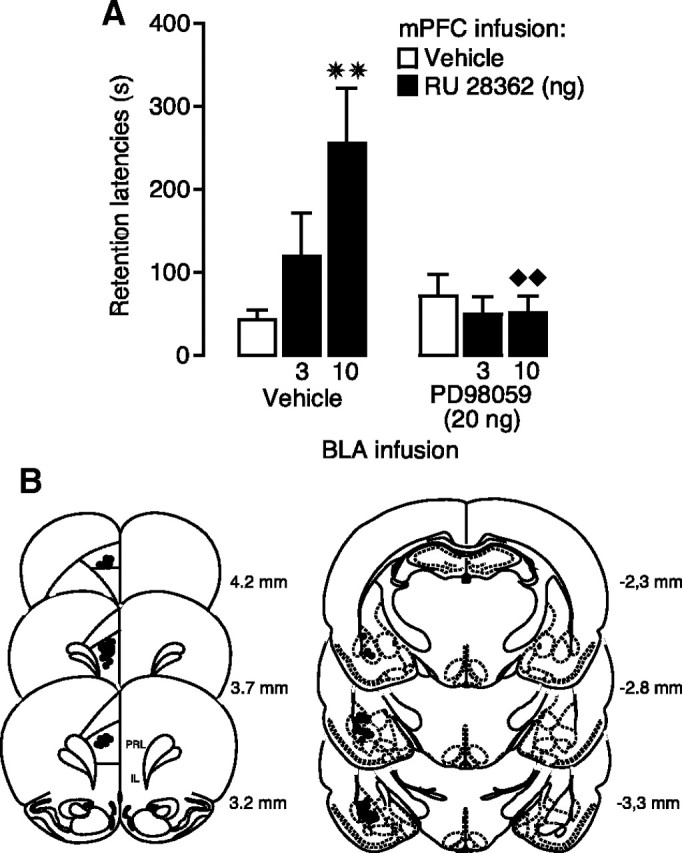
A MEK inhibitor infused into the BLA blocks inhibitory avoidance retention enhancement induced by posttraining GR agonist infusions into the mPFC. A, Step-through latencies (mean + SEM) in seconds on the 48 h inhibitory avoidance retention test of rats given immediate posttraining infusions of the GR agonist RU 28362 (3 or 10 ng in 0.5 μl) into the left mPFC either alone or together with the MEK inhibitor PD98059 (20 ng in 0.2 μl) into the ipsilateral BLA. ✷✷p < 0.01 compared with the corresponding vehicle group; ♦♦p < 0.01 compared with the corresponding saline–RU 28362 group (n = 10–13 per group). B, Injection needle tips in the left mPFC and BLA of 30 randomly selected rats included in the experiment.
Figure 3B shows the location of infusion needle tips in the left mPFC and BLA of 30 randomly selected animals.
GR agonist infusions into the BLA after inhibitory avoidance training increase pErk1/2 levels in the mPFC
In the first experiments above, we found that immediate posttraining manipulation of GR activity in the mPFC with the receptor agonist RU 28362 enhanced 48 h inhibitory avoidance retention performance via an influence involving the BLA. However, as the BLA is also known to project to the mPFC, it is possible that the well known memory enhancement induced by GR agonist infusions into the BLA may also depend on concurrent changes induced in the mPFC. To examine whether posttraining infusions of RU 28362 into the BLA increase pErk1/2 activity in the mPFC, RU 28362 (1 or 3 ng) or vehicle was infused into the left BLA immediately after inhibitory avoidance training and rats were killed 15 min later for quantification of pErk1/2 levels in the ipsilateral and contralateral mPFC. Figure 4A illustrates the ratio of pErk1/2 protein levels, normalized to total MAP kinase, in the mPFC across the two hemispheres. One-way ANOVA revealed a significant difference across groups (F(2,19) = 6.04; p < 0.01). For rats given either dose of RU 28362 into the left BLA, the ratio of pErk1/2 protein levels in the mPFC between the ipsilateral and contralateral side was significantly greater than that in rats administered vehicle (both doses, p < 0.01). In contrast, in rats that were not trained on the inhibitory avoidance task, pErk1/2 levels in the mPFC did not increase after infusions of RU 28362 into the BLA (F(2,12) = 0.18; p = 0.84). Figure 4B shows a representative example of an immunoblot illustrating pErk1/2 and total MAP kinase levels in the ipsilateral and contralateral mPFC of the different experimental groups.
Figure 4.

