

Abstract
Alloimmunity to mismatched donor HLA-antigens and autoimmunity to self-antigens have been hypothesized to play an important role in immunopathogenesis of chronic rejection of transplanted organs. However, it is not known what role, if any, alloimmune response plays in inducing autoimmunity. To test whether antibody developed post-transplantation to mismatched donor-MHC induces autoimmunity and chronic rejection, we developed a murine model wherein anti-MHC class I antibodies or control (C1.18.4/anti-keratin) were administered intra-bronchially into native lungs. Animals receiving anti-MHC class I, but not control antibodies, developed marked cellular infiltration around vessels and bronchiole of lung by day 15 followed by epithelial hyperplasia, fibrosis and occlusion of the distal airways similar to chronic rejection following human lung transplantation. Lungs of mice receiving anti-MHC class I showed increased expression of chemokines, their receptors and growth factors and induced IL-17 as well as de novo antibodies to self-antigens, K-α1 tubulin and collagenV. IL-17 neutralization by anti-IL-17 resulted in reduction of autoantibody and lesions induced by anti-MHC class I antibodies. Thus, our results indicate that antibodies to donor-MHC can induce autoimmunity, mediated by IL-17, which plays a pivotal role in chronic rejection post-lung transplantation. Therefore, approaches to prevent autoimmunity should be considered for the treatment of chronic rejection post-lung transplantation.
Keywords: chronic rejection, autoimmunity, antibodies, MHC, pathogenesis
Introduction
Transplantation is the current treatment of choice for many end stage renal, hepatic, cardiac and pulmonary diseases. Improvement in surgical techniques, post operative management and immunosuppression have all resulted in marked improvement in allograft survival. However, long term functions of the transplanted organs are often limited due to development of chronic rejection. This is particularly true following lung transplantation (LTx) where chronic rejection referred to as bronchiolitis obliterans syndrome (BOS) occurs in 50% of the transplants within 3 years post-transplant. A retrospective analysis of 2,190 transplants performed at Washington University/Barnes Hospital demonstrated that nearly all of the transplanted organs succumb to chronic rejection during the course of 10 years post-transplant (1). It is generally accepted that chronic rejection is a manifestation of alloimmune responses against the transplanted organ (2, 3). More recently, strong correlation has been noted for the development of anti-MHC class I antibodies (Abs) during the post-transplant period with development of chronic rejection (4, 5). A longitudinal study conducted over a ten year period for HLA Abs demonstrated a strong association between the production of HLA Abs after renal transplantation and graft failure (6). Similar association of post-transplant Abs with rejection of heart (7) and LTx (8) have also been reported. Based upon these reports, it is generally believed that HLA Ab monitoring during the post-transplant period is an effective way to predict impending chronic rejection of solid organs.
Induction of autoimmunity following allo-transplantation has also been demonstrated after heart and LTx (9, 10). It has been postulated that auto-reactive T cells to self antigens occurs after an allo-response, and once activated they can induce pathology of chronic rejection (11). Cellular auto reactivity against heat shock protein 60 (HSP-60) has been demonstrated in peripheral blood and graft infiltrating lymphocytes following renal transplantation (12). These cells produced both inflammatory and regulatory cytokines in response to HSP-60 and IL-10 predominance was noted in the late period post-transplant suggesting a regulatory role for HSP-60 autoreactivity. Cardiac myosin reactive T and B cells have been demonstrated following murine cardiac allograft rejection (9). Further, anti-cardiac myosin immune response has been shown to contribute to the graft rejection in that pre-transplant modulation of this response in recipient mice can affect cardiac allograft survival (13). It has been demonstrated that anti-cardiac myosin reactivity persists in the late post-transplant period in the absence of an alloimmune response suggesting that the auto-immune response to myosin may be associated with the pathogenesis of chronic rejection (14). Further, pre-transplant sensitization with cardiac myosin resulted in accelerated rejection of the graft and more significantly under the same condition, even a syngenic graft was rejected within 40 days (9). Using a rat model of LTx, evidence has been obtained for an immune response to a self antigen, collagen V (ColV), in the pathogenesis of lung allograft rejection (15). Two lines of evidence supporting the view that autoreactive T cells specific to ColV are sufficient to cause rejection of the transplants have been presented; 1) instillation of syngenic BAL cells pulsed with ColV was sufficient to cause rejection of lung allograft, and 2) adoptive transfer of anti-ColV cell lines into naïve rats caused peribronchiolar and perivascular inflammation in lung isografts (16). Taken together, the above experiments using animal models of transplantation strongly support the conclusion that the induction of an autoimmune response after transplantation may lead to chronic rejection of allografts.
