

Abstract
The alternative pathway (AP) of complement alone is capable of mediating immune complex-induced arthritis in the collagen antibody-induced arthritis (CAIA) model in mice. Whether the classical pathway (CP) or lectin pathway (LP) alone can mediate CAIA is not known. Using mice genetically deficient in different complement components, our results reported herein establish that the CP and LP alone are each incapable of mediating CAIA. A lower level or absence of C3 and/or C5 activation by the CP may be possible explanations for the importance of the AP in CAIA and in many murine models of disease. In addition, other investigators have reported that CP C5 convertase activity is absent in mouse sera. To address these questions, we employed an in vitro system of adherent immunoglobulin (Ig)G-induced complement activation using plates coated with murine anti-collagen monoclonal antibody (mAb). These experiments used complement-deficient mouse sera and wild-type mouse or normal human sera under conditions inactivating either the CP (Ca++ deficiency) or the AP (mAb inhibitory to factor B). Robust generation of both C3a and C5a by either the AP or CP alone were observed with both mouse and human sera, although there were some small differences between the species of sera. We conclude that neither the CP nor LP alone is capable of mediating CAIA in vivo and that mouse sera exhibits a high level of IgG-induced C5a generation in vitro through either the CP or AP.
Keywords: alternative pathway, arthritis, complement, immune complexes, inflammation
Introduction
The complement system, a part of the innate immune system, plays a key role in host resistance to infection and interacts with both the humoral and cellular arms of the adaptive immune system [1–3]. The complement system can be pathogenic in human disease through generation of potent anaphylatoxins such as C3a or C5a and through the lysis of cells or tissue injury mediated by the terminal complement C5b–9 complex (TCC) [4–6].
The complement system is activated through three pathways, all of which converge on C3 with subsequent cleavage of C5 and generation of the TCC. The classical pathway (CP) is initiated by binding of C1q to immunoglobulin (Ig)M or IgG antibody bound to an antigen on a target surface, followed by proteolysis of associated C1r and C1s. Activated C1s then cleaves C4 and C2, generating the CP C3 convertase (C4b2a). Cleavage of C3 generates the active anaphylatoxin C3a and the fragment C3b that forms an ester link with C4b to form the CP C5 convertase C4b2a3b.
The alternative pathway (AP) is activated continually at a low level in plasma by the ‘tickover’ mechanism, where spontaneous hydrolysis of the thioester bond in native C3 generates a C3b-like molecule, C3(H20) [7,8]. Factor B then binds to this C3b-like molecule in solution with cleavage of factor B by activated factor D, generating an AP C3 convertase [C3(H20)Bb], with further cleavage of C3. C3b has a short half-life in solution and binds quickly to nearby surfaces, including adherent IgG. The AP is enhanced or inhibited by binding of properdin or factor H, respectively, to adherent C3b. The AP may function primarily as an amplification loop of C3b following initiation by the CP, AP or lectin pathway (LP). In this manner, the AP amplification loop is initiated by the covalent binding of a small amount of C3b to hydroxyl or amino groups on cell surface carbohydrates and proteins followed by the binding of factor B. The AP C5 convertase (C3b)2Bb is created by two C3b molecules linked covalently. The AP may also be initiated by IgG [9] or by properdin binding directly to cell surfaces [10–12].
The LP is initiated by binding to terminal L-fucose, D-mannose or N-acetyl-D-glucosamine residues on the surface of various targets by a complex of mannose-binding lectin (MBL) and MBL-associated proteases (MASP-1 and -2) [13]. The proteases in the LP resemble C1r and C1s of the CP in cleaving C2 and C4 to generate the CP C3 convertase C4b2a, and subsequently the CP C5 convertase C4b2a3b. MBL may also activate C3 and the AP amplification loop in the absence of C2 and C4 [14]. In the absence of C3, thrombin can substitute for the CP C5 convertase with direct cleavage of C5 into C5a and C5b [15].
The AP may play a central role in many diseases, both in experimental animal models and in humans [8]. The association of the AP with human disease such as age-related macular degeneration, membranoproliferative glomerulonephritis and atypical haemolytic uraemic syndrome, is due to mutations or polymorphisms in factor H that lead to inadequate regulation of the AP [16,17]. The AP is required in an experimental animal model of inflammatory arthritis induced in mice by the active immunization with bovine collagen type II (CII) (collagen-induced arthritis, CIA) or by the passive infusion of a cocktail of monoclonal antibody (mAb) to CII (collagen antibody-induced arthritis, CAIA) [18–21]. The results of studies from our laboratory indicated that CAIA was decreased markedly in mice deficient in factor B or C3, but not in mice deficient in C4, C1q, MBL-A and -C, or in both C1q and MBL. We concluded that the AP amplification loop was absolutely necessary for the development of CAIA [21]. The levels of C3 deposition in the joints were absent in mice deficient in C3, 70% decreased in mice deficient in factor B, but actually increased in mice deficient in C4, C1q, MBL or in both C1q and MBL. We hypothesized that in the absence of the AP, the level of C3 deposition in the joint was not adequate to lead to clinical disease. However, the disease activity or level of C3 deposition in the joints in mice expressing only the CP or LP are not known. In addition, the possibility exists that differences in the levels of generation of C3a or C5a could also contribute to the extent of disease activity.