Infusions of the GR agonist RU 28362 (1 or 3 ng in 0.2 μl) into the BLA after inhibitory avoidance training increase pErk1/2 levels in the mPFC. A, Mean ratio (+SEM) of pErk1/2 in the mPFC ipsilateral to BLA infusions versus pErk1/2 measured in the contralateral mPFC is significantly greater in rats given RU 28362 than the ratio of the comparison across two hemispheres of vehicle-treated rats. ✷✷p < 0.01 (n = 7–8 per group). No significant differences were observed across hemispheres in rats that were not trained on the inhibitory avoidance task (n = 4–6 per group). B, Density of pErk1/2 bands was normalized to density of total MAP kinase bands on the same blot. The left band is ipsilateral (i) to intra-BLA infusion, and right is contralateral (c). C, Immunohistochemical demonstration of location of pErk1/2 within the mPFC. Positive cells were present in neurons in both the prelimbic and infralimbic regions of the ipsilateral mPFC, particularly in pyramidal cells of layers 2 and 3. Note pErk1/2 immunoreactivity in apical dendrites in this area.
To qualitatively confirm the Western blot data, we determined immunoreactivity levels of pErk1/2 in the mPFC 15 min after a unilateral infusion of RU 28362 (3 ng) or vehicle into the left BLA. As is shown in Figure 4C, RU 28362 treatment in the BLA increased the density of pErk1/2-positive neurons in both the prelimbic and infralimbic regions of the ipsilateral mPFC compared with rats given a vehicle infusion, although pErk1/2-immunopositive neurons were also present in this group. The increased density of pErk1/2 cells was predominantly found in layers 2 and 3, in cells with a long apical dendrite. These cells most likely represent pyramidal cells.
A MEK inhibitor infused into the mPFC blocks retention enhancement induced by posttraining infusion of a GR agonist into the BLA
To determine whether this increased pErk1/2 activity in the mPFC is involved in mediating the memory-enhancing effect of GR agonist infusions into the BLA, we examined whether a MEK inhibitor infused into the mPFC blocked this retention enhancement. Two-way ANOVA for entrance latencies on the training trial, before footshock and drug treatment, did not show significant differences between groups (p > 0.13 for all comparisons) (data not shown). Figure 5 shows 48 h inhibitory avoidance retention latencies of rats given immediate posttraining infusions of the MEK inhibitor PD98059 (50 ng) or vehicle into the left mPFC together with the GR agonist RU 28362 (1 or 3 ng) or vehicle into the ipsilateral BLA. Two-way ANOVA for retention latencies revealed a significant RU 28362 effect (F(2,63) = 3.75; p < 0.05), a significant PD98059 effect (F(1,63) = 8.37; p < 0.01), as well as a significant interaction between both factors (F(2,63) = 4.67; p < 0.05). Posttraining infusion of the higher dose of RU 28362 (3 ng) into the BLA enhanced retention of rats given vehicle into the mPFC, compared with the retention of corresponding vehicle-treated rats (p < 0.01). Infusion of PD98059 into the ipsilateral mPFC did not impair retention latencies when administered alone (p = 0.95) but blocked the retention-enhancing effects of RU 28362 administered into the BLA. Furthermore, retention latencies of RU 28362-treated rats given PD98059 into the mPFC were significantly shorter than retention latencies of RU 28362-treated rats given vehicle into the BLA (3 ng, p < 0.01).
Figure 5.

A MEK inhibitor infused into the mPFC blocks inhibitory avoidance retention enhancement induced by posttraining GR agonist infusions into the BLA. Step-through latencies (mean + SEM) in seconds on the 48 h inhibitory avoidance retention test of rats given immediate posttraining infusions of the GR agonist RU 28362 (1 or 3 ng in 0.2 μl) into the left BLA either alone or together with the MEK inhibitor PD98059 (50 ng in 0.5 μl) into the ipsilateral mPFC. ✷✷p < 0.01 compared with the corresponding vehicle group; ♦♦p < 0.01 compared with the corresponding vehicle–RU 28362 group (n = 10–13 per group).
GR agonist infusions into the mPFC or BLA enhance memory for similar components of inhibitory avoidance training
If, as suggested by the findings described above, the mPFC and BLA function as a circuit in enhancing GR agonist effects on memory consolidation, then it may be expected that the BLA and mPFC enhance memory of the same components of information acquired during inhibitory avoidance training. In inhibitory avoidance, rats learn both that they received a footshock as well as where they received the footshock. However, with traditional one-trial inhibitory avoidance training, it is not possible to determine whether the GR agonist infusions into the BLA or mPFC specifically enhanced memory of the context exposure, the footshock experience, or of both components of training. Recently, a modified, two-phase inhibitory avoidance procedure was developed to separate the learning about the context from the learning about the footshock. On the first day, animals freely explore the inhibitory avoidance apparatus to learn about the context and 24 h later are very briefly placed back into the shock compartment to receive footshock (Malin and McGaugh, 2006). In the present experiment, rats were trained on the two-phase inhibitory avoidance procedure and given bilateral infusions of the GR agonist RU 28362 into either the BLA (1 or 3 ng in 0.2 μl) or mPFC (3 or 10 ng in 0.5 μl) after the context training or footshock training.
