

Abstract
Mutations in the signal transducer and activator of transcription 3 (STAT3) were reported to cause hyperimmunoglobulin E syndrome (HIES). The present study investigates T helper type 17 (Th17) responses triggered by the relevant stimuli Staphylococcus aureus and Candidia albicans in five ‘classical’ HIES patients, and a family with three patients who all had a milder HIES phenotype. We demonstrate that patients with various forms of HIES have different defects in their Th17 response to S. aureus and C. albicans, and this is in line with the clinical features of the disease. Interestingly, a partial deficiency of interleukin (IL)-17 production, even when associated with STAT3 mutations, leads to a milder clinical phenotype. We also observed defective Th17 responses in patients with the ‘classical’ presentation of the disease but without STAT3 mutations. These data demonstrate that defective IL-17 production in response to specific pathogens can differ between patients with HIES and that the extent of the defective Th17 response determines their clinical phenotype.
Keywords: C. albicans, HIES, IL-17, S. aureus, STAT3
Introduction
Hyperimmunoglobulin E syndrome (HIES) is a primary immunodeficiency disorder characterized by recurrent staphylococcal skin abscesses, pulmonary infections, mucocutaneous candidiasis, skeletal and dental abnormalities and elevated serum immunoglobulin E (IgE) concentrations [1,2]. Although most cases of HIES are sporadic, familial cases are encountered, mainly with an autosomal dominant mode of inheritance [3].
Recently, mutations in the evolutionarily conserved SH2 and DNA-binding domains of the signal transducer and activator of transcription 3 (STAT 3) were found to be present in approximately 60% of the patients with HIES [4,5]. STAT3 is an important component of receptor signalling pathways for several cytokines, including interleukin-6 (IL-6) and IL-10, and patients with HIES have shown defective responses to these cytokines [4]. The role of STAT3 for normal signalling of the IL-6 receptor has important consequences for normal host defence. Together with other cytokines such as IL-1β and IL-23, the IL-6/STAT3 pathway is crucial for the normal development of CD4+–T helper type 17 (Th17) cells [6,7]. Because IL-17 has an important role in the activation of neutrophil-dependent immunity [8], defective Th17 generation as a result of STAT3 mutation may play an important role in the pathogenesis of HIES.
In a recent paper, Milner et al. have demonstrated that T lymphocytes from patients with HIES are unable to differentiate into Th17 after mitogenic stimulation [9]. These data were supported by two reports that also showed defective generation of Th17 when anti-CD3/anti-CD28/IL-2 or cytokine cocktails were used [10,11]. These studies reported the defective generation of Th17 using mitogenic cocktails in patients with established mutations in the SH2 and DNA-binding domains of STAT3. In contrast, patients with atopic dermatitis and high IgE, but without skin and respiratory infections and without STAT3 mutations, had normal Th17 responses [9,12].
In the present paper, we aimed to extend these initial findings by investigating the generation of Th17 cells and IL-17 production by relevant microbial stimuli for HIES. In addition, we assessed Th17 profiles in three distinct groups of patients: ‘classical’ HIES patients with STAT3 mutations in the SH2/DNA-binding domains, ‘classical’ HIES without STAT3 mutations and a family with ‘variant’ HIES that we described as having a milder clinical phenotype [13], with deletion of a triplet in the linker domain. The differences in the degree of IL-17 production defects after stimulation with Staphylococcus aureus or Candida albicans determined the severity of the clinical phenotype.
Materials and methods
Patients and controls
Eight patients with a clinical diagnosis of HIES at the out-patient clinic for infectious diseases and immunodeficiencies of the Department of General Internal Medicine of Radboud University Nijmegen Medical Centre were enrolled into the study. Three of these patients were family members. After informed consent, blood was collected from eight healthy, non-smoking volunteers who were free of infectious or inflammatory disease and the enrolled HIES patients by venipuncture into 10 ml ethylenediamine tetraacetic acid (EDTA) syringes (Monoject; BD Vacutainer, Plymouth, UK). STAT3 mutation analysis was kindly performed in the Laboratory of Human Molecular Biology and Genetics, Catholic University of the Sacred Heart, Milan, Italy (head Professor Roberto Colombo).