The possibility has been suggested that the mouse is not an adequate experimental model for human disease because of a deficient complement system, separate from the well-described C5 deficiency that is present in many mouse strains, but not in C57BL/6. The older literature contains several studies suggesting that in haemolytic assays mouse C4 is devoid of classical pathway C5 convertase activity because of weak binding of mouse C4b to C5 (reviewed in [22]). However, recent studies using human serum with a neutralizing mAb to factor D indicate that the AP contributes 80% of the total generation of C5a and TCC when activated by aggregated IgG [23]. Nevertheless, the possibility exists that inflammatory and autoimmune disease in experimental models in mice may appear to be dependent upon the AP because of absent or weak CP C5 convertase activity.
The objectives of our studies were to examine disease activity and C3 deposition in the joints in CAIA using mice expressing only the CP or LP, and to explore the ability of mouse and human serum to generate C3a or C5a in vitro after complement activation by adherent IgG.
Materials and methods
Sera from wild-type (WT) and complement-deficient mice
Sera from C57BL/6 mice deficient in genes for specific complement components were obtained from the following sources: Df−/−, MBL−/−/Df−/− (deficient in both MBL-A and MBL-C and in factor D); C1q−/−/MBL−/− and C1q−/−/Df−/− from the laboratories of authors outside Denver or from a colony at the University of Colorado Denver (UCD); and C3−/− and Bf−/− from breeding colonies at UCD. Control sera were obtained from WT C57BL/6 and NOD mice (Jackson Laboratories, Bar Harbor, ME, USA). All animals were kept in a barrier animal facility at UCD with a climate-controlled environment having 12-h light/dark cycles. Filter top cages were used with three mice in each cage. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Colorado Denver.
Collagen antibody-induced arthritis
CAIA was induced in Df−/−, MBL−/−/Df−/− and C1q−/−/Df−/− mice and in age- and sex-matched WT C57BL/6 mice (Jackson Laboratories), as described [20]. Eight mg per mouse of a cocktail of four murine mAb to conserved epitopes on the CB11 peptide of CII (Arthrogen, Chondrex, Redmond, WA, USA) was injected intraperitoneally (i.p.) into four or five separate mice of each genotype. On day 3, 50 µg per mouse of lipopolysaccharide (LPS) from Escherichia coli strain 011B4 was injected i.p. to synchronize the development of arthritis. The mice were examined daily for signs of arthritis by two trained observers who were blinded to the type of mouse, and clinical disease activity scores were determined [20]. Clinical disease activity was scored on a three-point scale per paw: 0 = normal joint; 1 = slight inflammation and redness; 2 = severe erythema and swelling affecting the entire paw with inhibition of use; and 3 = deformed paw or joint with ankylosis, joint rigidity and loss of function. The maximum score was 12, based on analysis of all four paws. Incidence was defined as a mouse with a score of at least one in any joint. The histopathology scores in mice with CAIA and levels of C3 deposition in the joints were determined by immunohistochemistry, as described [20].
Complement protein levels in mouse sera
The levels of C1q, factor D, factor B, C3 and C4 in sera from WT mice or from mice deficient in complement components were determined by enzyme-linked immunosorbent assay (ELISA), as described [21].
Activation of C3 and C5 in vitro using mouse or human sera
To explore the relative contributions of the CP or AP in activation of C3 and C5 in mouse or human serum, an in vitro system was developed using plates coated with four IgG mAb to CII, as described previously [21]. Adherent immune complexes (IC) were not used for these studies as the plates coated with CII alone, in the absence of the IgG mAb, activated C5. Mouse blood was drawn by intra-orbital bleeding according to Institutional Animal Care and Use Committee (IACUC) guidelines and placed immediately on ice. After clotting, serum was separated by centrifugation at 4°C and stored at −80°C for future experimental use. Human peripheral venous blood was obtained from normal healthy volunteers and serum was obtained as described above for mouse blood. All experiments were performed with 1:10 dilutions of sera in veronal saline buffer (VSB) and the diluted sera were incubated on the IgG-coated plates for 1 h at 37°C. In some experiments, the dilutions of mouse or human sera were incubated for 30 min on ice with a mAb to factor B to inhibit the AP before they were added to the plates. Anti-factor mAb was used at 4 µg/10 µl of mouse serum and at 8 µg/10 µl of human serum. It has been shown that, at the concentrations specified, this mAb completely neutralizes both mouse and human factor B [24]. Mouse or human sera were also diluted 1:10 in Ca++ deficient buffer [phosphate-buffered saline (PBS) with 5 mM MgCl2 and 10 mM ethylene glycol tetraacetic aid (EGTA)] to inhibit the CP and were compared with Ca++ sufficient buffer (VSB). Experiments with human sera were approved by the Colorado Multiple Institutional Review Board.
Measurement of C3a and C5a levels generated in vitro using mouse sera
To measure levels of C3a and C5a in culture supernatants, plates were coated with 100 µl of rat anti-mouse capture mAb specific for C3a or C5a (BD Biosciences, San Jose, CA, USA) at 1:250 dilutions in 0·1 M sodium carbonate buffer overnight at 4°C. Supernatants from the incubations of mouse sera on the IgG-coated plates were diluted 1:100 for C3a and 1:50 for C5a in VSB, added to the plates coated with capture antibody and incubated at room temperature for 2 h. After washing four times, 100 µl of biotinylated anti-C3a or anti-C5a Ab (BD Biosciences) at a 1:500 dilution in PBS with 1% bovine serum albumin (BSA) and 0·05% Tween was added to each well with incubation at room temperature for 1 h. The wells were washed again and then incubated with a 1:1000 dilution of streptavidin/horseradish peroxidase (HRP) (R&D Systems, Minneapolis, MN, USA) for 1 h at room temperature. The wells were washed and 100 µl of substrate solution was added to each well. Colour was developed for 3–5 min with the reaction stopped by adding 2N H2SO4. Absorbance was read at 450 nm with correction for absorbance at 550 nm. The background controls for C3a and C5a were the optical density (OD) values obtained with mouse serum in the presence of Ca++ deficient buffer and the mAb to factor B. Six experiments were performed for C5a generation and three experiments for C3a generation, using different sets of eight or nine mouse sera.