Figure 6A shows 48 h retention latencies of animals given GR agonist infusions into the BLA immediately after context or shock training of inhibitory avoidance. One-way ANOVAs for retention latencies revealed significant group effects of rats given intra-BLA infusions of RU 28362 immediately after either context (F(2,32) = 6.55; p < 0.005) or shock training (F(2,35) = 5.52; p < 0.01). Retention latencies of rats given intra-BLA infusions of the 1 or 3 ng dose of RU 28362 immediately after either context or shock training were significantly longer than those of their respective vehicle controls (for both context and shock: 1 ng, p < 0.05; 3 ng, p < 0.01). Retention latencies of animals given intra-mPFC infusions of RU 28362 or vehicle immediately after context or shock training of inhibitory avoidance are shown in Figure 6B. Very similar to the effects of BLA infusions, one-way ANOVAs revealed significant differences in retention latencies of animals given the GR agonist after either context (F(2,49) = 4.02; p < 0.05) or shock training (F(2,40) = 3.55; p < 0.05). Retention latencies of rats given either the 3 or 10 ng dose of RU 28362 immediately after either context or shock training were significantly longer than those of their respective vehicle controls (after context: 3 ng, p < 0.05; 10 ng, p < 0.01; after shock: both doses, p < 0.05). Importantly, RU 28362 administered into the BLA (3 ng) or the mPFC (10 ng) after either context or shock training to animals that did not receive the other component of training did not enhance 48 h retention latencies, indicating that the expression of the enhanced memory depends on learning about both context and footshock (Table 1).
Figure 6.

GR agonist infusions into the BLA or mPFC enhance memory for similar components of inhibitory avoidance training. Step-through latencies (mean + SEM) in seconds on the 48 h retention test of rats given bilateral infusions of the GR agonist RU 28362 into the BLA or mPFC immediately after either context or shock training on the two-phase modified inhibitory avoidance task. A, Posttraining infusions of RU 28362 (1 or 3 ng in 0.2 μl) into the BLA enhanced inhibitory avoidance retention latencies when administered after either the context exposure or the shock experience. B, Posttraining infusions of the GR agonist RU 28362 (3 or 10 ng in 0.5 μl) into the mPFC enhanced inhibitory avoidance retention latencies when administered after either the context exposure or the shock experience. ✷p < 0.05; ✷✷p < 0.01 compared with the corresponding vehicle group (n = 10–18 per group).
Table 1.
Effect of RU 28362 after context or shock exposure alone
| Context exposure alone |
Footshock exposure alone |
|||||
|---|---|---|---|---|---|---|
| Vehicle | RU 28362 | Vehicle | RU 28362 | |||
| BLA | 10.1 ± 3.0 (7) | 7.3 ± 0.7 (7) | p = 0.38 | 8.9 ± 2.9 (7) | 14.7 ± 7.0 (7) | p = 0.46 |
| mPFC | 13.7 ± 1.7 (7) | 13.8 ± 3.5 (7) | p = 0.97 | 13.2 ± 6.1 (5) | 13.9 ± 4.2 (7) | p = 0.92 |
Shown are retention latencies (mean + SEM) in seconds. The numbers of animals per group are indicated in parentheses. The dose of RU 28362 administered into the BLA was 3 ng in 0.2 μl and that into the mPFC was 10 ng in 0.5 μl.
Discussion
These findings provide evidence that glucocorticoid effects on the consolidation of memory for inhibitory avoidance training depend on functional interactions between the BLA and mPFC. The memory-enhancing effect of an immediate posttraining infusion of the GR agonist RU 28362 administered into the mPFC depends on an increased phosphorylation of Erk1/2 in the BLA, and, similarly, the memory enhancement induced by intra-BLA administration of RU 28362 requires elevated pErk1/2 levels in the mPFC. Moreover, the finding that the GR agonist infused into either the BLA or the mPFC enhances the consolidation of memory of both the contextual and aversively motivated aspects of information acquired during inhibitory avoidance training strongly suggests that these two brain regions function as a bidirectional circuit in emotional regulation of memory consolidation.
