

Abstract
We investigated Toll-like receptors (TLR-3, -4 and -7) expression in circulating mononuclear cells of patients with immunoglobulin A nephropathy (IgAN), a disease with debated relationships with mucosal immunity. TLR-4 expression (detected by fluorescence activated cell sorter) and mRNA transcriptional levels (Taqman) were significantly higher in patients with IgAN than in healthy controls (P = 0·00200 and P = 0·0200). TLR-3 and TLR-7 were not modified significantly. In IgAN patients proteinuria was correlated significantly with TLR-4 expression (P = 0·0312). In a group of nephrotic syndromes, TLR-3, -4 and -7 expression was similar to healthy controls. A significant difference in TLR-4 expression and mRNA levels was found between very active IgAN patients (proteinuria > 1 g/1·73 m2/day in association with severe microscopic haematuria) and inactive patients (proteinuria < 0·5 g/1·73 m2/day, with absent or minimal haematuria). No correlation with levels of aberrantly glycosylated IgA1, age, renal biopsy features or therapy was found. This study shows for the first time an up-regulation of TLR-4 in circulating mononuclear cells of patients with IgAN, particularly in association with proteinuria and heavy microscopic haematuria.
Keywords: aberrantly glycosylated IgA, IgA nephropathy, proteinuria, Toll-like receptor
Introduction
Immunoglobulin A nephropathy (IgAN), a glomerular disease characterized by prevalent mesangial IgA deposits, presents frequently with macroscopic haematuria coincident with mucosal infections [1]. A role of mucosal immunity has been suspected following several observations, such as the prevalence of polymeric IgA (a typical products of mucosal immune system) and the detection of secretory IgA in mesangial deposits [2], the presence of IgA reacting with environmental antigens in sera and in glomeruli [3,4] and the cross-reactivity of tonsillar IgA with IgAN deposits [5]. IgA mesangial deposits were reproduced in experimental animals triggering mucosal immunity by using oral immunization with gliadin or other alimentary antigens [6,7], intranasal administration of Sendai virus, a common respiratory pathogen [8,9] or oral immunization with Haemophilus parainfluenzae antigens [10], and these antigens were detected in renal tissue of patients with IgAN [11]. Mice strains with persistent parvovirus infections (Aleutian mice) develop glomerular changes with IgA deposits [12]. In mice prone to develop IgAN (ddY), infection with Coxsackie B4 increased mesangial proliferation and matrix expansion [13]. The antigen of the Staphylococcus aureus cell envelope induced experimental IgA deposits in mice [14]. In humans, besides cases of methycillin-resistant S. aureus (MRSA) infection [15], Staphylococcus antigens have been reported in 50% of kidneys of patients with IgAN [16]. Relationships between IgAN and infection have been claimed for other pathogens, including cytomegalovirus, Epstein–Barr virus, enterovirus, Helicobacter pylori and others [1]. All these data suggest that exogenous antigens derived from pathogens could play a role in the pathogenesis of IgAN, although the deep mechanisms through which these antigens trigger IgAN are still undefined.
In mesangial deposits and in sera of patients with IgAN IgA1 presents with an abnormal glycosylation [17–21], which has been proved to be consequent to abnormal systemic responses to mucosally encountered antigen [22]. The mucosal surfaces are in continuous contact with environmental antigens or microbes, either pathogens or not, with an individual variability in immune response, either innate or adaptive. The first immune reaction is driven by innate immunity and one of the major actors are the Toll-like receptors (TLRs). TLRs sense pathogen-associated molecular patterns (PAMPs) of bacterial or viral origin, and also endogenous host ligands, including damage-associated molecular patterns (DAMPs) released from necrotic cells or in inflammatory environments [23–25]. Upon activation, most TLRs induce a common intracellular signalling pathway that culminates in the activation of the interferon regulatory factor (IRF)-3/IRF-7 and nuclear factor kappa B (NF-κB) transcription factors, with consequent induction of cytokines, chemokines, cell surface adhesion molecules and co-stimulatory molecules, promoting not only innate but also adaptive immune response and inflammation [26]. Hence, TLR ligation may exacerbate glomerulonephritis by activating neutrophils, macrophages or other cells of the innate immune system to increase glomerular inflammation and renal damage [24,25]. Alternatively, inflammation products can activate TLRs present on intrinsic renal cells, including mesangial cells. TLRs are considered a link between innate and adaptive immunity played at mucosal and systemic level.
In IgAN, several indications suggest a dysregulation of processing exogenous antigens derived from common pathogens, which can lead to abnormal immune response, aberrant IgA synthesis and renal damage. We hypothesized that TLR activation might be triggered by a defective mucosal control of exogenous antigens, and we speculated that this activation may condition IgA synthesis, IgA renal deposits formation and renal inflammation. Hence, we aimed at investigating TLR expression in circulating mononuclear cells of patients with IgAN. We focused upon TLR-3 (activated mainly by viral dsRNA), TLR-7 (receptor for viral ssRNA) and TLR-4 [activated by various ligands, including Gram-negative bacterial lipopolysaccharide (LPS), heat shock proteins of bacterial and host origin, fibrinogen and fibronectin and several DAMPs derived from host cells] looking for correlations with patients' clinical and histological features. TLR-3 and TLR-7 were selected due to a possible role of viral infections in IgAN and TLR-4 because it can be triggered by a large variety of exogenous and endogenous agonists, and it has significant cross-talk with other TLRs, including TLR-2 [27,28].
Finally, levels of aberrantly glycosylated IgA1, which are thought currently to originate from the mucosal immune system, were measured.
Materials and methods
Subjects
The study enrolled 47 patients of Caucasian origin with biopsy-proven diagnosis of primary IgAN from three participating centres upon written informed consent and approval of local Ethical Committees. Table 1 reports clinical and histological details. At time of blood withdrawal no patient had fever or active urinary or respiratory tract infections.
Table 1.