Microorganisms
C. albicans American Type Culture Collection (ATCC) MYA-3573 (UC820), a strain well described elsewhere [14], was used. C. albicans was grown overnight in Sabouraud broth at 37°C, cells were harvested by centrifugation, washed twice and resuspended in culture medium (RPMI-1640 Dutch modification; ICN Biomedicals, Aurora, OH, USA) [15]. C. albicans was heat-killed for 1 h at 100°C and all experiments were performed with heat-killed C. albicans. The clinical isolate of S. aureus was heat-killed and used at a dosage of 107/ml.
In vitro cytokine production
Separation and stimulation of peripheral blood mononuclear cells (PBMCs) was performed as described previously [16]. Briefly, the PBMC fraction was obtained by density centrifugation of diluted blood (one part blood to one part pyrogen-free saline) over Ficoll-Paque (Pharmacia Biotech, Uppsala, Sweden). PBMCs were washed twice in saline and suspended in culture medium supplemented with gentamycin 1%, L-glutamine 1% and pyruvate 1%. The cells were counted in a Bürker counting chamber, and cell numbers were adjusted to 5 × 106 cells/ml; 5 × 105 PBMCs in a volume of 100 µl per well were incubated at 37°C in round-bottomed 96-well plates (Greiner, Nuremberg, Germany) in the presence of 10% human pooled serum with stimuli or culture medium alone, and where indicated with the cytokines IL-6 and IL-10 (100 ng/ml). After 5 days of incubation, supernatants were collected and stored at −20°C until assayed.
Cytokine assays
IL-1β and IL-17 concentrations were measured by commercial enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems); interferon (IFN)-γ and IL-10 (Pelikine Compact, Sanquin, Amsterdam, the Netherlands), according to the manufacturer's instructions.
Intracellular cytokine staining
PBMC cells were stimulated as described above and restimulated for 4–6 h with phorbol myristate acetate (PMA) (50 ng/ml; Sigma) and ionomycin (1 µg/ml; Sigma, St. Louis, MO, USA) in the presence of Golgiplug (BD Biosciences, Dendermonde, Belgium), according to the manufacturer's protocol. Cells were first stained extracellularly using an anti-CD4 allophycocyanin (APC) antibody (BD Biosciences). Subsequently the cells were fixed and permeabilized with Cytofix/Cytoperm solution (BD Biosciences) and then stained intracellularly with anti-IFN-γ phycoerythrin (PE) (eBiosciences, Hatfield, UK) and anti-IL-17 fluorescein isothiocyanate (FITC) (eBiosciences). Samples were measured on a fluorescence activated cell sorter (FACS)Calibur and data were analysed using CellQuest-Pro software (BD Biosciences).
Statistical analyses
The differences between groups were analysed using the Mann–Whitney U-test, and considered statistically significant when P ≤ 0·05. Data are presented as the cumulative result of all experiments performed, unless indicated otherwise. Data are given as median or mean ± standard error of the mean (SEM) unless indicated otherwise.
Results
Patients
The clinical description of patients with HIES are summarized in Table 1. All patients were of Dutch ancestry. In Fig. 1 the pedigrees of the HIES family are presented. Of note, the clinical data of the HIES family have been published elsewhere [13,17]. Blood sampling and Th17 profile were assessed in cells isolated from three HIES patients in the third generation of the family and five patients with ‘classical’ HIES. In the large HIES family with milder HIES (variant HIES) the STAT3 gene had deletion of a triplet, leading to the deletion of serine in position 560 of the linker domain. Three of the five ‘classical’ HIES patients had known STAT3 mutations (R382W twice and V463del) [5] (Table 1). Two of the patients with ‘classical’ HIES had no STAT3 mutation.
Table 1.