Considerable evidence from electrophysiological studies indicates that the mPFC and BLA interact (Timms, 1977; Pérez-Jaranay and Vives, 1991; Rosenkranz and Grace, 2002; Maroun and Richter-Levin, 2003; Likhtik et al., 2005) and that mPFC–BLA interactions regulate performance on affectively motivated tasks (Schoenbaum et al., 1998; Bechara et al., 1999). For example, several studies implicate mPFC–BLA interactions in controlling the expression and extinction of conditioned fear (Garcia et al., 1999; Milad and Quirk, 2002). Moreover, we previously reported that lesions of the BLA block working memory impairment induced by intra-mPFC infusions of a GR agonist (Roozendaal et al., 2004). The present experiments investigated whether the mPFC and BLA interact in regulating the consolidation of memory of emotionally arousing training experiences. The first experiment determined that a lesion of the BLA made ipsilaterally to the mPFC infusion site blocked the GR agonist-induced memory enhancement, indicating that an intact and functional BLA is required for mediating the modulatory influences of intra-mPFC glucocorticoid administration on memory consolidation. As a contralateral BLA lesion did not block the GR agonist-induced memory enhancement, evidence indicates that the interaction is mediated directly through neural pathways connecting these two brain regions (Krettek and Price, 1977; Russchen, 1982) rather than by an effect of GR manipulation on hypothalamic–pituitary–adrenocortical axis activity. These findings are very similar to those of previous studies indicating that a disruption of BLA activity blocks memory enhancement induced by the administration of a glucocorticoid or other memory-enhancing compound into the hippocampus, dorsal striatum, entorhinal cortex, insular cortex, or anterior cingulate cortex (Roozendaal and McGaugh, 1997a; Roesler et al., 2002; Miranda and McGaugh, 2004; Malin et al., 2007) and further support the view that the BLA regulates memory consolidation via interactions with many brain regions (McGaugh, 2002).
Whereas previous findings suggest that the BLA regulates neuroplasticity and memory consolidation primarily through efferent projections from the BLA to these other brain regions (McGaugh, 2002), the present results indicate that interactions between the BLA and mPFC in modulating memory consolidation are bidirectional and, thus, that BLA activity does not simply regulate information storage processes in the mPFC. Our finding that the GR agonist infused unilaterally into the mPFC immediately after inhibitory avoidance training elevated pErk1/2 levels in the ipsilateral BLA shows that a manipulation of GR activity in the mPFC modulates BLA activity. The observation that the GR agonist also increased pErk1/2 levels in the central part of the lateral division of the central amygdala, intercalated mass, and to a lesser extent the medial amygdala is consistent with evidence that the mPFC has projection neurons to these other regions (Cassell and Wright, 1986). We measured pErk1/2 levels to assess BLA activity because Erk is a rapidly activated protein that has been implicated in neuroplasticity and memory consolidation (Atkins et al., 1998; Schafe et al., 2000). Moreover, and most importantly, as the specific MEK inhibitor PD98059 infused selectively into the BLA blocked this memory enhancement, these findings suggest that this increased phosphorylation of Erk1/2 in the BLA is essential in mediating the memory-modulatory effect of intra-mPFC GR agonist administration. As some evidence suggests that the medial amygdala, but not the central amygdala, is also involved in regulating stress hormone effects on memory consolidation (Roozendaal and McGaugh, 1996), it is possible that mPFC influences on pErk1/2 activity within the medial amygdala normally contribute to the enhanced retention initiated by GR agonist infusion into the mPFC. Highly comparable with the effects of GR agonist infusions into the mPFC, RU 28362 administration into the BLA after inhibitory avoidance training induced a rapid increase in pErk1/2 levels in both the prelimbic and infralimbic regions of the mPFC. Furthermore, an immediate posttraining infusion of a MEK inhibitor into the mPFC blocked the memory enhancement induced by intra-BLA administration of the GR agonist. Thus, the findings indicate that a GR agonist infusion into the mPFC influences BLA activity, whereas a GR agonist infusion into the BLA alters mPFC activity. Interference with this process at either site prevents the enhancing effect of glucocorticoid administration on memory consolidation, likely by disturbing the processing of information storage occurring in distal brain regions (McGaugh, 2002; Roozendaal, 2002). To date, it is not known whether glucocorticoid modulation of memories in these distal brain regions also depends on increased pErk1/2 levels in the mPFC and/or BLA.
Interestingly, several findings indicate that glucocorticoid effects on memory consolidation depend on pErk1/2-MAP kinase activation. For example, Revest et al. (2005) reported that stress or corticosterone induced a slow-onset (after 2 h) increase in the expression and enzymatic activity of the MAP kinase pathway in the hippocampus. Blockade of this effect with intrahippocampal infusions of a MEK inhibitor prevented glucocorticoid effects on fear memory consolidation. Our data appear to differ in two important ways: First, our findings clearly demonstrate that GR agonist administration into either the mPFC or BLA induces pErk1/2 activation in the other brain site. Second, our data indicate elevated pErk1/2 levels in the mPFC or BLA within 15 min after GR agonist administration. Such a time frame strongly suggests the involvement of a nongenomic glucocorticoid action, possibly through an activation of membrane-associated receptors (Dallman, 2005; Johnson et al., 2005). Such findings are consistent with evidence of rapid interactions of glucocorticoids or GR agonist with arousal-induced noradrenergic and endocannabinoid mechanisms in influencing neuroplasticity and memory consolidation (Quirarte et al., 1997; Roozendaal et al., 2006; Pu et al., 2007; Campolongo et al., 2009). Also, the findings that the GR agonist did not alter pErk1/2 levels when infused into either the mPFC or BLA of nontrained control animals support the view that glucocorticoids affect BLA–mPFC circuitry and enhance memory consolidation only under conditions of emotional arousal.