Relevant clinical data of the 47 patients with immunoglobulin A nephropathy (IgAN) and 40 healthy controls (HC) investigated.
| IgAN | HC | |
|---|---|---|
| Gender: males/females | 39/8 | 33/7 |
| Age (years) | 22·17 ± 15·3 (5·4–72) | 25·4 ± 14·7 (5·5–70) |
| Time (years) from renal biopsy | 2·8 ± 3·2 (0–12) | – |
| Proteinuria (g/day/1·73 m2) | 1·2 ± 1·5 (0–9·44) | – |
| Creatinine clearance (ml/min/1·73m2) | 104·2 ± 43·7 (25–189) | – |
| CKD I | 29 | |
| CKD II | 11 | |
| CKD III | 6 | |
| CKD IV | 1 | |
| Histological grade | – | |
| G1 mild | 14 | |
| G2 moderate | 21 | |
| G3 severe | 12 |
Data are expressed as means ± standard deviations and ranges in brackets. CKD: chronic kidney disease stages. Histological grade (G) (according to reference [38]) (G1, normal or mild mesangial proliferation and matrix increase; G2, moderate focal or diffuse mesangial proliferation, endocapillary proliferation and crescents in <50% of glomeruli; G3, severe lesions with sclerosis involving > 30% of glomeruli or crescents in >50% of glomeruli). At time of blood withdrawal no patient had fever or active urinary or respiratory tract infections.
The histological classification of IgAN in three grades of renal lesions (grade 1: mild; grade 2: moderate; grade 3: severe) was performed according to a recent report [29]. Patients with Henoch–Schoenlein purpura nephritis, lupus, coeliac disease, chronic liver disease or diabetes mellitus were excluded.
As the renal disease control population, we tested 27 samples from 18 young subjects with idiopathic nephrotic syndrome (NS) (six had focal segmental glomerulosclerosis at renal biopsy, three minimal change disease and others were children and adolescents with steroid-sensitive NS). A group of 10 children and adolescents (mean age 9·5 ± 2·5 years) with respiratory or gastrointestinal infections without any detectable urinary involvement was also investigated: six cases had acute febrile gastrointestinal diseases (stool culture negative for bacteria), three had viral pneumonia and one infectious mononucleosis. As a healthy control population (HC), a group of 40 sex- and age-matched Caucasian healthy subjects was selected.
Creatinine clearance (CrCl) and proteinuria were calculated from 24 h collection and corrected for the body surface area in both children and adults, in an effort to homogenize units of measurement in various ages [30]. We considered clinically inactive the IgAN patients who had proteinuria < 0·5 g/1·73 m2/day and microscopic haematuria 0 or 1+ at dipstick in the morning urine sample (the dipstick measure range was 0–3+). Very active IgAN patients were considered those with proteinuria > 1 g/1·73 m2/day and microscopic haematuria 2+ or 3+ at dipstick in the morning urine sample.
Flow cytometry
Peripheral blood mononuclear cells (PBMC) (5 × 106 cells), isolated by Ficoll-Hypaque density gradients (Sigma Co., St Louis, MO, USA) washed with ice-cold phosphate-buffered saline (PBS), were double-stained with 5 ml of anti-human CD14-phycoerythrin (PE) (mouse monoclonal IgG2ak; BD Pharmingen, San Diego, CA, USA) and 5 ml of TLR-4 fluorescein isothiocyanate (FITC) mouse monoclonal antibody (IgG2a; Abcam, Cambridge, UK) for 30 min at room temperature (RT). After PBS washes, cells were analysed by flow cytometry (Beckam-Coulter Epics XL, Miami, FL, USA). Acquired data were analysed with EXPO32 software (Beckman-Coulter). Results were expressed as percentage of cells positive for both TLR-4 and CD14 and as log mean fluorescence intensity (log MFI).
To detect intracellular TLR-3 and -7, PBMC were permeabilized with the Fix & Perm Cell Kit (Caltag Laboratories, Carlsbad, CA, USA) and stained with mouse monoclonal anti-TLR-3–FITC (IgG1 subclass; Abcam) or primary rabbit polyclonal antibody to TLR-7 (IgG; Abcam) followed by fluoresceinated goat anti-rabbit IgG (Abcam). No selection was made for CD14+ cells. As negative control, appropriate isotype controls were used (BD Pharmingen and Abcam).
Reverse transcription
Total RNA was extracted from PBMC using the TRI-Reagent kit (Sigma Co.) according to the manufacturer's protocol and 1 mg of total RNA was reverse-transcribed as detailed in Coppo et al.[31] in a GeneAmp PCR system 9700 Thermal Cycle (Applied Biosystems, Foster City, CA, USA) under the following conditions: 10 min at 25°C, 60 min at 48°C and 5 min at 95°C for the inactivation of enzyme; the cDNAs were stored at −80°C.
Relative quantification by real-time PC
Relative quantification of mRNA expression of selected genes was achieved by means of Taqman amplification and normalization to Abelson murine leukaemia viral oncogene homologue 1 (Abl), chosen as a reference gene, using the ABI PRISM 7700 Sequence Detection System (PE; Applied Biosystems). For quantification of TLR-3, -4 and -7 mRNA expression we used a set of primers and a fluorogenic oligonucleotide probe designed to hybridize to the specific target sequence (Assay-on-Demand: TLR-3 hs-00152933, TLR-4 hs-00152937, TLR-7 hs-00152971, and ABL hs-00245445; Applied Biosystems). Relative quantification of target gene expression in patients compared with normal samples was performed with the ΔΔCt method and the relative TLRs fold changes were determined as detailed previously [31].
Isolation of IgA1
Immunoglobulins were isolated by ammonium sulphate precipitation at 4°C for 12 h and dissolved in 0·175 M Tris-HCl, pH 7·5. The fractions obtained were incubated with agarose (Sigma) conjugated with the lectin Jacalin (specific for GalNAc-Gal residues) at RT for 12 h. After washing in Tris-HCl, the IgA1 fraction was eluted by 0·8 M Gal and dialyzed. Purified IgA1 samples were stored at −80°C until use.
Detection of aberrantly glycosylated IgA1
Aberrantly glycosylated IgA1 were detected by their increased binding to lectins specific for GalNAc residues (Helix aspersa: HA and Vicia villosa: VV). Microplates were coated with preparations of purified IgA1 isolated by Jacalin–agarose columns. After washes, 50 µl of biotinylated lectins HA (Sigma Co.), 2 µg/ml, VV (Vector, Burlingame, CA, USA) was added and incubated for 2 h at RT. Then, 50 µl of alkaline phosphatase–streptavidin conjugate (Sigma Co.) (2·5 µg/ml) was added and incubated for 2 h at RT. After washes, 50 µl of phosphatase substrate (P-nitrophenyl phosphate; Sigma Co.) was added and the absorbance at 405 nm was read on a Biorad microplate reader in the linear phase of the enzymatic reaction. The method has been detailed elsewhere [32].