IL-17 has been to shown to be a potent pro-inflammatory cytokine that acts on epithelial cells, endothelial cells (ECs), fibrocytes and stromal cells leading to a pro-inflammatory signal which results in enhanced pro-inflammatory cytokine and chemokine secretion (17). IL-17 has also been shown to play a crucial role in the induction of humoral auto-immune responses (18). Analysis of the T cell responses to ColV in LTx recipients has been shown to be dependent on CD4 T cells and monocytes (19) and this response was found to be dependent on IL-17, TNF-α and IL-1β. (19). Furthermore, this report demonstrated the interaction between IL-17 and monocytes is necessary for the induction of the ColV specific responses which correlates with the development of BOS. Studies in the literature have also demonstrated that a deficiency in the IL-17 or blocking of IL-17 results in a decrease in the production of auto-Abs (20, 21). IL-17 has been also shown to play a crucial role in the formation of germinal centers (GCs) and regulate the movement of the B cells in the GCs as mediated by chemokines CXCL12 and CXCL13. Blocking of the IL-17 has been shown to result in a reduction in the levels of GCs and decreases the titer of auto-Abs (22). Taken together, several body of evidence strongly supports the hypothesis that IL-17 may be an important contributor in the production of auto-Abs.
Although there is a strong evidence for both allo- and auto-immune responses in the pathogenesis of chronic rejection, the relationship between allo-immunity and auto-immunity and its role in the pathogenesis of chronic rejection still remains to be elucidated. In our study, we administered anti-MHC class I Abs intrabronchially into the native murine lung so that anti-MHC class I Abs will specifically ligate MHC molecules in the lung parenchyma. Using this novel model, we demonstrate for the first time that ligation of anti-MHC class I Abs to the lung parenchyma results in an auto-immune response leading to endotheliitis, cellular infiltration, epithelial hyperplasia and fibrosis as well as luminal occlusion of the small airways. Histopathology of the lesion closely resembles chronic rejection pathology noted following human LTx, thus providing support for both allo-immunity and auto-immunity in the immunopathogenesis of chronic rejection following LTx. Further, our data demonstrate that binding of MHC molecules with specific Abs can result in exposure of self antigens of the epithelial cells to immune system leading to both cellular and humoral auto-immunity and immunopathogenesis of chronic rejection that is predominantly mediated by the IL-17 producing cells.
Materials and Methods
Anti-MHC class I Abs induced OAD development
We developed a murine model in which OAD (obliterative airway disease), correlate of BOS, takes place in the distal airways following ligation of MHC molecules in the lung by a mAb to MHC class I antigens. Since we wanted to have good titer of anti-MHC class I Ab in the lung and not absorbed by other tissues, we opted to give anti-MHC class I Abs directly into the lung. All experiments were performed in compliance with the guidelines of the Institutional Laboratory Animal Care and Use Committee of Washington University School of Medicine (protocol 20070121). Murine mAb to H2Kd (BALB/c, 6 week, male, IgG2a) which has no detectable endotoxin as measured by LAL assay was given at a dose of 200 μg per administration into mice intubated with a 20 gauge catheter. Similarly, a murine mAB to H2Kb (C57/BL6, 6 weeks, male, IgG2a) as well as murine mAB against the HLA class I molecules (HLA-A2 transgenic C57/BL6 mice, 6 weeks, male) were administered endbronchially to test the ability of anti-MHC abs to induce lesions in other strains of mice. In brief, mice were anesthetized and tongue pulled out. A mosquito clamp was inserted into the mouth and a 20 gauge catheter was placed into the trachea. Antibody (200 μg) was administered into the lung on day 1, 2, 3, 6 and then weekly thereafter. C1.18.4, same isotype, was given on the days mentioned above as controls.