Measurement of C3a and C5a levels generated in vitro using human sera
Supernatants from the incubations of human sera on IgG-coated plates were diluted 1:5000 for C3a or 1:50 for C5a in ELISA diluent provided by the manufacturer (BD Biosciences) and 100 µl was added to each well. The ELISAs were then performed according to the manufacturer's instructions. The background controls for C3a and C5a levels were the OD values obtained using human serum without any incubation on anti-CII-coated plates. The same nine human sera were used in four repeat experiments.
Statistical analysis
An unpaired two-tailed Student's t-test was used to determine significance; preliminary analyses using a null hypothesis for w-statistics indicated that the data for all assays were distributed normally. P-values less than 0·05 were considered significant.
Results
Role of the CP or LP alone in CAIA
CAIA was induced in WT mice, in mice genetically deficient in factor D (Df−/−), deficient in MBL and factor D (MBL−/−/Df−/−), where only the CP was active, and deficient in C1q and factor D (C1q−/−/Df−/−), where only the LP was intact. In each experiment WT mice developed severe arthritis beginning on day 4 with 100% incidence by day 5 (Fig. 1a, c and d). Disease activity scores of 10·0 ± 1·4, 9·3 ± 1·1, and 11·8 ± 0·3 [mean ± standard error of the mean (s.e.m.), n = 4], respectively, were observed on day 10 in the three separate experiments (Fig. 1b, d and f). In contrast, only minimal arthritis was observed in any set of complement deficient mice (P < 0·0003 compared to WT mice on day 10). In vitro control experiments indicated that sera from the MBL−/−/Df−/− (CP only) and C1q−/−/Df−/− (LP only) mice exhibited the expected phenotypes. mAb-coated plates led to maximal levels of C3 deposition mediated by the CP (sera from MBL−/−/Df−/− mice), whereas the LP (sera from C1q−/−/Df−/− mice) did not mediate any detectable C3 activation; the opposite pattern was observed using mannan-coated plates (data not shown).
Fig. 1.
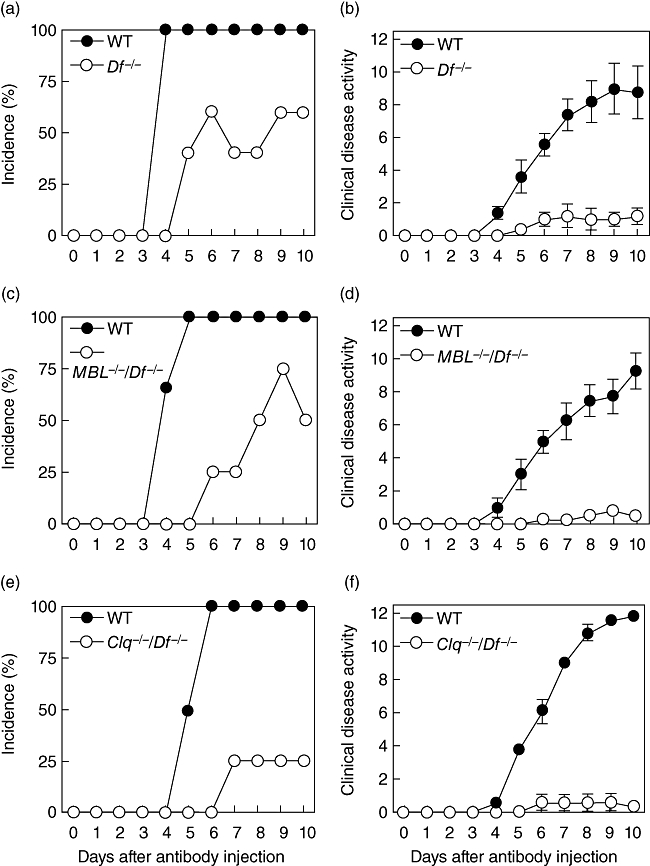
Collagen antibody-induced arthritis (CAIA) in wild-type (WT) mice, Df−/− mice (a, b), MBL−/−/Df−/−- mice (classical pathway only) (c, d) and C1q−/−/Df−/− mice (lectin pathway only) (e, f). The mice were injected intraperitoneally with a cocktail of four monoclonal antibodies (mAb) to type II collagen. Lipopolysaccharide (LPS) was administered on day 3 to synchronize the development of arthritis. The data are expressed as the incidence of arthritis (a, c, e) and clinical disease activity score (b, d, f) versus days after the mAb injection. The data represent the mean ± standard error of the mean based on n = 5 for the Df−/− mice, n = 4 for the MBL−/−/Df−/− and C1q−/−/Df−/− mice and n = 4 for each set of WT mice.
The histopathology scores paralleled the clinical disease activity scores, i.e. higher in the WT mice in comparison to each set of complement deficient mice (Table 1). However, the levels of C3 deposited in the joints were only partially reduced in the complement deficient mice with values of 54%, 58% and 26%, respectively, in the Df−/−, MBL−/−/Df−/−, and C1q−/−/Df−/− mice in comparison to WT mice.