Statistical analysis
The normal distribution of variables was tested by means of the one-sample Kolmogorov–Smirnov test. Data were expressed as mean ± standard deviation (s.d.). Differences between multiple groups were analysed by means of analysis of variance (anova) with the Newman–Keuls multi-comparison test. For comparison of the two groups the independent-sample t-test or the Mann–Whitney U-test were used when appropriate. Pearson's test was used to correlate the two data series. A P-value of <0·05 was considered significant. Statistical software spss version 14·0 was used to elaborate data (SPSS Inc., Chicago, IL, USA).
Results
The study enrolled 47 patients with primary IgAN, who contributed 74 blood samples (15 patients had two to four determinations over a period of 2 months–3 years) and 40 age- and sex-matched healthy controls (HC) (relevant clinical data shown in Table 1). Histological grading showed the prevalence of moderate lesions. Mean proteinuria was 1·2 ± 1·5 (ranging from 0 to 9·4) g/24 h/1·73 m2. Blood samples were obtained from 19 patients considered to be very active (proteinuria > 1 g/1·73 m2/day in association with haematuria 2+ or 3+ at dipstick in the morning urine), while 15 samples were obtained from patients in inactive clinical phase (proteinuria < 0·5 g/1·73/m2/day and haematuria 0+ or 1+). Samples were obtained from patients receiving angiotensin II receptors blockers in 37 cases and prednisone or cytotoxic drugs in 26 cases; in 11 cases no treatment was given. A disease control group of 27 samples from 18 patients with idiopathic nephrotic syndrome (mean age 14·58 ± 11·06) was also investigated. They were in various phases of activity, with mean proteinuria of 7·07 ± 5·74 g/1·73 m2/day (range 0–17). In 18 cases patients were assuming steroids and in six cases immunosuppressive drugs. Their creatinine clearance was always >90 ml/min/1·73 m2. We also investigated 10 children or adolescents with respiratory or gastrointestinal infections without any detectable urinary involvement.
In patients with IgAN, circulating PBMC showed significantly increased surface expression of TLR-4 in the CD14-positive population (herein defined as monocytes) at double-staining (detailed results in Table 2), and mRNA expression analysis increased levels of TLR-4 mRNA in PBMC (Fig. 1 and Table 2). In pilot experiments mRNA expression analysis was also performed on the separate subpopulation of monocytes, confirming the same results; therefore, for all the experiments whole PBMC were used due to higher RNA recovery.
Table 2.
Expression of Toll-like receptor (TLR)-4, TLR-3 and TLR-7 in peripheral blood mononuclear cells (PBMC) in patients with immunoglobulin A nephropathy (IgAN) and in healthy controls.
| IgAN patients | Healthy controls | P | ||
|---|---|---|---|---|
| TLR-4 flow cytometry | % positive cells† | 13·22 ± 21·91 | 4·29 ± 6·32 | 0·0113 |
| log MFI‡ | 3·52 ± 2·81 | 1·70 ± 1·46 | 0·0073 | |
| TLR-4 mRNA | Fold increase§ | 2·18 ± 2·39 | 1·42 ± 0·66 | 0·0200 |
| TLR-3 flow cytometry | % positive cells† | 9·99 ± 19·09 | 5·57 ± 3·06 | n.s. |
| log MFI‡ | 3·82 ± 2·26 | 1·54 ± 0·32 | 0·0246 | |
| TLR-3 mRNA | Fold increase§ | 0·71 ± 0·83 | 1·27 ± 0·99 | n.s. |
| TLR-7 flow cytometry | % positive cells† | 1·21 ± 1·47 | 1·09 ± 1·03 | n.s. |
| log MFI‡ | 1·20 ± 0·75 | 1·26 ± 0·20 | n.s. | |
| TLR-7 mRNA | Fold increase§ | 0·75 ± 0·83 | 0·90 ± 0·39 | n.s. |
Data are expressed as means ± standard deviations and ranges in brackets. P: significance of the difference (Student's t-test); n.s.: not significant.
% positive cells: percentage of cells positive for surface TLR staining. Fluorescence activated cell sorter (FACS) analysis. TLR-4 expression was assessed in CD14 positive cells, while other data refer to PBMC.
Log MFI: mean fluorescence intensity of surface TLR staining (FACS). TLR-4 expression was assessed in CD14 positive cells, while other data refer to PBMC.
Fold increase: semiquantitative analysis of TLR mRNA in PBMC expressed in arbitrary units, as TLRs mRNA fold changes to the reference mRNA (reverse transcription–polymerase chain reaction, Taqman).
Fig. 1.
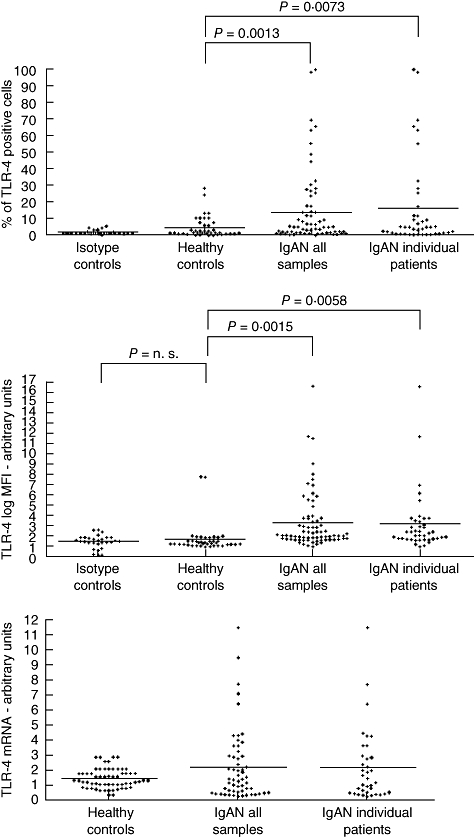
Expression of Toll-like receptor (TLR)-4 in peripheral blood mononuclear cells in patients with immunoglobulin A nephropathy (IgAN). Percentage of cells positive for surface TLR-4 staining for CD14+cells. Fluorescence activated cell sorter (FACS) analysis. Isotype control: appropriate isotype controls. Healthy controls: 40 samples from 40 healthy subjects. IgAN all samples: 74 samples from 47 patients. IgAN individual patients: first sample obtained from 47 patients during the follow-up. Mean fluorescence intensity (log MFI) of surface TLR-4 staining for CD14+ cells (FACS). Isotype control: appropriate isotype controls. Healthy controls: 40 samples from 40 healthy subjects. IgAN all samples: 74 samples from 47 patients. IgAN individual patients: first sample obtained from 47 patients during the follow-up. Semiquantitative analysis of TLR-4 mRNA in peripheral blood mononuclear cells expressed as TLRs mRNA fold changes to the reference mRNA (reverse transcription–polymerase chain reaction, Taqman). Healthy controls: 40 samples from 40 healthy subjects. IgAN all samples: 74 samples from 47 patients. IgAN individual patients: first sample obtained from 47 patients during the follow-up. P: significance of the difference between groups.