Clinical characteristics of the patients with hyperimmunoglobuline E syndrome (HIES).
| Infections |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Localization |
Aetiology |
||||||||
| HIES patients | Gender/age (years) | Pulmonary | Skin | Other | Bacteria | Fungi | Other abnormalities | IgE IU/l | STAT3 mutation |
| ‘Variant’ | |||||||||
| 1 | Female/45 | No | Yes | S. aureus | C. albicans | Palatoschizis Fractures | 900 | S560del | |
| 2 | Male/46 | No | Yes | S. aureus | C. albicans | Abnormal dentition | 16 700 | S560del | |
| 3 | Female/38 | No | Yes | S. aureus | C. albicans | Hypertension | 7 210 | S560del | |
| ‘Classical’ | |||||||||
| 1 | Female/27 | Yes lobectomy for Aspergillus pneumonia | Yes | Bartholinitis | S. aureus | Aspergillus | 3 600 | V463del | |
| 2 | Male/40 | Yes | Yes | S. aureus | Aspergillus | Giant chalazia | 17 200 | R382W | |
| 3 | Male/39 | Yes | Yes | Osteomyelitis | S. aureus | 1 762 | R382W | ||
| 4 | Male/43 | Yes | Yes | C. albicans | 2 340 | None | |||
| 5 | Male/36 | Yes | Yes | Spondylodiscitis | S. aureus | 16 000 | None | ||
IgE, immunoglobulin E; STAT 3, signal transducer and activator of transcription 3; C. albicans, Candida albicans; S aureus, Staphylococcus aureus.
Fig. 1.
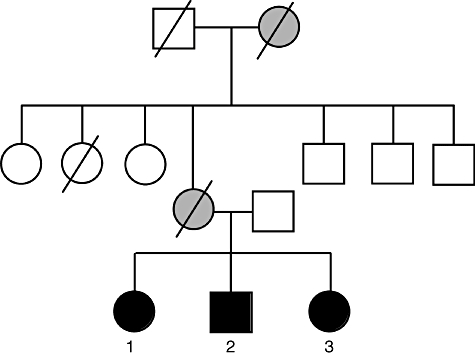
The pedigree of a family with hyperimmunoglobulin E syndrome (HIES) of Dutch ancestry. Clinical symptoms characteristic for HIES were recorded in a large kindred, with patients affected [closed black symbols are family members affected signal transducer and activator of transcription 3 (STAT 3) mutation] from several generations. Closed grey symbols represent suspicion for the STAT3 mutation, but not proven. Line through symbol represents deceased family member.
Defective IL-17 and IFN-γ production in HIES patients in response to S. aureus and C. albicans
To investigate the immunological functional properties with respect to Th17 responses in HIES patients with different mutations, PBMC from healthy volunteers, ‘classical’ HIES patients and three members from a HIES family with ‘variant’ HIES were assessed for the capacity to mount IL-17 responses. We have developed a new methodology of Th17 generation using human PBMC stimulated with whole microbial stimuli relevant for HIES: S. aureus and C. albicans[18]. HIES patients had a defective response to C. albicans, although IL-17 was measurable in all patients (Fig. 2a). Interestingly, IL-17 production was completely absent in PBMC stimulated with S. aureus in all ‘classical’ HIES patients (Fig. 2b). In contrast, PBMC isolated from the variant HIES patients, bearing the STAT3 mutations in the linker domain, were able to produce IL-17 in response to S. aureus, albeit at lower concentrations when compared to healthy volunteers (Fig. 2b and c). IFN-γ production was distorted in HIES patients when compared to healthy controls, while IL-10 was found to be elevated in HIES patients when stimulated with both S. aureus and C. albicans.
Fig. 2.