Table 1.
Histopathology scores and C3 deposition in the joints of wild-type (WT) and complement-deficient mice.
| Histopathology† | |||||
|---|---|---|---|---|---|
| Mouse type | Inflammation | Pannus | Cartilage damage | Bone damage | Total |
| WT (n = 4) | 2.70 ± 0.17 | 3.00 ± 0.12 | 2.45 ± 0.15 | 2.70 ± 0.13 | 10.9 ± 0.53 |
| Df−/− (n = 5) | 1.12 ± 0.17 | 1.44 ± 0.17 | 1.28 ± 0.26 | 1.32 ± 0.22 | 5.16 ± 0.79 |
| P | < 0.001 | < 0.001 | < 0.01 | < 0.001 | < 0.001 |
| WT (n = 4) | 1.35 ± 0.28 | 1.15 ± 0.29 | 1.20 ± 0.54 | 0.95 ± 0.12 | 4.65 ± 1.34 |
| MBL−/−/Df−/− (n = 4) | 0.40 ± 0.08 | 0.40 ± 0.08 | 0.50 ± 0.10 | 0.40 ± 0.08 | 1.70 ± 0.13 |
| P | < 0.01 | < 0.02 | < 0.12 | < 0.06 | < 0.04 |
| WT (n = 4) | 3.15 ± 0.05 | 2.60 ± 0.14 | 2.55 ± 0.05 | 2.60 ± 0.14 | 10.9 ± 0.26 |
| C1q−/−/Df−/− (n = 4) | 0.45 ± 0.10 | 0.40 ± 0.08 | 0.70 ± 0.24 | 0.40 ± 0.08 | 1.95 ± 0.43 |
| P | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 |
| C3 Deposition† | |||||
| Mouse type | Synovium | Cartilage | Total | ||
| WT (n = 4) | 2.75 ± 0.13 | 2.10 ± 0.17 | 4.85 ± 0.28 | ||
| Df−/− (n = 5) | 1.60 ± 0.14 | 1.04 ± 0.10 | 2.64 ± 0.21 | ||
| P | < 0.001 | < 0.001 | < 0.0003 | ||
| WT (n = 4) | 2.00 ± 0.29 | 1.30 ± 0.30 | 3.30 ± 0.58 | ||
| MBL−/−/Df−/− (n = 4) | 1.00 ± 0.08 | 0.90 ± 0.10 | 1.90 ± 0.13 | ||
| P | < 0.01 | < 0.13 | < 0.03 | ||
| WT (n = 4) | 2.60 ± 0.08 | 2.30 ± 0.29 | 4.90 ± 0.33 | ||
| C1q−/−/Df−/− (n = 4) | 0.80 ± 0.08 | 0.50 ± 0.06 | 1.30 ± 0.13 | ||
| P | < 0.0001 | < 0.001 | < 0.0001 | ||
Five joints were examined in each animal and the results expressed as mean ± standard error of the mean for the indicated number of mice.
These results indicate that neither the CP nor LP alone is capable of mediating CAIA. The presence of detectable C3 deposition in the joints of mice possessing only intact CP or LP suggests that these pathways are capable of activating C3 at low levels in vivo. However, in the absence of the AP and the amplification loop, the levels of tissue C3 deposition may not be adequate to produce clinical disease. The possibility also exists that differences in the levels of C3a or C5a generation through each pathway may explain, at least in part, this requirement for the AP.
Generation of C5a in vitro in sera from mice of various genotypes
Experiments were next performed to examine the relative roles of the AP or CP in generation of C5a levels in vitro using mouse sera. Plates coated with the same four mAb specific for bovine CII as were used in the CAIA experiments were incubated with WT mouse sera or with sera from mice deficient in various complement components. High levels of C5a generation were observed using WT sera or sera from mice lacking both C1q and MBL (AP only) (Fig. 2). The results for sera from MBL−/−/Df−/− and C1q−/−/Df−/− mice were 28·4% and 8·1%, respectively, of the results for WT sera (P < 0·0001 for each comparison). The OD values (mean ± s.e.m., n = 5) and ranges for these C5a data were: WT, 1·48 ± 0·06 (1·29–1·58); MBL−/−/Df−/−, 0·42 ± 0·15 (0·16–0·88); C1q−/−/Df−/−, 0·12 ± 0·01 (0·09–0·16); and C1q−/−/MBL−/−, 1·64 ± 0·10 (1·39–1·86). No C5a generation was present in the supernatants of antibody-coated plates incubated with sera from mice deficient in C3 or C5 (NOD). Generation of C3a was not determined in this experiment, although in a separate experiment measuring only C3a the levels were absent using sera from C3−/− mice and were not significantly different from WT using sera from mice with the other genotypes examined in Fig. 2 (data not shown).
Fig. 2.