To avoid the possible bias effect of multiple repeated sampling from the same patients we repeated the analysis considering one sample from each IgAN patient (the first obtained during the follow-up) obtaining the same significant difference versus controls (% positive cells: 15·80 ± 26·38 versus 4·29 ± 6·32 in HC, P = 0·0073; log MFI: 3·13 ± 2·85 versus 1·70 ± 1·47 in HC, P = 0·0058; TLR-4 mRNA 2·16 ± 2·42 versus 1·42 ± 0·66 in HC, P = 0·0283) (Fig. 1). A significant correlation was found between % positive TLR-4 cells and TLR-4 mRNA levels in monocytes of patients with IgAN (P = 0·0389).
TLR-3 showed only a trend towards increased mean expression in PBMC, without changes in TLR-3 mRNA values. Expression of intracellular TLR-7 and TLR-7 mRNA transcriptional levels in circulating mononuclear cells were almost identical in IgAN patients and in HC (details in Table 2).
No modifications in expression of TLR-3, -4 and -7 and their corresponding mRNA levels were detected in the disease control groups of patients with nephrotic syndrome or with common respiratory or gastrointestinal infections without renal involvement [fluorescence activated cell sorter (FACS) data not shown; Fig. 2 reports TLR mRNA transcriptional levels] compared to HC.
Fig. 2.
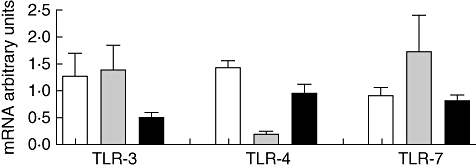
Levels of Toll-like receptor (TLR)-3, -4 and -7 mRNAs in peripheral mononuclear cells of control groups (columns indicate means and bars represent standard deviations). White columns: data in 40 healthy controls. Grey columns: data in 10 subjects with respiratory or gastrointestinal infections without any detectable urinary involvement. Black columns: data in 27 patients with nephrotic syndrome. No significant difference between the groups was observed.
In patients with IgAN, a significant correlation was found between proteinuria and TLR-4 expression (P = 0·0312). Proteinuria levels were significantly higher in patients with abnormally increased expression of TLR-4 (>95th percentile log MFI in healthy controls) than in those with normal values (P < 0·0001) (Fig. 3). No correlation between proteinuria and TLR expression was detected in patients with idiopathic nephrotic syndrome. During phases of clinical activity with high proteinuria and severe microscopic haematuria IgAN had significantly greater expression of TLR-4 on CD14+ PBMC than during phases of clinical inactivity (log MFI: 4·99 ± 4·22 versus 3·01 ± 2·74; mRNA expression 2·48 ± 2·66 versus 0·85 ± 0·72: P < 0·05) (Fig. 4).
Fig. 3.
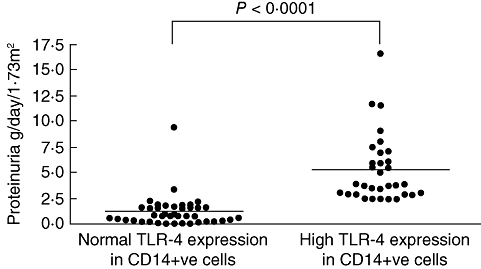
Relationship between Toll-like receptor (TLR)-4 expression and proteinuria in peripheral CD14+ cells of patients with immunoglobulin A nephropathy (IgAN). Significant difference in proteinuria levels between patients with normal or increased expression of TLR4 in CD14+ cells. Normal MFI values were considered those < 95th percentile in healthy controls (MFI < 2·3) (P < 0·0001).
Fig. 4.
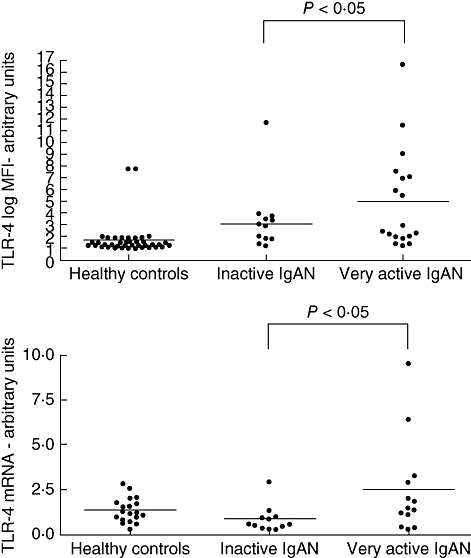
Expression of Toll-like receptor (TLR)-4 in peripheral CD14+ cells in healthy controls and in patients with immunoglobulin A nephropathy (IgAN) during different phases of clinical activity. Inactive IgAN: 15 patients presenting with proteinuria < 0·5 g/ 1·73 m2/day and microscopic haematuria 0 or 1+ at dipstick (range 0 to 3+) in the morning urine sample. Very active IgAN: 19 patients with proteinuria > 1 g/1·73m2/day in association with severe microscopic haematuria (2 or 3+ at dipstick). (a) Mean fluorescence intensity (log MFI) of surface TLR-4 staining for CD14+ cells. (b) Semiquantitative analysis of TLR-4 mRNA in peripheral blood mononuclear cells.