Immunohistochemical analysis
Frozen lung samples were embedded in Freez Tissue matrix (OCT), and sections were cut at 5 μm thickness. The sections were fixed in cold alcohol for 2 min (-20 °C) and air dried. The sections were treated with 3% H2O2 in EtOH for 10 min to block endogenous peroxidase activity. The sections were blocked with Biotin/Avidin system components for 15 min by blocking reagent (Avidin/Biotin Blocking Kit, Vecter laboratories, Burlingame, CA). Primary and secondary Abs were diluted with Ab dilution solution (BD biosciences, San Diego, CA). The sections were incubated for O/N at 4 °C with purified rat anti-mouse CD4, CD8, CD11b and CD19 mAbs (5.0 μg/mL, BD Pharmingen,) Then, the sections were incubated for 30 min at room temperature (RT) with diluted biotin-conjugated goat anti-rat IgG (1:50, BD Pharmingen). The sections were incubated with Streptavidin-HRP and incubated for 30 min at RT (BD Pharmingen). The presence of positive cells was detected with the DAB substrate kit (BD Pharmingen), counter-stained with hematoxylin, and examined using a light microscope. Positive cells were counted by random sampling.
Lungs were fixed in 10% formaldehyde. Sections were cut at 5 μm thickness and stained with Masson's trichrome and hematoxylin & eosin (H&E). Lesions that displayed cellular infiltration, epithelial abnormalities, and fibro proliferation were analyzed by random sampling. Percent fibrosis was calculated using Optimas software version 6.5.172 (Media Cybernetics). Percent fibrosis was quantified by dividing the total area enclosed by basement membrane. Percent cellular infiltration and epithelial abnormalities were calculated by dividing the total bronchiole and vessel, respectively
Gene expression array
To determine chemokines and their receptors, lungs were harvested at 4 and 15 days after treatment, respectively. Expression of 96 genes, including chemokines, their receptors, and growth factors, was analyzed by RT2 Profiler™ PCR Array for mouse Chemokines & Receptors and RT2 Profiler™ PCR Array for mouse growth factor (SuperArray, Bethesda, MD). Lung tissues were homogenized with TRIzol reagent (Life Technologies Gaithersburg, MD) and total RNA was isolated. cDNA was synthesized by extension of primers using the ReactionReady First Stand cDNA Synthesis Kit (SuperArray).
Isolation of lung infiltrating lymphocytes
Lung tissues were mechanically cut and stirred in a suspension of RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with HEPES buffer (25 mM), sodium pyruvate (1 mM), non-essential amino acid (100 μM), penicillin (100 units/ml) and streptomycin (100 μg/ml) along with 0.1 % collagenase type XI (Sigma, St. Louis, MO) and 0.002% DNAse (Sigma, St Louis, MO) for enzymatic digestion O/N at RT. The suspension was filtered thought a cell strainer and washed with PBS twice. Lymphocytes were separated using Ficoll-Paque density centrifugation. Buffy coat enriched in lymphocytes was collected and washed with PBS twice. Lymphocytes isolated from lung tissues were suspended in medium.
ELISPOT assay
ELISPOT assays were performed as described previously (23). Briefly, MultiScreen 96-well filtration plates (Millipore) were coated O/N at 4°C with 5.0 μg/ml capture mouse cytokine-specific mAb (BD Biosciences) in 0.05 M carbonate-bicarbonate buffer (pH 9.6). The plates were blocked with 5% BSA for 2 hr and washed 3 times with PBS. Subsequently, 3 × 105 cells were cultured in triplicate in the presence of ColV (20 μg/ml) or human K-α1 tubulin (1 μg/ml) and irradiated feeder autologous splenocytes (1:1 ratio). After 48-72hr, the plates were washed 3 times with PBS and 3 times with 0.05% PBS/Tween 20. Then, 2.0 μg/ml biotinylated mouse cytokine specific mAb (BD Biosciences) in PBS/BSA/Tween 20 was added to the wells. After an O/N incubation at 4°C, the plates were washed 3 times and HRP-labeled streptavidin (BD Biosciences) diluted 1/100 in PBS/BSA/Tween 20 was added to the wells. After 2 hr, the assay was developed with 3-amino-9-ethylcarbazole substrate reagent (BD Biosciences) for 5–10 min. The plates were washed with tap water to stop the reaction and air dried. Spots were analyzed in an ImmunoSpot Series I analyzer (Cellular Technology), and the results were expressed as spots per million cells (spm) ± standard error. Any spots obtained by culturing T-cell lines with APCs alone without ColV or human K-α1 tubulin were subtracted from the number of spots in the test cultures.