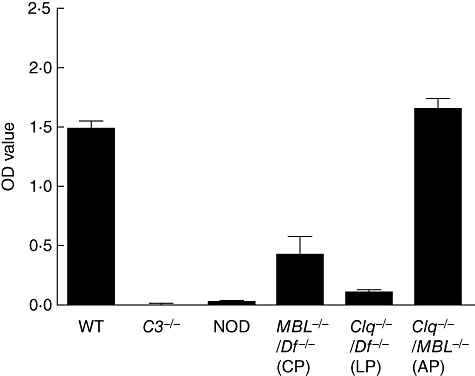
C5a generation using sera from mice genetically deficient in complement components. Plates coated with four monoclonal antibodies (mAb) specific for bovine type II collagen were incubated with 1:10 dilutions of wild-type (WT) mouse sera or with sera deficient in various complement components: C3−/− = sera deficient in C3; NOD = non-obese diabetic mouse, naturally deficient in C5; MBL−/−/Df−/− = deficient in both mannose-binding lectin (MBL) and factor D (only classical pathway intact); C1q−/−/Df−/− = deficient in both C1q and factor D (only lectin pathway intact); C1q−/−/MBL−/−- = deficient in both C1q and MBL (only altenative pathway intact). C5a generation was measured in the supernatants and expressed as optical density units. The data represent the mean ± standard error of the mean based on n = 5 for each type of mouse serum.
These results indicate that the AP alone is capable of generating C5a to a level equal to that observed with WT sera. However, the CP alone, in the absence of the AP, generated 71% less C5a than was observed with WT sera; the LP alone generated minimal C5a.
Levels of complement components in mouse sera
The protein levels of C1q, C3, C4, factor B and factor D were determined in the sera from WT mice and from mice genetically deficient in factor D, both MBL and factor D, and both C1q and factor D. C1q was present at equivalent levels in the WT, Df−/− and MBL−/−/Df−/− sera but was not detected in the sera from C1q−/−/Df−/− mice (Table 2). The levels of C3 were higher in sera from Df−/− and C1q−/−/Df−/− mice in comparison to WT. However, C3 levels in sera from MBL−/−/Df−/− mice were not different from WT. C4 was decreased significantly in the sera from C1q−/−/Df−/− mice in comparison to WT. Factor B levels were higher in Df−/− sera in comparison to WT. As expected, factor D levels were undetectable in the three sets of mice genetically deficient in this complement component. MBL levels in the mouse sera were not determined.
Table 2.
Levels of complement components in sera from wild-type (WT) and complement-deficient mice.
| Complement components† | |||||
|---|---|---|---|---|---|
| Mice | C1q | C3 | C4 | Factor B | Factor D |
| WT | 0.80 ± 0.06 | 1.63 ± 0.10 | 0.37 ± 0.06 | 0.91 ± 0.05 | 1.85 ± 0.02 |
| Df−/− | 0.90 ± 0.11 | 2.11 ± 0.09* | 0.26 ± 0.08 | 1.20 ± 0.07** | < 0.10 |
| MBL−/−/Df−/− | 0.86 ± 0.09 | 1.65 ± 0.04 | 0.19 ± 0.05 | 1.05 ± 0.05 | < 0.10 |
| C1q−/−/Df−/− | < 0.15 | 2.27 ± 0.07*** | < 0.10**** | 0.97 ± 0.04 | < 0.10 |
P < 0·004,
P < 0·005,
P < 0·0003,
P < 0·0001, each in comparison to WT.
The data are expressed as optical density units with mean ± standard error of the mean based on n = 6.
Generation of C3a and C5a in vitro in sera from mice lacking factor B
To examine further the relative roles of the CP and AP in generating C3a and C5a levels, an in vitro experiment was carried out using sera from WT mice or from mice genetically deficient in factor B, where the AP is inactive. The levels of C3a generated using the sera from Bf−/− mice were 75·5% of the levels observed with WT sera (Fig. 3a). The OD values (mean ± s.e.m., n = 9) and ranges for these C3a data were: WT, 1·51 ± 0·04 (1·29–1·69); and Bf−/−, 1·14 ± 0·09 (0·83–1·60) (P < 0·001). However, the levels of C5a generated using the sera from Bf−/− mice were decreased further to 17·1% of the levels seen with WT sera in this experiment (Fig. 3b). The OD values (mean ± s.e.m., n = 9) and ranges for these C5a data were: WT, 2·40 ± 0·04 (2·22–2·58); and Bf−/−, 0·41 ± 0·06 (0·18–0·63) (P < 0·0001). In four different experiments, the C5a level seen with sera from Bf−/− mice was 12·5% ± 3·5% (mean ± s.e.m.) of the value observed with sera from WT mice.
Fig. 3.
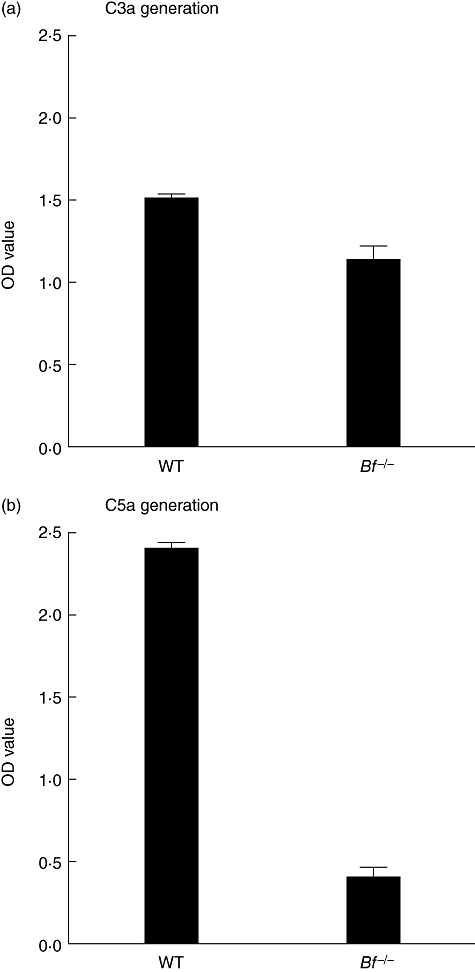
C3a generation and C5a generation using sera from wild-type (WT) mice or from mice deficient in factor B (Bf−/−). Plates coated with the four monoclonal antibodies (mAb) to type II collagen were incubated with 1:10 dilutions of sera from WT mice or from mice deficient in factor B. (a) C3a generation was measured in the supernatant and expressed in optical density (OD) units. (b) C5a generation was measured in the supernatant and expressed in OD units. The data represent the mean ± standard error of the mean based on n = 9 for each type of serum. This experiment was repeated four times.