No correlation was found between TLR expression in patients with IgAN and age, disease duration from onset or renal biopsy grades.
Some patients had repeated measures of TLR-4 in circulating CD+ 14 cells during various clinical phases of IgAN and both TLR-4 surface expression and mRNA levels showed changes consensual with the clinical activity. One example concerns a boy investigated during a phase of clinical and histological activity with gross haematuria, followed by clinical improvement, whose data are reported in Fig. 5.
Fig. 5.
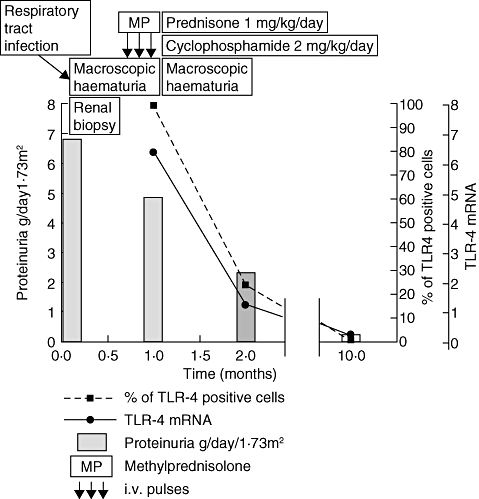
Correlation between clinical features and expression of Toll-like receptor (TLR)-4 in peripheral monocytes (CD14+ cells) in a representative case of immunoglobulin A nephropathy (IgAN). ST, a 9-year-old boy, had a macroscopic haematuria shortly following an upper respiratory tract infection. Proteinuria (grey bars) was concomitantly very high. Renal biopsy showed a severely proliferative IgAN with intra- and extracapillary proliferation (class 2 according to reference [29]). He was treated with three methylprednisolone pulses (MP), 10 mg/kg each, followed by oral prednisone (P) 1 mg/kg/day in association with cyclophosphamide 2 mg/kg/day for 8 weeks. One month after renal biopsy he had another episode of macroscopic haematuria: circulating mononuclear cells analysis showed 100% CD14 cells positive for TLR-4 with sixfold increased TLR-4 mRNA. During the follow-up the up-regulation of TLR-4 was reversed within normal limits in parallel with disappearance of proteinuria with residual moderate microscopic haematuria.
Serum levels of aberrantly glycosylated IgA were increased significantly in patients with IgAN compared to HC (binding to VV: IgAN: 0·24 ± 0·06, range: 0·18–0·41, HC: 0·20 ± 0·02, range: 0·16–0·27; P = 0·0003; binding to HA: IgAN: 0·46 ± 0·10, range 0·34–0·70, HC: 0·42 ± 0·04, range: 0·35–0·55; P = 0·0326). No correlation was found between levels of aberrantly glycosylated IgA1 and mononuclear cells TLR mRNA expression in patients with IgAN or in HC.
Discussion
This is the first report showing an increased expression of TLR-4 in circulating mononuclear cells of patients with IgAN. In allograft biopsies from patients with IgAN recurring after transplantation, increased expression of TLR-4 and TLR-9 mRNAs was observed recently [33], but no data in peripheral cells were reported. Activation of the TLR-9 and its intracellular adapter myeloid differentiation factor 88 (MyD88) pathway has been reported recently in splenocytes of ddY mice, which develop IgAN spontaneously [33]. In patients with IgAN the TLR-9 polymorphism was found to be associated with disease progression but not with disease incidence, leaving unclear the functional consequences of this polymorphism [33]. Until now, no report has existed on TLR-9 expression in circulating blood cells of patients with IgAN and our report does not exclude TLR-9 engagement in IgAN, which was not investigated in our protocol. Similarly, we cannot exclude engagements of other TLRs not investigated directly, e.g. TLR-2.
In our investigation, TLR-4 transcriptional levels and surface expression in peripheral monocytes were increased significantly in patients with IgAN, particularly in coincidence of signs of urinary activity. No significant modifications were found for TLR-3 and TLR-7 expression. The correlation with proteinuria was specific to IgAN and not observed in patients with idiopathic nephrotic syndrome. However, we did not attempt to prove the absolute specificity of TLR up-regulation in IgAN because it might be detected in other glomerular or tubulo-interstitial renal diseases, as suggested by several different experimental models and in various human settings [34,35]. The present report indicates that in IgAN the TLR pathway of the innate immunity is activated, which is in agreement with the old observation of bouts of gross haematuria coincident with mucosal infection. Most experimental models and clinical observations suggested a role for viral pathogens in IgAN and the results we observed, indicating an isolated up-regulation of TLR-4, were somewhat unexpected. TLR-4 recognizes exogenous ligands (LPS from Gram-negative bacteria, Chlamydophila pneumoniae, Chlamydial heat shock protein) as well as endogenous ligands (heat shock proteins HSP-60, fibronectin extra domain A, MM-LDL, hyaluronic acid oligosaccharides, heparin sulphate and DAMPs derived from damaged host cells) [23]. It is of interest that in a model of nephrotoxic nephritis, TLR-4 ligation is necessary for the full development of glomerular inflammation [36], with the contribution of both intrinsic renal cells and circulating leucocytes. LPS injection in nephrotoxic nephritis activates TLR-4 on mesangial cells and produces release of CXC chemokines which favours neutrophil influx and glomerulonephritis. Notably, in this model TLR-4 ligation in peripheral monocytes was proven to worsen antibody-mediated renal damage, via release of proinflammatory cytokines and adhesion molecules. This experimental observation in animals gives speculative relevance to our findings of increased TLR-4 expression in peripheral monocytes of patients with IgAN and correlation with urinary activity. Indeed, we observed a significant correlation between expression of TLR-4 on circulating mononuclear cells (CD14 positive cells) and amount of proteinuria, or phases of clinical activity in patients with IgAN. In respect of this finding the reported association of CD14 gene–159C polymorphism with progression of IgAN, in which heavy proteinuria plays a pivotal role, may also be of interest [37]. All these observations suggest that TLR-4 engagement in circulating mononuclear cells can play a role in the development of glomerular inflammation and a possible specific involvement in active and progressive IgAN.