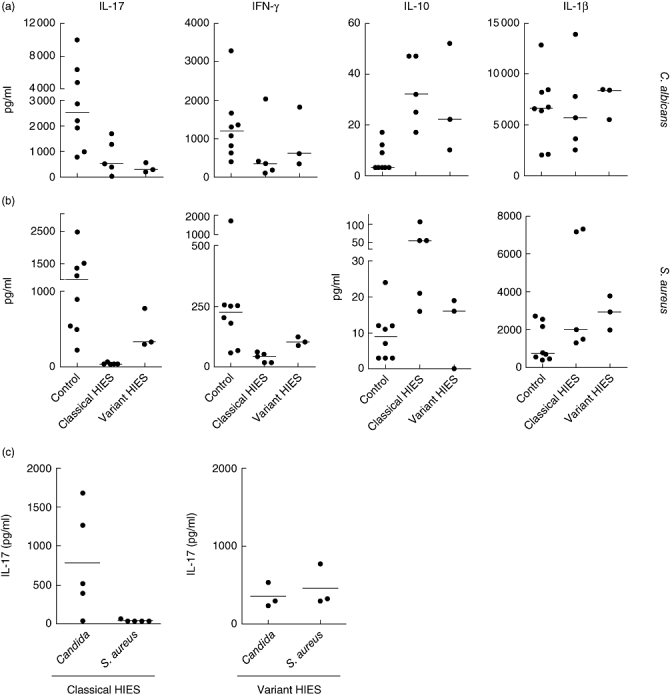
Defective interleukin (IL)-17 and interferon (IFN)-γ production in hyperimmunoglobulin E syndrome (HIES) patients in response to Staphylococcus aureus and Candida albicans. Human peripheral blood mononuclear cells (PBMCs) from healthy controls (n = 8), ‘classical’ HIES (n = 5) and three members of the HIES family bearing the STAT3 linker domain mutation were stimulated for 5 days with C. albicans (a) or S. aureus (b). Cytokines were measured by enzyme-linked immunosorbent assay. (c) IL-17 production by PBMC stimulated as in (a) and (b) from ‘classical’ HIES patients and familial HIES patients.
Normal induction of Th17 cells in familial variant HIES patients
The in vitro stimulations described above suggest that HIES patients have a significant defect in the generation of Th17 cells. This was indeed the case for the patients with ‘classical’ HIES, either bearing STAT3 mutations or not (Fig. 3). Surprisingly, when the familial variant HIES patients were challenged with disease-related microorganisms, they showed a clear induction of single IL-17-positive and IL-17/IFN-γ-positive CD4+ cells compared to normal controls (Fig. 3).
Fig. 3.
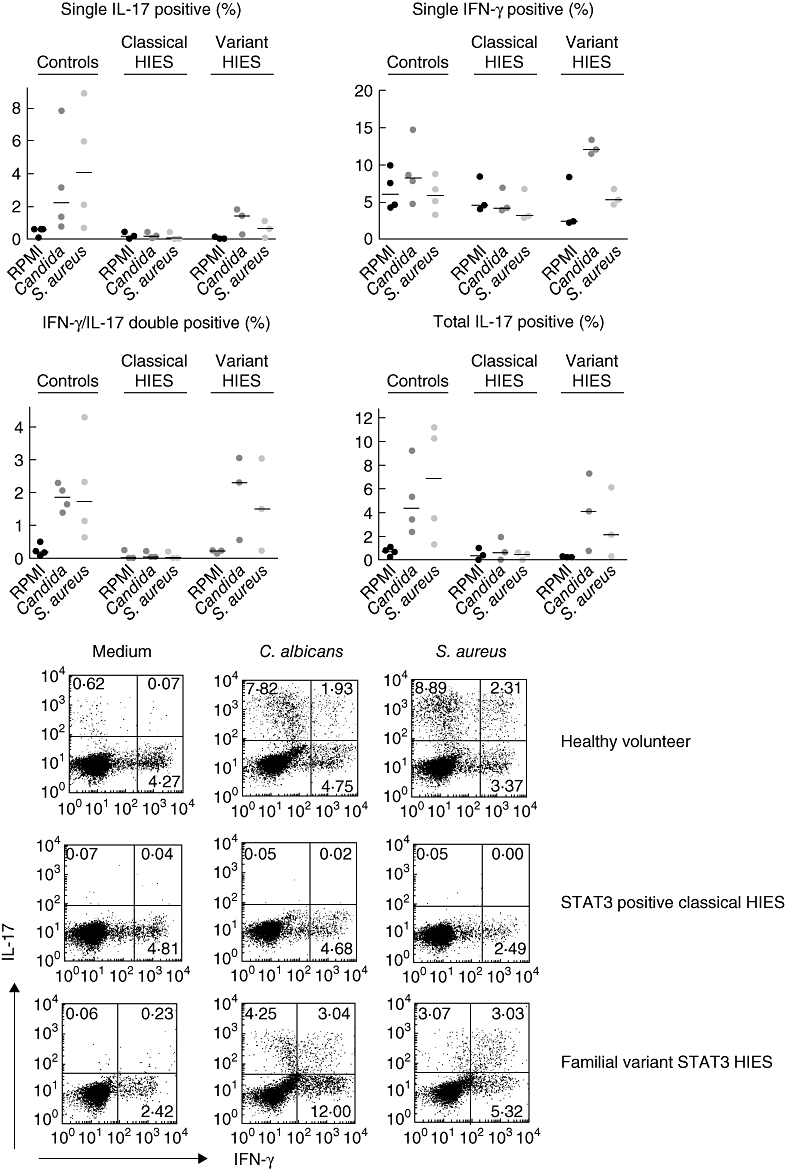