The results of these experiments with sera from Bf−/− mice indicate that whereas the CP alone, in the absence of the AP, generated 75% of the level of C3a observed with WT sera, the level of C5a generated was 17% or less.
C3a and C5a levels generated in vitro in normal mouse and human sera
Generation of C3a and C5a in vitro was next examined using plates coated with adherent IgG mAb to CII as a mimic of adherent IC under conditions where both the CP and AP were active, the AP alone was active (Ca++ deficient buffer) or the CP alone was active (presence of a mAb to factor B). Six experiments were performed with mouse and human sera and the data were expressed as OD values because of a lack of accurate standards for C3a and C5a in ELISA. However, the OD values for C3a and C5a could not be compared accurately between mouse and human sera as the sensitivities of the ELISAs may have varied. To permit compilation of the data between experiments, and to allow a comparison between species, the results were normalized to 100% of the values obtained with intact complement activation pathways (Ca++ sufficient buffer in the absence of mAb to factor B).
The normalized data indicate that higher levels of C3a were generated by either the AP alone or by the CP alone in human sera compared to mouse sera (Table 3). The level of C3a generated by the AP alone in mouse sera (76·6) was significantly higher than the level generated by the CP alone (60·5) (P < 0·002). The same was observed with human sera, where the level of C3a generated by the AP alone (88·9) was significantly higher than the level generated by the CP alone (71·0) (P < 0·001). Although the level of C5a generated by the AP alone in mouse sera was higher than the value in human sera, the CP alone generated higher levels of C5a in human sera. However, in the absence of an active AP, the CP alone in mouse sera generated a level of C5a that was 46·6% of WT. The level of C5a generated by the AP alone in mouse sera (79·1) was significantly higher than the level generated by the CP alone (46·6) (P < 0·0001). However, the opposite was observed with human sera, where the level of C5a generated by the AP alone (62·8) was significantly lower than the level generated by the CP alone (74·3) (P < 0·015).
Table 3.
Normalized data for C3a and C5a levels using mouse and human sera.†
| Mouse sera (%) | Human sera (%) | |
|---|---|---|
| C3a levels | ||
| No mAb to factor B with: | ||
| Ca++ sufficient buffer (both CP and AP) | 100 | 100 |
| Ca++ deficient buffer (AP only) | 76.6 ± 3.6* | 88.9 ± 4.5* |
| (range) | (33–106) | (45–140) |
| mAb to factor B with: | ||
| Ca++ sufficient buffer (CP only) | 60.5 ± 3.3** | 71.0 ± 2.8** |
| (range) | (33–91) | (43–102) |
| Ca++ deficient buffer (no CP or AP) | 0 | 5.3 ± 1.1 |
| C5a levels | ||
| No mAb to factor B with: | ||
| Ca++ sufficient buffer (both CP and AP) | 100 | 100 |
| Ca++ deficient buffer (AP only) | 79.1 ± 3.6*** | 62.8 ± 3.8*** |
| (range) | (14–116) | (18–110) |
| mAb to factor B with: | ||
| Ca++ sufficient buffer (CP only) | 46.6 ± 2.8**** | 74.3 ± 2.2**** |
| (range) | (4–92) | (48–94) |
| Ca++ deficient buffer (no CP or AP) | 0 | 0.8 ± 0.6 |
P-values for mouse versus human sera:
P = 0·04;
P = 0·02;
P = 0·003;
P < 0·0001.
The data for up to six experiments were normalized to 100% of the values obtained in the absence of monoclonal antibody (mAb) to factor using Ca++ sufficient buffer.
The data are expressed as mean ± standard error of the mean based on n = 34 human sera for both assays, n = 45 murine sera for C5a levels and n = 24 murine sera for C3a levels. AP, alternative pathway; CO, classical pathway.
Discussion
The first objective to the studies reported herein was to examine the abilities of the CP or LP alone to mediate CAIA. The results indicated clearly that neither of these complement pathways alone is capable of mediating more than minimal CAIA, confirming our earlier observation that this disease model is dependent upon the AP [20,21]. The second objective of the current studies was to explore the abilities of the CP and AP in mouse and human serum to activate C3 and C5 in vitro. Our results show some small differences between mouse and human sera. However, the most important result is that both the CP and AP in mouse serum can generate C5a.
The results of our earlier studies led to the hypothesis that the dependency of CAIA on the AP was due to the ability of the AP amplification loop to enhance C3 deposition initiated by any of the three pathways of complement activation [20,21]. We have shown previously that C3 deposition in the joints was not detectable in mice genetically deficient in C3 and that the levels of C3 deposition in the joints of Bf−/− mice were reduced to 30% of the levels seen in WT mice. This hypothesis is explored further in the current studies, where the levels of C3 deposition in the joints of mice expressing only the CP (MBL−/−/Df−/−) and only the LP (C1q−/−/Df−/−) were 58% and 26%, respectively, of the levels observed in WT mice (Table 1). The modest decrease in C3 deposition in the joints in CP only mice, which exhibited a near absence of clinical disease, suggest that these mice may possess an additional deficiency such as the ability to generate adequate levels of C3a or C5a.