Additional support for a necessary interaction between the BLA and mPFC in regulating glucocorticoid effects on memory consolidation in efferent brain regions was provided by our finding that both brain regions played a role in GR agonist effects on the consolidation of memory of contextual and aversively motivated aspects of inhibitory avoidance training. As noted above, BLA activation is known to enhance the consolidation of memory of many different training experiences via interactions with different brain regions (McGaugh, 2002). The present demonstration that the GR agonist administered into the mPFC, like infusions into the BLA, enhanced retention when administered after either the context training or footshock experience indicate that both brain regions share a functional commonality and strongly support the view that both brain regions cooperate in regulating memory consolidation. In contrast, previous experiments investigating the effect of drug administration into the hippocampus, anterior cingulate cortex, or dorsal striatum after training on the two-phase inhibitory avoidance task indicated that those brain regions are dedicated to the selective modulation of memory of specific components of the training experience (Malin and McGaugh, 2006; Medina et al., 2007; Roozendaal et al., 2008). These findings not only substantiate the unique roles of the BLA and mPFC, and their interaction, in emotional regulation of memory consolidation of many different types of training experiences (Tronel and Sara, 2003; Xiang and Brown, 2004; Touzani et al., 2007; McDonald et al., 2008), but also strongly suggest that drug infusions into these other brain regions do not simply enhance memory consolidation by activating the BLA and mPFC. Rather, these findings are consistent with our view that the BLA–mPFC circuit regulates memory consolidation in these other, distal brain regions. Several studies indicate that such interactions between the mPFC and BLA may be essential for an adaptive regulation of affect and memory (Amat et al., 2005) and to exert a cognitive and emotional control to downregulate limbic circuitry (Burgos-Robles et al., 2007; Mueller et al., 2008).
In summary, we demonstrate a crucial role for bidirectional interactions between the BLA and mPFC in regulating glucocorticoid effects on memory consolidation. Abnormalities in the BLA and mPFC are consistently noted in affective disorders such as post-traumatic stress disorder and major depressive illness (Bremner et al., 1999; Johnstone et al., 2007). The identification of the nature and functional dynamics of this BLA–mPFC circuit that modulate emotional arousal effects on memory consolidation may lead to an improved understanding of how this system may be involved in the aberrant cognitive and emotional responses observed in affective disorders.
Footnotes
This work was supported by National Science Foundation Grant IOB-0618211 (B.R.) and National Institute of Mental Health Grant MH12526 (J.L.M.). We thank Laura Stillman and Kathleen Perez for excellent technical assistance.
References
- Akirav I, Maroun M. Ventromedial prefrontal cortex is obligatory for consolidation and reconsolidation of object recognition memory. Cereb Cortex. 2006;16:1759–1765. doi: 10.1093/cercor/bhj114. [DOI] [PubMed] [Google Scholar]
- Amat J, Baratta MV, Paul E, Bland ST, Watkins LR, Maier SF. Medial prefrontal cortex determines how stressor controllability affects behavior and dorsal raphe nucleus. Nat Neurosci. 2005;8:365–371. doi: 10.1038/nn1399. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Damasio AR, Lee GP. Different contributions of the human amygdala and ventromedial prefrontal cortex to decision-making. J Neurosci. 1999;19:5473–5481. doi: 10.1523/JNEUROSCI.19-13-05473.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Staib LH, Kaloupek D, Southwick SM, Soufer R, Charney DS. Neural correlates of exposure to traumatic pictures and sound in Vietnam combat veterans with and without posttraumatic stress disorder: a positron emission tomography study. Biol Psychiatry. 1999;45:806–816. doi: 10.1016/s0006-3223(98)00297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ. Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron. 2007;53:871–880. doi: 10.1016/j.neuron.2007.02.021. [DOI] [PubMed] [Google Scholar]
- Campolongo P, Roozendaal B, Trezza V, Hauer D, Schelling G, McGaugh JL, Cuomo V. Endocannabinoids in the rat basolateral amygdala enhance memory consolidaton and enable glucocortiocid modulation of memory. Proc Natl Acad Sci U S A. 2009;106:4888–4893. doi: 10.1073/pnas.0900835106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassell MD, Wright DJ. Topography of projections from the medial prefrontal cortex to the amygdala in the rat. Brain Res Bull. 1986;17:321–333. doi: 10.1016/0361-9230(86)90237-6. [DOI] [PubMed] [Google Scholar]