T helper type 17 (Th17) cell generation in hyperimmunoglobulin E syndrome (HIES) patients. Human peripheral blood mononuclear cells (PBMCs) from healthy controls (n = 4), ‘classical’ HIES (n = 3) and three members of the HIES family bearing the signal transducer and activator of transcription 3 (STAT 3) linker domain mutation were stimulated for 5 days with Candida albicans or Staphylococcus aureus. The interleukin (IL)-17- or interferon (IFN)-γ-producing Th17 cells were assessed by intracellular staining (see description in the Methods).
HIES patients have a defect in IL-6, but not in IL-10, signalling
IL-6 augmented IL-17 production induced by C. albicans and S. aureus in cells isolated from healthy controls (Fig. 4a). No effect was apparent in the HIES patients, independently of the type of STA3 mutation. In contrast to IL-6, IL-10 reduced the amount of IL-17, and this effect was observed both in healthy controls and HIES patients (Fig. 4b).
Fig. 4.
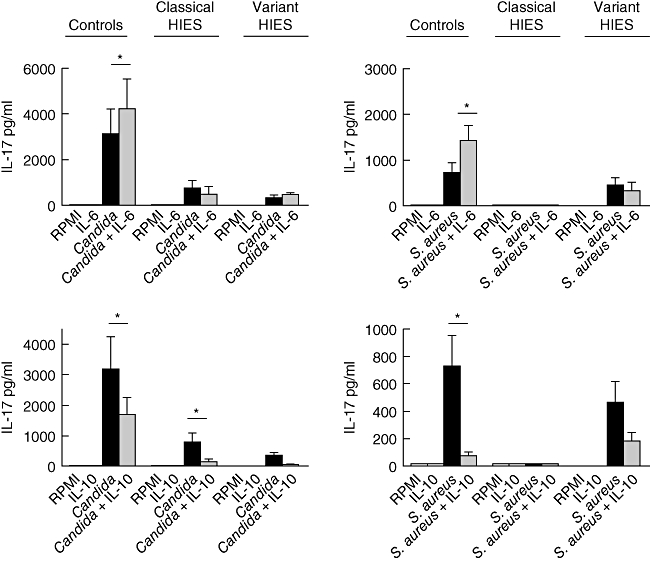
Defective interleukin (IL)-6, but not IL-10, pathway in hyperimmunoglobulin E syndrome (HIES) patients. Human peripheral blood mononuclear cells (PBMCs) from healthy controls (n = 8), ‘classical’ HIES (n = 5) and three members of the HIES family bearing the signal transducer and activator of transcription 3 (STAT 3) linker domain mutation were stimulated for 5 days with Candida albicans or Staphylococcus aureus. The effect of either IL-6 or IL-10 on the induction of IL-17 was assessed by enzyme-linked immunosorbent assay. *P < 0·05.