ELISA assay
ELISA plate (Nunc) was coated with pure ColV (BD Biosciences) (1μg/ml) in PBS O/N at 4°C. Serum samples were collected from mice treated with anti-H2kd Ab or Isotype Ab and non treated mice. Serum samples were tested (1:250 and 1:500) for binding to ColV. Detection was done with anti mouse IgG, IgM –HRP and development using TMB substrate and read at 450nm. A sample was considered as positive if the values were over an average cutoff values obtained from normal sera from 10 different mice. Concentration of Abs was calculated based on a standard curve using the binding of known concentration of anti-ColV Abs (Santacruz)
ELISA to detect anti-K-α1 tubulin Abs was done in a similar manner using purified recombinant human K-α1 tubulin (1μg/ml). Concentration of anti-K-α1 tubulin Abs was expressed as μg/ml based on a standard curve using the binding of known concentration of anti-K-α1 tubulin Abs (SantacruzBiotech).
Neutralization of IL-17
In order to study the role of IL-17 in induction of autoimmunity following ligation of anti-MHC class I Abs, we administered on day 1, 3, 5 and weekly thereafter 50 μg of anti-IL-17 mAb (eBioscience, San Diego, CA) intravenously into BALB/c mice treated with anti-MHC class I Ab on days 1, 2, 3, 6 and weekly thereafter (24). Serum from these mice was collected on day 30 following anti-MHC class I Ab treatment to quantitate the levels of Abs against K-α1 tubulin and ColV as described earlier. Histopathological analysis of the lungs was also performed to determine the levels of cellular infiltration and epithelial hyperplasia by H&E staining. Fibrosis in the lungs was analyzed by trichrome staining.
Results
Ligation of MHC class I molecules of the lung parenchyma by specific anti-MHC class I Abs results in auto-immune cellular infiltration, endotheliitis, epithelial hyperplasia and fibrosis
Development of anti-donor MHC Abs have been strongly associated with early development of chronic rejection of kidney (6), heart (7) and lung (8) allografts. In order to determine the consequences of anti-MHC class I Ab binding to the organ we administered intrabronchially into the lungs of BALB/c mice mAb with specificity to H2Kd (anti-MHC class I Ab). C1.18.4 control Ab of the same isotype was administered in control animals.
Fifteen and 30 days after administration of Abs, the lung tissue was harvested and histopathological analysis was performed. Lungs from animals administered with anti-MHC class I Ab after 15 days demonstrated significant inflammatory cells around the vessels and bronchiole (Figure1A). In addition, there was epithelial hyperplasia (Figure 1D) and an increase in fibrosis. Morphometric analysis of these lesions demonstrated 51.4% cellular infiltration around the vessels, 71.1% cellular infiltration around the bronchiole, 71.1% epithelial hyperplasia and 15.2% fibrosis (Table 1). In contrast, C1.18.4 or anti-keratin ab administered lungs did not show any significant lesions (Figure 1B, 1E, and 1C, 1F).
Figure 1. Administration of anti-MHC class I Abs developed significant cellular infiltration around vessel and bronchiole as well as hyperplasia of the bronchial epithelium (H&E stain).