The activation of TLRs has been supposed recently to influence the switch from IgM to IgA and the development of experimental IgAN. Interferon (IFN)-γ and -α responses, triggered by TLR activation, induce overexpression of B cell activation factor (BAFF) in dendritic cells, favouring B cell expansion and increasing IgA synthesis [38]. Notably, transgenic mice overexpressing BAFF develop IgAN [39], and in patients with IgAN a recent study reported an increase in BAFF and IFN-γ production by tonsillar mononuclear cells treated with cytosine-phosphate-guanosine–oligodeoxynucleotides (CpG–ODN) [40]. Hence, a relationship between activation of TLRs and aberrant glycosylation of IgA1 in IgAN has been suggested. The IgA1 O-glycans are based on a core N-acetyl galactosamine (GalNAc) usually extended with galactose (Gal) under the effect of a β1,3 Gal transferase (C1GALT1) working with its chaperone Cosmc [core 1 -1-phosphate uridyltransferase (GALT)-specific molecular chaperone][41]. It is of interest that activation of TLR-4 by bacterial LPS induces methylation of Cosms leading to reduced glycosylation of IgA1 molecules [42]. In our study we failed to detect a simple relationship between TLR-4 up-regulation in peripheral mononuclear cells and levels of aberrantly glycosylated IgA1, which might be due to differences in synchronism of the two phenomena. Finally, in future it might be of interest to explore possible correlations between levels of IgA/fibronectin complexes, which have been detected in sera of patients with IgAN [43], and TLR-4 expression in monocytes, considering that the extra domain A of fibronectin (FN-EDA) is one of its specific ligands.
The major source of aberrantly glycosylated IgA in patients with IgAN are B cells from bone marrow, but it is likely that these cells have encountered the antigen at mucosal sites and are then relocated to the bone marrow [22,44]. In IgAN there is a reduced mucosal response to neoantigens [45,46], which may result in impaired elimination of mucosal antigens, prolonged antigen exposure to B cells and increase in immunological memory. Indeed, these patients have a greater response to mucosal and systemic antigenic challenge in comparison to healthy controls [47]. According to this hypothesis, in subjects with IgAN the mucosal immune system is dealing with continuous antigenic challenge which leads to the production of nephritogenic IgA, and common microbial or food antigens may play the role of eliciting agents. TLR activation may represent the final common pathway for exogenous antigens which have a defective mucosal handling in patients with IgAN. Because we detected hyperexpression of TLR-4 in circulating mononuclear cells of patients with IgAN, whose major ligand is LPS, it is tempting to speculate a role for intestinal bacteria [48,49]. This may reinforce the relationships between chronic intestinal diseases with oral tolerance breakdown and IgAN, which have never been elucidated fully. Approximately 4% of patients with IgAN have coeliac disease, compared with 0·5–1% in the healthy population, and anti-gliadin antibodies are detectable in one-third of patients with IgAN without coeliac disease [50,51]. We have reported previously that a gluten-free diet was able to reduce the levels of anti-gliadin IgA [51], and we reproduced IgA mesangial deposits in mice after a chronic gluten-rich diet [7]. A recent investigation detected rectal mucosal hypersensitivity to gluten in one-third of patients with IgAN without manifest coeliac disease [52]. This subclinical inflammation may increase the intestinal permeability to common intestinal flora pathogens and trigger mucosal innate immunity. Of interest, in coeliac disease antibodies against a particular gluten fraction recognize the viral protein VP-7, suggesting a possible involvement of rotavirus infection in the pathogenesis of the disease through a mechanism of molecular mimicry, which also activates the engagement of TLR-4 [53]. In coeliac disease the growing role of the innate immunity is being recognized and the increased expression of some Toll-like receptors appears to delineate a new inherent defect in this branch of innate immunity. A similar conclusion may be derived for patients with IgAN from the results of our study. The newly observed up-regulation of TLR-4 in circulating mononuclear cells in patients with IgAN and some correlation with clinical activity suggests further investigation in this somewhat unexplored area.
Disclosure
All the authors declare no interests to disclose.
References
- 1.Schena FP, Coppo R. Primary IgA nephropathy. In: Davidson AM, editor. Oxford textbook of clinical nephrology. 3rd edn. Oxford: Oxford University Press; 2006. pp. 469–501. [Google Scholar]
- 2.Oortwijn BD, van der Boog PJ, Roos A, et al. A pathogenic role for secretory IgA in IgA nephropathy. Kidney Int. 2006;69:1131–8. doi: 10.1038/sj.ki.5000074. [DOI] [PubMed] [Google Scholar]
- 3.Russell MW, Spotswood MF, Julian BA, Galla JH, Mestecky J. Detection of food antigen-specific IgA immune complexes in human sera. Adv Exp Med Biol. 1987;216A:813–20. doi: 10.1007/978-1-4684-5344-7_94. [DOI] [PubMed] [Google Scholar]
- 4.Coppo R. The pathogenetic potential of environmental antigens in IgA nephropathy. Am J Kidney Dis. 1988;12:420–4. doi: 10.1016/s0272-6386(88)80038-6. [DOI] [PubMed] [Google Scholar]
- 5.Tomino Y, Sakai H, Endoh M, et al. Cross-reactivity of IgA antibodies between renal mesangial areas and nuclei of tonsillar cells in patients with IgA nephropathy. Clin Exp Immunol. 1983;5:605–10. [PMC free article] [PubMed] [Google Scholar]