Our studies used three different approaches to examine the abilities of the AP or CP to activate C3 or C5 in vitro after induction by adherent IgG mAb to CII. The first approach utilized sera of mice genetically deficient in various complement components (Fig. 2). The second approach used sera from mice lacking factor B (Fig. 3). The third approach compared mouse and human sera under Ca++ deficient conditions to inactivate the CP, or in the presence of a neutralizing mAb to factor B to inactivate the AP; these data were normalized to allow compilation of data from six experiments (Table 3). The overall results indicate that IgG-induced C3a and C5a generation are robust in both mouse and human sera in the presence of either the AP or CP alone. The results using the three different approaches were comparable, except for C5a generation by the CP in mouse sera. Sera from MBL−/−/Df−/− or Bf−/− mice gave C5a values of 28·6% (Fig. 2) and 12·5% (Fig. 3), respectively, of WT compared to 46·6% of WT when mouse sera was used in the presence of a mAb to factor B (Table 3). However, using each of these three different experimental approaches the ranges were large, making the observed differences in mean values less striking.
These data show that CP C5 convertase activity in mouse sera was not completely deficient, but was present at a lower level in comparison to human sera. Earlier investigations suggested that CP C5 convertase activity might be absent in mice [22]. The differences between our results and those of others may be due to the utilization of different assay systems. We examined in vitro complement activation by adherent anti-CII IgG mAb, conditions simulating those of IC adherent to the cartilage or other surfaces in vivo in animal models of disease or in human diseases. Using a one-step heterologous haemolytic assay with mouse sera as a source of C4 and guinea pig sera as a source of C5, Ebanks and Isenman observed CP-dependent C4 haemolytic activity ∼6% of WT [22]. However, with a multi-step haemolytic assay the residual C4 haemolytic activity was shown to be due to the AP amplification loop, leading these authors to conclude that mouse C4 was devoid of CP C5 convertase activity. Our data are not consistent with that conclusion.
IgG initiates complement activation in both assays, bound either to a solid surface in our assay or as an antibody to sheep erythrocytes in the haemolytic assay. We have demonstrated recently that adherent anti-CII IgG mAb can initiate complement activation in vitro by the AP in the absence of the CP, although when all three complement pathways are intact the CP may predominate [9]. After initiation of complement activation by any pathway, the C3 convertases C4bC2a (CP or LP) and C3bBb (AP) are assembled on cell surfaces or tissue substrates with C3b binding to the cell through a metastable thioester site. However, the C3 convertases exhibit weak cleavage of C5. Additional C3b molecules are generated in waves around the initial C3 convertase sites, leading to the formation of C4b–C3b and C3b–C3b dimers that form high-affinity C5 binding sites with functional C5 convertase activity for the CP and AP, respectively [25]. Through the AP amplification loop, every C3b molecule generated by each of the three complement activation pathways has the capacity to generate an AP C5 convertase complex. In most situations more AP C5 convertase sites are generated than CP conversion sites [23]. In addition, the C3b conjugated to the heavy chains of IgG by ester linkages acquires a conformation that enhances binding of factor B and inhibits binding of factor H, a major regulatory protein of the AP [26]. Thus, IgG provides a protected surface for amplification of the AP.
The binding of C4 to C5 may be weaker when the C5 binding site is generated by the CP alone rather than when the AP amplification loop is active because of a higher density of C3b deposition generated by the AP. A greater enhancement in the development of AP C5 convertase sites may occur with the adherent IgG in our experimental system in comparison to IgG-coated erythrocytes in a haemolytic system because of differences in the involved surface. Such differences may include charge, density of bound IgG or the degree and pattern of binding of the inhibitor factor H. Lastly, measurement of C5a levels in our studies, with adherent IgG inducing continuous complement activation, may be a more sensitive indicator of C5 convertase activity than a one-hit haemolytic assay using reagents from multiple species. These reasons may explain, at least in part, why we observed robust CP C5 convertase activity in mouse serum.
We observed no detectable C4 in sera lacking both C1q and factor D. The results of previous studies indicated that serum levels of C4 were decreased 20% in the sera from C3−/− mice, 80% in the sera from C1q−/− mice and 90% in the sera from C1q−/−/MBL−/− mice, all in comparison to WT mice [21]. The levels of C4 in the sera from Bf−/− mice were 30% lower in comparison to WT sera, although this difference was not statistically different [21]. The reasons for the decreases in levels of C4 in mice genetically deficient in complement components, particularly C1q, remain unexplained. However, these decreases in C4 levels did not influence our results as they occurred primarily in the absence of C1q, where the CP was already inactive.
We also observed increases in levels of C3 in sera from mice deficient in factor D or in both C1q and factor D, but not in sera from mice deficient in both MBL and factor D (Table 2). C3 levels may be increased in Df−/− mice because of a lack of AP activation through the tickover mechanism, but why this appears to be abrogated by MBL deficiency is unknown.