Discussion
Mutations in the SH2 and DNA-binding domain of STAT3 have been reported to be the cause of disease in a large proportion of HIES patients [4]. These mutations function as dominant-negative mutations [4] and result in a defective Th17 response in these patients [9,10], explaining many of the clinical features of HIES. In the present study we confirm, on one hand, the relationship between HIES and defective Th17 responses; on the other hand, we also refine this notion to include the relationships between the type of STAT3 mutation, immunological response to relevant microbial stimulation and clinical phenotype of the patients.
All studies to date have used an artificial methodology to generate Th17 cells and induce IL-17 production in cells from HIES patients, using either mitogenic antibodies such as anti-CD3 and anti-CD28 [9,10] or recombinant cytokine cocktails [11]. Recently we have developed a novel method to induce IL-17 production and generate Th17 cells using exclusively microbial stimulation [18], a method that mimics much more closely the in vivo conditions during infection. Although we can confirm defective Th17 generation and IL-17 production by cells isolated from patients with HIES [9–11], several important aspects are now apparent when using this improved methodology. First, defective IL-17 induction differs between stimulation with S. aureus or C. albicans. When Th17 responses were assessed both these microorganisms, which are the most important in HIES patients, were equally defective in generating CD4+ IL-17+ cells. Surprisingly, however, C. albicans was still capable of stimulating approximately 20–30% of normal IL-17 production, while S. aureus was completely defective as an IL-17 stimulus in HIES patients (Fig. 1c). This finding is important as it may explain why it is mainly mucosal; nailbed infection is the most common Candida complication in HIES patients (83% in one large study), while systemic candidiasis is relatively rare [3]. Notably, patients with chronic mucocutaneous candidiasis who have the same clinical spectrum of Candida infection [19] have also been reported to have a specific defect in Candida-induced IL-17 production [20]. This supports the conclusion that IL-17 is important in mucosal anti-Candida host defence and that the lower IL-17 found in our patients is indeed clinically relevant.
Secondly, an important observation of our study is represented by indistinguishable immunological responses in patients with the ‘classical’ clinical form of HIES, independent of the presence or absence of STAT3 mutations. All the patients who had a strong phenotype of the disease displayed similar defects in IL-17 production and Th17 generation. Our data are supported by the report of one HIES patient without STAT3 mutation and defective Th17 responses [21], and suggests strongly that in patients with the ‘classical’ presentation of HIES, but in which no STAT3 mutation is found, defects in the same immunological pathways are the most probable cause of the disease. This may also imply that defective Th17 responses are a more sensitive diagnostic tool for HIES.
Thirdly, one of the most interesting findings of our study is the description of a clear association of a milder phenotype of the disease in a Dutch family with a less severe defect in IL-17 production, due probably to the linker domain triplet that did not lead to a frameshift [13]. Patients from this family suffer from skin infections with S. aureus, candidiasis of the nailbeds (but not of the mucosae), dermatitis, hyper-IgE and eosinophilia, but they lack any respiratory infections (either with S. aureus or other pathogens). With regard to non-infectious manifestations, persistent primary dentition, a common sign in HIES, was seen in one patient of this family, one patient displayed scoliosis, cheilognathopalatoschisis and pathological fractures, and one patient suffered from an aortic aneurysm. The defects in IL-17 responses to S. aureus in cells isolated from this family were milder compared to the ‘classical’ HIES patients, as they were still able to release approximately 30% of the normal IL-17 production. In line with the presence of candidiasis as a clinical symptom in the family, IL-17 production after C. albicans stimulation was equally defective compared to the other patients.