- 6.Emancipator SN, Gallo GR, Lamm ME. Experimental IgA nephropathy induced by oral immunization. J Exp Med. 1983;157:572–82. doi: 10.1084/jem.157.2.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coppo R, Mazzucco G, Martina G, et al. Gluten-induced experimental IgA glomerulopathy. Lab Invest. 1989;60:499–506. [PubMed] [Google Scholar]
- 8.Jessen RH, Emancipator SN, Jacobs GH, Nedrud JG. Experimental IgA–IgG nephropathy induced by a viral respiratory pathogen. Dependence on antigen form and immune status. Lab Invest. 1992;67:379–86. [PubMed] [Google Scholar]
- 9.Amore A, Coppo R, Nedrud JG, Sigmund N, Lamm ME, Emancipator SN. The role of nasal tolerance in a model of IgA nephropathy induced in mice by Sendai virus. Clin Immunol. 2004;113:101–8. doi: 10.1016/j.clim.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto C, Suzuki S, Kimura H, Yoshida H, Gejyo F. Experimental nephropathy induced by Haemophilus parainfluenzae antigens. Nephron. 2002;90:320–7. doi: 10.1159/000049068. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki S, Nakatomi Y, Sato H, Tsukada H, Arakawa M. Haemophilus parainfluenzae antigen and antibody in renal biopsy samples and serum of patients with IgA nephropathy. Lancet. 1994;343:12–16. doi: 10.1016/s0140-6736(94)90875-3. [DOI] [PubMed] [Google Scholar]
- 12.Portis JL, Coe JE. Deposition of IgA in renal glomeruli of mink affected with Aleutian disease. Am J Pathol. 1979;96:227–36. [PMC free article] [PubMed] [Google Scholar]
- 13.Kawasaki Y, Mitsuaki H, Isome M, Nozawa R, Suzuki H. Renal effects of Coxsackie B4 virus in hyper-IgA mice. J Am Soc Nephrol. 2006;17:2760–9. doi: 10.1681/ASN.2006050495. [DOI] [PubMed] [Google Scholar]
- 14.Sharmin S, Shimizu Y, Hagiwara M, Hirayama K, Koyama A. Staphylococcus aureus antigens induce IgA-type glomerulonephritis in Balb/c mice. J Nephrol. 2004;17:504–11. [PubMed] [Google Scholar]
- 15.Satoskar AA, Nadasdy G, Plaza JA, et al. Staphylococcus infection-associated glomerulonephritis mimicking IgA nephropathy. Clin J Am Soc Nephrol. 2006;1:1179–86. doi: 10.2215/CJN.01030306. [DOI] [PubMed] [Google Scholar]
- 16.Koyama A, Sharmin S, Sakurai H, et al. Staphylococcus aureus cell envelope antigen is a new candidate for the induction of IgA nephropathy. Kidney Int. 2004;66:121–32. doi: 10.1111/j.1523-1755.2004.00714.x. [DOI] [PubMed] [Google Scholar]
- 17.Allen AC, Harper SJ, Feehally J. Galactosylation of N-and O-linked carbohydrate moieties of IgA1 and IgG in IgA nephropathy. Clin Exp Immunol. 1995;100:470–4. doi: 10.1111/j.1365-2249.1995.tb03724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomana M, Matousovic K, Julian BA, Radl J, Konecny K, Mestecky J. Galactose-deficient IgA1 in sera of IgA nephropathy patients is present in complexes with IgG. Kidney Int. 1997;52:509–16. doi: 10.1038/ki.1997.361. [DOI] [PubMed] [Google Scholar]
- 19.Hiki Y, Tanaka A, Kokubo T, et al. Analysis of IgA1 hinge glycopeptides in IgA nephropathy by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J Am Soc Nephrol. 1998;9:577–82. doi: 10.1681/ASN.V94577. [DOI] [PubMed] [Google Scholar]
- 20.Amore A, Cirina P, Conti G, Brusa P, Peruzzi L, Coppo R. Glycosylation of circulating IgA in patients with IgA nephropathy modulates proliferation and apoptosis of mesangial cells. J Am Soc Nephrol. 2001;12:1862–71. doi: 10.1681/ASN.V1291862. [DOI] [PubMed] [Google Scholar]
- 21.Coppo R, Amore A. Aberrant glycosylation in IgA nephropathy (IgAN) Kidney Int. 2004;65:1544–7. doi: 10.1111/j.1523-1755.2004.05407.x. [DOI] [PubMed] [Google Scholar]
- 22.Smith AC, Molyneux K, Feehally J, Barratt J. O-glycosylation of serum IgA1 antibodies against mucosal and systemic antigens in IgA nephropathy. J Am Soc Nephrol. 2006;17:3520–8. doi: 10.1681/ASN.2006060658. [DOI] [PubMed] [Google Scholar]
- 23.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 24.Anders HJ, Schlöndorff D. Toll-like receptors: emerging concepts in kidney disease. Curr Opin Nephrol Hypertens. 2007;16:177–83. doi: 10.1097/MNH.0b013e32803fb767. [DOI] [PubMed] [Google Scholar]
- 25.Tipping PG. Toll-like receptors: the interface between innate and adaptive immunity. J Am Soc Nephrol. 2006;17:1769–71. doi: 10.1681/ASN.2006050489. [DOI] [PubMed] [Google Scholar]
- 26.O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. 2008;226:10–18. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh TK, Mickelson DJ, Solberg JC, Lipson KE, Inglefield JR, Alkan SS. TLR–TLR cross talk in human PBMC resulting in synergistic and antagonistic regulation of type-1 and 2 interferons, IL-12 and TNF-alpha. Int Immunopharmacol. 2007;7:1111–21. doi: 10.1016/j.intimp.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 28.Skinner NA, MacIsaac CM, Hamilton JA, Visvanathan K. Regulation of Toll-like receptor (TLR)-2 and TLR-4 on CD14dimCD16+ monocytes in response to sepsis-related antigens. Clin Exp Immunol. 2005;141:270–8. doi: 10.1111/j.1365-2249.2005.02839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manno C, Strippoli GF, D'Altri C, Torres D, Rossini M, Schena FP. A novel simpler histological classification for renal survival in IgA nephropathy: a retrospective study. Am J Kidney Dis. 2007;49:763–75. doi: 10.1053/j.ajkd.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Coppo R, Peruzzi L, Amore A, et al. IgACE: a placebo-controlled, randomized trial of angiotensin-converting enzyme inhibitors in children and young people with IgA nephropathy and moderate proteinuria. J Am Soc Nephrol. 2007;18:1880–8. doi: 10.1681/ASN.2006040347. [DOI] [PubMed] [Google Scholar]