- Dallman MF. Fast glucocorticoid actions on brain: back to the future. Front Neuroendocrinol. 2005;26:103–108. doi: 10.1016/j.yfrne.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Davidson RJ. Anxiety and affective style: role of prefrontal cortex and amygdala. Biol Psychiatry. 2002;51:68–80. doi: 10.1016/s0006-3223(01)01328-2. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Oitzl MS, Joëls M. Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci. 1999;22:422–426. doi: 10.1016/s0166-2236(99)01438-1. [DOI] [PubMed] [Google Scholar]
- Dolcos F, LaBar KS, Cabeza R. Interaction between the amygala and the medial temporal lobe system predicts better memory for emotional events. Neuron. 2004;42:855–863. doi: 10.1016/s0896-6273(04)00289-2. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Nader K, LeDoux JE. Activation of extracellular signal-regulated kinase–mitogen-activated protein kinase cascade in the amygdala is required for memory reconsolidation of auditory fear conditioning. Eur J Neurosci. 2005;21:283–289. doi: 10.1111/j.1460-9568.2004.03824.x. [DOI] [PubMed] [Google Scholar]
- Garcia R, Vouimba RM, Baudry M, Thompson RF. The amygdala modulates prefrontal cortex activity relative to conditioned fear. Nature. 1999;402:294–296. doi: 10.1038/46286. [DOI] [PubMed] [Google Scholar]
- Huff NC, Frank M, Wright-Hardesty K, Sprunger D, Matus-Amat P, Higgins E, Rudy JW. Amygdala regulation of immediate-early gene expression in the hippocampus induced by contextual fear conditioning. J Neurosci. 2006;26:1616–1623. doi: 10.1523/JNEUROSCI.4964-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo LA, Barros DM, da Costa JC, Furini C, Zinn C, Cammarota M, Bevilaqua LR, Izquierdo I. A link between role of two prefrontal areas in immediate memory and in long-term memory consolidation. Neurobiol Learn Mem. 2007;88:160–166. doi: 10.1016/j.nlm.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Johnson LR, Farb C, Morrison JH, McEwen BS, LeDoux JE. Localization of glucocorticoid receptors at postsynaptic membranes in the lateral amygdala. Neuroscience. 2005;136:289–299. doi: 10.1016/j.neuroscience.2005.06.050. [DOI] [PubMed] [Google Scholar]
- Johnstone T, van Reekum CM, Urry HL, Kalin NH, Davidson RJ. Failure to regulate: counterproductive recruitment of top-down prefrontal-subcortical circuitry in major depression. J Neurosci. 2007;7:8877–8884. doi: 10.1523/JNEUROSCI.2063-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krettek JE, Price JL. Projections from the amygdaloid complex in the cerebral cortex and thalamus in the rat and cat. J Comp Neurol. 1977;172:687–722. doi: 10.1002/cne.901720408. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, Grace AA. Cannabinoids potentiate emotional learning plasticity in neurons of the medial prefrontal cortex through basolateral amygdala inputs. J Neurosci. 2006;26:6458–6468. doi: 10.1523/JNEUROSCI.0707-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviolette SR, Lipski WJ, Grace AA. A subpopulation of neurons in the medial prefrontal cortex encodes emotional learning with burst and frequency codes through a dopamine D4 receptor-dependent basolateral amygdala input. J Neurosci. 2005;25:6066–6075. doi: 10.1523/JNEUROSCI.1168-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Pelletier JG, Paz R, Paré D. Prefrontal control of the amygdala. J Neurosci. 2005;25:7429–7437. doi: 10.1523/JNEUROSCI.2314-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons DM, Lopez JM, Yang C, Schatzberg AF. Stress-level cortisol treatment impairs inhibitory control of behavior in monkeys. J Neurosci. 2000;20:7816–7821. doi: 10.1523/JNEUROSCI.20-20-07816.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin EL, McGaugh JL. Differential involvement of the hippocampus, anterior cingulate cortex, and basolateral amygdala in memory for context and footshock. Proc Natl Acad Sci U S A. 2006;103:1959–1963. doi: 10.1073/pnas.0510890103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin EL, Ibrahim DY, Tu JW, McGaugh JL. Involvement of the rostral anterior cingulate cortex in consolidation of inhibitory avoidance memory: interaction with the basolateral amygdala. Neurobiol Learn Mem. 2007;87:295–302. doi: 10.1016/j.nlm.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroun M, Richter-Levin G. Exposure to acute stress blocks the induction of long-term potentiation of the amygdala-prefrontal cortex pathway in vivo. J Neurosci. 2003;23:4406–4409. doi: 10.1523/JNEUROSCI.23-11-04406.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald RJ, King AL, Foong N, Rizos Z, Hong NS. Neurotoxic lesions of the medial prefrontal cortex or medial striatum impair multiple-location place learning in the water maze: evidence for neural structures with complementary roles in behavioural flexibility. Exp Brain Res. 2008;187:419–427. doi: 10.1007/s00221-008-1314-z. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Time-dependent processes in memory storage. Science. 1966;153:1351–1358. doi: 10.1126/science.153.3742.1351. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Memory consolidation and the amygdala: a systems perspective. Trends Neurosci. 2002;25:456–461. doi: 10.1016/s0166-2236(02)02211-7. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, Roozendaal B. Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol. 2002;12:205–210. doi: 10.1016/s0959-4388(02)00306-9. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, Introini-Collison IB, Nagahara AH. Memory-enhancing effects of posttraining naloxone: involvement of beta-noradrenergic influences in the amygdaloid complex. Brain Res. 1988;446:37–49. doi: 10.1016/0006-8993(88)91294-2. [DOI] [PubMed] [Google Scholar]
- McIntyre CK, Miyashita T, Setlow B, Marjon KD, Steward O, Guzowski JF, McGaugh JL. Memory-influencing intra-basolateral amygdala drug infusions modulate expression of Arc protein in the hippocampus. Proc Natl Acad Sci U S A. 2005;102:10718–10723. doi: 10.1073/pnas.0504436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina AC, Charles JR, Espinoza-González V, Sánchez-Resendis O, Prado-Alcalá RA, Roozendaal B, Quirarte GL. Glucocorticoid administratium into the dorsal striatum facilitates memory consolidation of inhibitory avoidance training but not of the context or footshock components. Learn Mem. 2007;14:673–677. doi: 10.1101/lm.654407. [DOI] [PubMed] [Google Scholar]
- Merino SM, Maren S. Hitting Ras where it counts: Ras antagonism in the basolateral amygdala inhibits long-term fear memory. Eur J Neurosci. 2006;23:196–204. doi: 10.1111/j.1460-9568.2005.04546.x. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Miranda MI, McGaugh JL. Enhancement of inhibitory avoidance and conditioned taste aversion memory with insular cortex infusions of 8-Br-cAMP: involvement of the basolateral amygdala. Learn Mem. 2004;11:312–317. doi: 10.1101/lm.72804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller D, Porter JT, Quirk GJ. Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J Neurosci. 2008;28:369–375. doi: 10.1523/JNEUROSCI.3248-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego: Academic; 2005. [DOI] [PubMed] [Google Scholar]
- Pérez-Jaranay JM, Vives F. Electrophysiological study of the response of medial prefrontal cortex neurons to stimulation of the basolateral nucleus of the amygdala in the rat. Brain Res. 1991;564:97–101. doi: 10.1016/0006-8993(91)91357-7. [DOI] [PubMed] [Google Scholar]
- Pu Z, Krugers HJ, Joëls M. Corticosterone time-dependently modulates beta-adrenergic effects on long-term potentiation in the hippocampal dentate gyrus. Learn Mem. 2007;14:359–367. doi: 10.1101/lm.527207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirarte GL, Roozendaal B, McGaugh JL. Glucocorticoid enhancement of memory storage involves noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci U S A. 1997;94:14048–14053. doi: 10.1073/pnas.94.25.14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Gehlert DR. Inhibition of the amygdala: key to pathological states? Ann N Y Acad Sci. 2003;985:263–272. doi: 10.1111/j.1749-6632.2003.tb07087.x. [DOI] [PubMed] [Google Scholar]
- Revest JM, Di Blasi F, Kitchener P, Rougé-Pont F, Desmedt A, Turiault M, Tronche F, Piazza PV. The MAPK pathway and Egr-1 mediate stress-related behavioral effects of glucocorticoids. Nat Neurosci. 2005;8:664–672. doi: 10.1038/nn1441. [DOI] [PubMed] [Google Scholar]
- Roesler R, Roozendaal B, McGaugh JL. Basolateral amygdala lesions block the memory-enhancing effect of 8-Br-cAMP infused into the entorhinal cortex of rats after training. Eur J Neurosci. 2002;15:905–910. doi: 10.1046/j.1460-9568.2002.01924.x. [DOI] [PubMed] [Google Scholar]
- Roozendaal B. Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology. 2000;25:213–238. doi: 10.1016/s0306-4530(99)00058-x. [DOI] [PubMed] [Google Scholar]
- Roozendaal B. Stress and memory: opposing effects of glucocorticoids on memory consolidation and memory retrieval. Neurobiol Learn Mem. 2002;78:578–595. doi: 10.1006/nlme.2002.4080. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McGaugh JL. Amygdaloid nuclei lesions differentially affect glucocorticoid-induced memory enhancement in an inhibitory avoidance task. Neurobiol Learn Mem. 1996;65:1–8. doi: 10.1006/nlme.1996.0001. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McGaugh JL. Basolateral amygdala lesions block the memory-enhancing effect of glucocorticoid administration in the dorsal hippocampus of rats. Eur J Neurosci. 1997a;9:76–83. doi: 10.1111/j.1460-9568.1997.tb01355.x. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McGaugh JL. Glucocorticoid receptor agonist and antagonist administration into the basolateral but not central amygdala modulates memory storage. Neurobiol Learn Mem. 1997b;67:176–179. doi: 10.1006/nlme.1996.3765. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Portillo-Marquez G, McGaugh JL. Basolateral amygdala lesions block glucocorticoid-induced modulation of memory for spatial learning. Behav Neurosci. 1996;110:1074–1083. doi: 10.1037//0735-7044.110.5.1074. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Quirarte GL, McGaugh JL. Glucocorticoids interact with the basolateral amygdala β-adrenoceptor-cAMP/PKA system in influencing memory consolidation. Eur J Neurosci. 2002;15:553–560. doi: 10.1046/j.0953-816x.2001.01876.x. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McReynolds JR, McGaugh JL. The basolateral amygdala interacts with the medial prefrontal cortex in regulating glucocorticoid effects on working memory impairment. J Neurosci. 2004;24:1385–1392. doi: 10.1523/JNEUROSCI.4664-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Okuda S, Van der Zee EA, McGaugh JL. Glucocorticoid enhancement of memory requires arousal-induced noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci U S A. 2006;103:6741–6746. doi: 10.1073/pnas.0601874103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Lengvilas R, McGaugh JL, Civelli O, Reinscheid RK. Orphanin FQ/nociceptin interacts with the basolateral amygdala noradrenergic system in memory consolidation. Learn Mem. 2007;14:29–35. doi: 10.1101/lm.403607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Barsegyan A, Lee S. Adrenal stress hormones, amygdala activation, and memory for emotionally arousing experiences. Prog Brain Res. 2008;167:79–97. doi: 10.1016/S0079-6123(07)67006-X. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA. Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo. J Neurosci. 2002;22:324–337. doi: 10.1523/JNEUROSCI.22-01-00324.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runyan JD, Dash PK. Intra-medial prefrontal administration of SCH-23390 attenuates ERK phosphorylation and long-term memory for trace fear conditioning in rats. Neurobiol Learn Mem. 2004;82:65–70. doi: 10.1016/j.nlm.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Russchen FT. Amygdalopetal projections in the cat. I. Cortical afferent connections. A study with retrograde and anterograde tracing techniques. J Comp Neurol. 1982;206:159–179. doi: 10.1002/cne.902060206. [DOI] [PubMed] [Google Scholar]
- Sandi C, Pinelo-Nava MT. Stress and memory: behavioral effects and neurobiological mechanisms. Neural Plast. 2007;2007:78970. doi: 10.1155/2007/78970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J Neurosci. 2000;20:8177–8187. doi: 10.1523/JNEUROSCI.20-21-08177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenbaum G, Chiba AA, Gallagher M. Orbitofrontal cortex and basolateral amygdala encode expected outcomes during learning. Nat Neurosci. 1998;1:155–159. doi: 10.1038/407. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Carew TJ. Inclusion of phosphatase inhibitors during Western blotting enhances signal detection with phospho-specific antibodies. Anal Biochem. 2002;307:187–189. doi: 10.1016/s0003-2697(02)00008-8. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Sherff CM, Shobe J, Bagnall MW, Sutton MA, Carew TJ. Differential role of mitogen-activated protein kinase in three distinct phases of memory for sensitization in Aplysia. J Neurosci. 2003;23:3899–3907. doi: 10.1523/JNEUROSCI.23-09-03899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teutsch G, Costerousse G, Deraedt R, Benzoni J, Fortin M, Philibert D. 17alpha-alkynyl-11beta,17-dihydroxyandrostane derivates: a new class of potent glucocorticoids. Steroids. 1981;38:651–665. doi: 10.1016/0039-128x(81)90084-2. [DOI] [PubMed] [Google Scholar]
- Timms RJ. Cortical inhibition and facilitation of the defense reaction. J Physiol. 1977;266:98P–99P. [PubMed] [Google Scholar]
- Touzani K, Puthanveettil SV, Kandel ER. Consolidation of learning strategies during spatial working memory task requires protein synthesis in the prefrontal cortex. Proc Natl Acad Sci U S A. 2007;104:5632–5637. doi: 10.1073/pnas.0611554104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronel S, Sara SJ. Blockade of NMDA receptors in prelimbic cortex induces an enduring amnesia for odor-reward associative learning. J Neurosci. 2003;2:5472–5476. doi: 10.1523/JNEUROSCI.23-13-05472.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoory MM, Vouimba RM, Akirav I, Kavushansky A, Avital A, Richter-Levin G. Amygdala modulation of memory-related processes in the hippocampus: potential relevance to PTSD. Prog Brain Res. 2008;167:35–51. doi: 10.1016/S0079-6123(07)67003-4. [DOI] [PubMed] [Google Scholar]
- Xiang JZ, Brown MW. Neuronal responses related to long-term recognition memory processes in prefrontal cortex. Neuron. 2004;42:817–829. doi: 10.1016/j.neuron.2004.05.013. [DOI] [PubMed] [Google Scholar]