In addition to IL-17, other defects in the cytokine response of HIES patients have also been reported, such as a defective IFN-γ production [17,22], and increased granulocyte–macrophage colony-stimulating factor (GM-CSF) [23]. In line with these previous studies, in our study IFN-γ production was decreased in HIES patients, while IL-10 release was significantly higher compared to controls. Production of IFN-γ was defective in response to both C. albicans and S. aureus. IFN-γ is the prototype of Th1 cytokines and plays a crucial role in activation of the innate and adaptive host response against these pathogens [24]. Therefore, the defective IFN-γ response could be at least as relevant as the defect found in IL-17. Furthermore, it should be kept in mind that IFN-γ therapy is a relatively safe therapeutic option [25] and it has been reported that recombinant IFN-γ can enhance neutrophil chemotactic responses in patients with HIES [26]. Together, these data argue strongly for a dysbalance of Th subsets in patients with HIES, with defective responses of the proinflammatory subsets Th1 and Th17, and increased function of the anti-inflammatory Th2 subset.
In contrast to Th-derived cytokines, the release of IL-1β was normal in HIES patients. As IL-1β is important for the generation of Th17 cells [27], this result suggests that it is not a defective IL-1β/IL-1RI axis that is responsible for the defects of IL-17 production in HIES patients. This hypothesis is sustained by the normal generation of Th17 responses in individuals with MyD88 or IRAK4 mutations that are defective in the IL-1RI signalling [as well as Toll-like receptor (TLR) and IL-18R pathways][11].
The defective generation of Th17 responses in HIES must therefore be located at the level of another immunological pathway, the most obvious being the IL-6/STAT3 axis [6]. To test this hypothesis, we investigated the effect of IL-17 co-stimulation with microbial stimuli in combination with IL-6. While IL-6 potentiated the production of IL-17 induced by C. albicans or S. aureus in healthy individuals, no such effect was observed in either the ‘classical’ HIES or the family with the variant HIES. The striking observation that the members of the HIES family were able to generate Th17 cells upon contact with pathogens suggests that the linker domain of the STAT3 gene is involved in another unknown function of the STAT3 molecule, and highlights the observation that a mutation at a different location in the STAT3 gene leads to a different functional and clinical outcome. In contrast to the defective responses to IL-6, the inhibitory effects of IL-10 on IL-17 production were similar in healthy volunteers or HIES patients, suggesting that STAT3 is redundant for IL-10 signalling leading to reduced IL-17 production.
In conclusion, the present study demonstrates that patients with HIES have differential defects in IL-17 responses to the two main pathogens associated with the disease, S. aureus and C. albicans, and this is comparable with the clinical features of this syndrome. In addition, the extent of the Th17 defect is due to the location of the STAT3 mutation, and is associated with the clinical phenotype in these patients. Furthermore, defective Th17 responses are a more sensitive marker of the disease in HIES patients than STAT3 mutations.
Acknowledgments
M. G. N. was supported by a Vidi Grant of the Netherlands Organization for Scientific Research. These studies were supported by donations collected by one of the HIES patients.
Disclosure
None declared.
References
- 1.Davis SD, Schaller J, Wedgwood RJ. Job's syndrome: recurrent ‘cold’ staphylococcal abcesses. Lancet. 1966;1:1013–15. doi: 10.1016/s0140-6736(66)90119-x. [DOI] [PubMed] [Google Scholar]
- 2.Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics. 1972;49:59–70. [PubMed] [Google Scholar]
- 3.Grimbacher B, Holland SM, Gallin JI, et al. Hyper-IgE syndrome with recurrent infections – an autosomal dominant multisystem disorder. N Engl J Med. 1999;340:692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- 4.Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–62. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 5.Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 6.Nishihara M, Ogura H, Ueda N, et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int Immunol. 2007;19:695–702. doi: 10.1093/intimm/dxm045. [DOI] [PubMed] [Google Scholar]