- 31.Coppo R, Camilla R, Alfarano A, et al. Upregulation of the immunoproteasome in peripheral blood mononuclear cells of patients with IgA nephropathy. Kidney Int. 2009;75:536–41. doi: 10.1038/ki.2008.579. [DOI] [PubMed] [Google Scholar]
- 32.Coppo R, Amore A, Chiesa M, et al. Serological and genetic factors in early recurrence of IgA nephropathy after renal transplantation. Clin Transplant. 2007;21:728–37. doi: 10.1111/j.1399-0012.2007.00730.x. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki H, Suzuki Y, Narita I, et al. Toll-like receptor 9 affects severity of IgA nephropathy. J Am Soc Nephrol. 2008;19:2384–95. doi: 10.1681/ASN.2007121311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu J, Yang Y, Sun J, et al. Expression of Toll-like receptors and their association with cytokine responses in peripheral blood mononuclear cells of children with acute rotavirus diarrhoea. Clin Exp Immunol. 2006;144:376–81. doi: 10.1111/j.1365-2249.2006.03079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Migita K, Miyashita T, Maeda Y, et al. Toll-like receptor expression in lupus peripheral blood mononuclear cells. J Rheumatol. 2007;34:493–500. [PubMed] [Google Scholar]
- 36.Brown HJ, Lock HR, Wolfs TG, Buurman WA, Sacks SH, Robson MG. Toll-like receptor 4 ligation on intrinsic renal cells contributes to the induction of antibody-mediated glomerulonephritis via CXCL1 and CXCL2. J Am Soc Nephrol. 2007;18:1732–9. doi: 10.1681/ASN.2006060634. [DOI] [PubMed] [Google Scholar]
- 37.Yoon HJ, Shin JH, Yang SH, et al. Association of the CD14 gene −159C polymorphism with progression of IgA nephropathy. J Med Genet. 2003;40:104–8. doi: 10.1136/jmg.40.2.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCarthy DD, Chiu S, Gao Y, Summers-deLuca LE, Gommerman JL. BAFF induces a hyper-IgA syndrome in the intestinal lamina propria concomitant with IgA deposition in the kidney independent of LIGHT. Cell Immunol. 2006;241:85–94. doi: 10.1016/j.cellimm.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 39.Goto T, Bandoh N, Yoshizaki T, et al. Increase in B-cell-activation factor (BAFF) and IFN-gamma productions by tonsillar mononuclear cells stimulated with deoxycytidyl-deoxyguanosine oligodeoxynucleotides (CpG-ODN) in patients with IgA nephropathy. Clin Immunol. 2008;126:260–9. doi: 10.1016/j.clim.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Inoue T, Sugiyama H, Kikumoto Y, et al. Downregulation of the beta1,3- galactosyltransferase gene in tonsillar B lymphocytes and aberrant lectin bindings to tonsillar IgA as a pathogenesis of IgA nephropathy. Contrib Nephrol. 2007;157:120–4. doi: 10.1159/000102315. [DOI] [PubMed] [Google Scholar]
- 41.Ju T, Cummings RD. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc Natl Acad Sci USA. 2002;99:16613–18. doi: 10.1073/pnas.262438199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin W, Zhong X, Fan JM, Zhang YJ, Liu XR, Ma XY. External suppression causes the low expression of the Cosmc gene in IgA nephropathy. Nephrol Dial Transplant. 2008;23:1608–14. doi: 10.1093/ndt/gfm781. [DOI] [PubMed] [Google Scholar]
- 43.Cederholm B, Wieslander J, Bygren P, Heinegård D. Circulating complexes containing IgA and fibronectin in patients with primary IgA nephropathy. Proc Natl Acad Sci USA. 1988;88:4865–8. doi: 10.1073/pnas.85.13.4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suzuki Y, Tomino Y. The mucosa–bone-marrow axis in IgA nephropathy. Contrib Nephrol. 2007;157:70–9. doi: 10.1159/000102307. [DOI] [PubMed] [Google Scholar]
- 45.de Fijter JW, Eijgenraam JW, Braam CA, et al. Deficient IgA1 immune response to nasal cholera toxin subunit B in primary IgA nephropathy. Kidney Int. 1996;50:952–61. doi: 10.1038/ki.1996.396. [DOI] [PubMed] [Google Scholar]
- 46.Roodnat JI, de Fijter JW, van Kooten C, Daha MR, van Es LA. Decreased IgA1 response after primary oral immunization with live typhoid vaccine in primary IgA nephropathy. Nephrol Dial Transplant. 1999;14:353–9. doi: 10.1093/ndt/14.2.353. [DOI] [PubMed] [Google Scholar]
- 47.Leinikki PO, Mustonen J, Pasternack A. Immune response to oral polio vaccine in patients with IgA glomerulonephritis. Clin Exp Immunol. 1987;68:33–8. [PMC free article] [PubMed] [Google Scholar]
- 48.Shang L, Fukata M, Thirunarayanan N, et al. Toll-like receptor signaling in small intestinal epithelium promotes B-cell recruitment and IgA production in lamina propria. Gastroenterology. 2008;135:529–38. doi: 10.1053/j.gastro.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boirivant M, Amendola A, Butera A. Intestinal microflora and immunoregulation. Mucosal Immunol. 2008;1(Suppl 1):S47–9. doi: 10.1038/mi.2008.52. [DOI] [PubMed] [Google Scholar]
- 50.Collin P, Syrjänen J, Partanen J, Pasternack A, Kaukinen K, Mustonen J. Celiac disease and HLA DQ in patients with IgA nephropathy. Am J Gastroenterol. 2002;97:2572–6. doi: 10.1111/j.1572-0241.2002.06025.x. [DOI] [PubMed] [Google Scholar]
- 51.Coppo R, Roccatello D, Amore A, et al. Effects of a gluten-free diet in primary IgA nephropathy. Clin Nephrol. 1990;33:72–86. [PubMed] [Google Scholar]
- 52.Smerud HK, Fellström B, Hällgren R, Osagie S, Venge P, Kristjánsson G. Gluten sensitivity in patients with IgA nephropathy. Nephrol Dial Transplant. 2009;24:2476–81. doi: 10.1093/ndt/gfp133. [DOI] [PubMed] [Google Scholar]
- 53.Zanoni G, Navone R, Lunardi C, et al. In celiac disease, a subset of autoantibodies against transglutaminase binds toll-like receptor 4 and induces activation of monocytes. PLoS Med. 2006;3:e358. doi: 10.1371/journal.pmed.0030358. [DOI] [PMC free article] [PubMed] [Google Scholar]