In conclusion, the CP or LP alone cannot mediate CAIA, confirming further our previous findings that the AP is both necessary and sufficient for this disease [20,21]. Our data suggest that the dependency of CAIA on the AP may be due primarily to a lower level or to a different distribution of C3 deposition by the CP in the absence of the AP amplification loop. The clinical relevance of these observations in a murine model of IC-induced arthritis and in comparable in vitro studies awaits the use of pathway and mechanism-specific complement inhibitors in patients with various diseases [27].
Acknowledgments
This work was supported by NIH Grant no. AR-51749.
Disclosure
VMH has patent rights related to a subset of the compounds included in this article and has share ownership in as well as has served as a Consultant for Taligen Therapeutics. The other authors have nothing to disclose.
References
- 1.Walport MJ. Complement. N Engl J Med. 2001;344:1058–66. 1140–4. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 2.Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–6. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 3.Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7:9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
- 4.Guo R-F, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–52. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 5.Haas PJ, van Striip J. Anaphylatoxins: their role in bacterial infection and inflammation. Immunol Res. 2007;37:161–75. doi: 10.1007/BF02697367. [DOI] [PubMed] [Google Scholar]
- 6.Malmsten M, Schmidtchen AS. Antimicrobial C3a – biology, biophysics, and evolution. Adv Exp Med Biol. 2007;598:141–58. doi: 10.1007/978-0-387-71767-8_11. [DOI] [PubMed] [Google Scholar]
- 7.Pangburn MK, Schreiber RD, Müller-Eberhard HJ. Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med. 1981;154:856–67. doi: 10.1084/jem.154.3.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176:1305–10. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- 9.Banda NK, Wood AK, Takahashi K, et al. Initiation of the alternative pathway of murine complement by immune complexes is dependent on N-glycans in IgG antibodies. Arthritis Rheum. 2008;58:3081–9. doi: 10.1002/art.23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol. 2007;179:2600–8. doi: 10.4049/jimmunol.179.4.2600. [DOI] [PubMed] [Google Scholar]
- 11.Kemper C, Mitchell LM, Zhang L, Hourcade DE. The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci USA. 2008;105:9023–8. doi: 10.1073/pnas.0801015105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kemper C, Hourcade DE. Properdin: new roles in pattern recognition and target clearance. Mol Immunol. 2008;45:4048–56. doi: 10.1016/j.molimm.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujita T. Evolution of the lectin-complement pathway and its role in innate immunity. Nat Rev Immunol. 2002;2:346–53. doi: 10.1038/nri800. [DOI] [PubMed] [Google Scholar]
- 14.Selander B, Mårtensson U, Weintraub A, et al. Mannan-binding lectin activates C3 and the alternative complement pathway without involvement of C2. J Clin Invest. 2006;116:1425–34. doi: 10.1172/JCI25982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hubert-Lang M, Sarma JV, Zetoune FS, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–7. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 16.Rodrigez de Córdoba S, Goicoechea de Jorge E. Genetics and disease associations of human complement factor H. Clin Exp Immunol. 2008;151:1–13. doi: 10.1111/j.1365-2249.2007.03552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pickering MC, Cook HT. Renal diseases associated with complement factor H: novel insights from humans and animals. Clin Exp Immunol. 2008;151:210–30. doi: 10.1111/j.1365-2249.2007.03574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hietala MA, Johnson I-M, Tarkowski A, Kleinau S, Pekna M. Complement deficiency ameliorates collagen-induced arthritis in mice. J Immunol. 2002;169:454–9. doi: 10.4049/jimmunol.169.1.454. [DOI] [PubMed] [Google Scholar]
- 19.Hietala MA, Nundakumar KS, Persson L, et al. Complement activation by both classical and alternative pathways is critical for the effector phase of arthritis. Eur J Immunol. 2004;34:1208–16. doi: 10.1002/eji.200424895. [DOI] [PubMed] [Google Scholar]
- 20.Banda NK, Thurman JM, Kraus D, et al. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J Immunol. 2006;177:1904–12. doi: 10.4049/jimmunol.177.3.1904. [DOI] [PubMed] [Google Scholar]
- 21.Banda NK, Takahashi K, Wood AK, Holers VM, Arend WP. Pathogenic complement activation in collagen antibody-induced arthritis in mice requires amplification by the alternative pathway. J Immunol. 2007;179:4101–9. doi: 10.4049/jimmunol.179.6.4101. [DOI] [PubMed] [Google Scholar]
- 22.Ebanks RO, Isenman DE. Mouse complement component C4 is devoid of classical pathway C5 convertase subunit activity. Mol Immunol. 1996;33:297–309. doi: 10.1016/0161-5890(95)00135-2. [DOI] [PubMed] [Google Scholar]
- 23.Harboe M, Ulvund G, Vein L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. 2004;138:439–46. doi: 10.1111/j.1365-2249.2004.02627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thurman JM, Kraus DM, Girardi G, et al. A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody-induced pregnancy loss in mice. Mol Immunol. 2005;42:87–97. doi: 10.1016/j.molimm.2004.07.043. [DOI] [PubMed] [Google Scholar]
- 25.Rawal N, Pangburn MK. Structure/function of C5 convertases of complement. Int Immunopharmacol. 2001;1:415–22. doi: 10.1016/s1567-5769(00)00039-4. [DOI] [PubMed] [Google Scholar]
- 26.Reiter J, Fishelson Z. Targeting of complement to tumor cells by heteroconjugates composed of antibodies and of the complement component C3b. J Immunol. 1989;142:2771–7. [PubMed] [Google Scholar]
- 27.Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25:1265–73. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]