- 7.Yang XO, Panopoulos AD, Nurieva R, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–63. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 8.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–67. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milner JD, Brenchley JM, Laurence A, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–6. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma CS, Chew GY, Simpson N, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205:1551–7. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Beaucoudrey L, Puel A, Filipe-Santos O, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. 2008;205:1543–50. doi: 10.1084/jem.20080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mezger M, Kneitz S, Wozniok I, Kurzai O, Einsele H, Loeffler J. Proinflammatory response of immature human dendritic cells is mediated by dectin-1 after exposure to Aspergillus fumigatus germ tubes. J Infect Dis. 2008;197:924–31. doi: 10.1086/528694. [DOI] [PubMed] [Google Scholar]
- 13.Netea MG, Schneeberger PM, de Vries E, Kullberg BJ, van der Meer JW, Koolen MI. Th1/Th2 cytokine imbalance in a family with hyper-IgE syndrome. Neth J Med. 2002;60:349–53. [PubMed] [Google Scholar]
- 14.Lehrer RI, Cline MJ. Interaction of Candida albicans with human leukocytes and serum. J Bacteriol. 1969;98:996–1004. doi: 10.1128/jb.98.3.996-1004.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Graaf CA, Netea MG, Verschueren I, van der Meer JW, Kullberg BJ. Differential cytokine production and Toll-like receptor signaling pathways by Candida albicans blastoconidia and hyphae. Infect Immun. 2005;73:7458–64. doi: 10.1128/IAI.73.11.7458-7464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Netea MG, Gow NA, Munro CA, et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J Clin Invest. 2006;116:1642–50. doi: 10.1172/JCI27114. Epub 18 May 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Netea MG, Kullberg BJ, van der Meer JW. Severely impaired IL-12/IL-18/IFNgamma axis in patients with hyper IgE syndrome. Eur J Clin Invest. 2005;35:718–21. doi: 10.1111/j.1365-2362.2005.01564.x. [DOI] [PubMed] [Google Scholar]
- 18.van de Veerdonk FL, Marijnissen RJ, Kullberg BJ, et al. The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe. 2009;5:329–40. doi: 10.1016/j.chom.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Lilic D. New perspectives on the immunology of chronic mucocutaneous candidiasis. Curr Opin Infect Dis. 2002;15:143–7. doi: 10.1097/00001432-200204000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Eyerich K, Foerster S, Rombold S, et al. Patients with chronic mucocutaneous candidiasis exhibit reduced production of Th17-associated cytokines IL-17 and IL-22. J Invest Dermatol. 2008;128:2640–5. doi: 10.1038/jid.2008.139. [DOI] [PubMed] [Google Scholar]
- 21.Renner ED, Rylaarsdam S, Anover-Sombke S, et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol. 2008;122:181–7. doi: 10.1016/j.jaci.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shirafuji Y, Matsuura H, Sato A, Kanzaki H, Katayama H, Arata J. Hyperimmunoglobulin E syndrome: a sign of Th1/Th2 imbalance. Eur J Dermatol. 1999;9:129–31. [PubMed] [Google Scholar]
- 23.Vargas L, Patino PJ, Rodriguez MF, et al. Increase in granulocyte–macrophage-colony-stimulating factor secretion and the respiratory burst with decreased l-selectin expression in hyper-IgE syndrome patients. Ann Allergy Asthma Immunol. 1999;83:245–51. doi: 10.1016/S1081-1206(10)62648-8. [DOI] [PubMed] [Google Scholar]
- 24.Billiau A. Interferon-γ: biology and role in pathogesis. Adv Immunol. 1996;61:61–130. doi: 10.1016/s0065-2776(08)60428-9. [DOI] [PubMed] [Google Scholar]
- 25.Hubel K, Dale DC, Liles WC. Therapeutic use of cytokines to modulate phagocyte function for the treatment of infectious diseases: current status of granulocyte colony-stimulating factor, granulocyte–macrophage colony-stimulating factor, macrophage colony-stimulating factor, and interferon-gamma. J Infect Dis. 2002;185:1490–501. doi: 10.1086/340221. [DOI] [PubMed] [Google Scholar]
- 26.Jeppson JD, Jaffe HS, Hill HR. Use of recombinant human interferon gamma to enhance neutrophil chemotactic responses in Job syndrome of hyperimmunoglobulinemia. J Pediatr. 1991;118:383–7. doi: 10.1016/s0022-3476(05)82151-1. [DOI] [PubMed] [Google Scholar]
- 27.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–9. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]