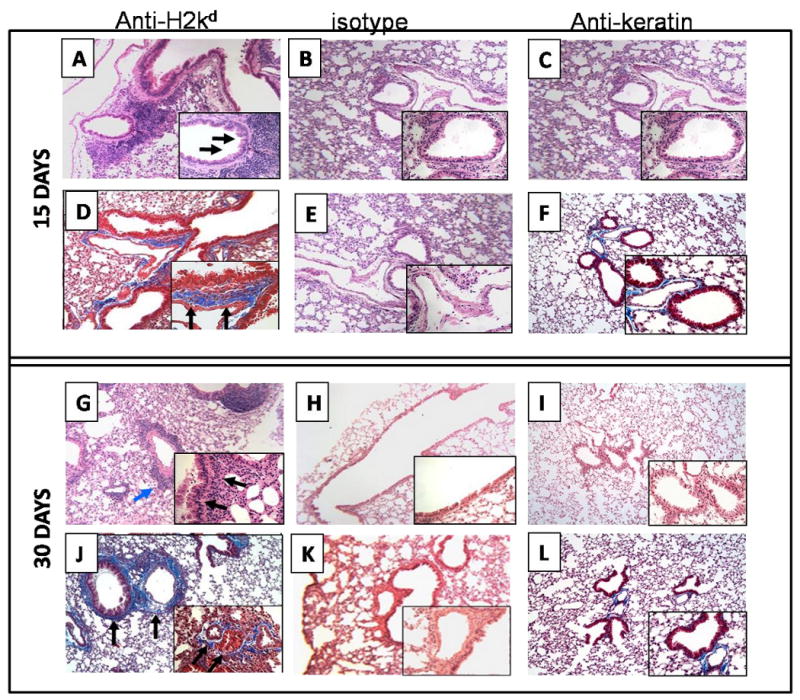
Anti-H2kd or control (C1.18.4) Ab or anti-keratin antibody was administered endobronchially in BALB/c mice on days 1, 2, 3, 6 and weekly thereafter. The lungs were harvested on day 15 or 30 and analyzed by H&E and trichrome staining. A representative picture from data obtained from 5 mice is presented in the figure. Original magnification: × 100, insets: ×400. (A, B, C, G, H and I are sections stained with H& E; D, E, F, J, K, and L are trichrome staining. A, and D: anti-H2Kd treated mice on day 15; G and J : anti-H2Kd treated mice on day 30; B, E: C1.18.4 Ab treated mice on day 15; H, K: C1.18.4 Ab treated mice on day 30; C,F: anti-keratin Ab treated mice on day 15; I, L: anti-keratin Ab treated mice on day 30. Lungs from the anti-H2kd Ab treated mice showed significant cellular infiltration around bronchia and vessel (A) and hyperplasia of the bronchial epithelium (G) are marked by the arrows. A significant increase in the fibroproliferation, collagen deposition and luminal occlusion was observed in the lungs of the anti-H2kd Ab treated mice (J). Mice treated with isotype control Ab or anti-keratin Ab showed no significant morphological changes compared to the anti-H2Kd Ab treated mice (B, C, E, F, H, I, K and L).
Table 1. Morphometric analysis of lesions developed in lungs by anti-MHC class I antibody.
| Day | Antibody | Cellular Infiltration Around Bronchiole | Cellular Infiltration Around Vessel | Epithelial Hyperplasia | Luminal Occlusion | Fibrosis Area | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD11b | CD4 | CD8 | Total | CD11b | CD4 | CD8 | Total | |||||
| 15 | H2Kd | 34.3* (± 17.9) |
33.0* (± 5.6) |
2.0* (± 0.4) |
71.1% (± 15.3) |
48.0* (± 2.8) |
145.5* (± 16.3) |
2.0* (± 0.3) |
51.4% (14.7) |
66.1% (± 15.9) |
0.0% | 15.2% (± 6.1) |
| C1.18.4 | 18.3* (± 4.2) |
17.7* (± 13.9) |
2.0* (± 0.2) |
15.4% (± 5.9) |
17.5* (± 4.9) |
18.0* (± 4.4) |
1.0* (± 0.4) |
15.1% (± 5.8) |
2.4% (± 1.1) |
0.0% | 3.4% (± 2.8) |
|
| 30 | H2Kd | 54.3* (± 17.9) |
74.0* (± 20.9) |
6.7* (± 1.2) |
81.8% (± 12.2) |
52.0* (± 9.2) |
50.0* (± 16.7) |
13.0* (± 2.1) |
93.1% (± 5.7) |
40.5% (± 20.2) |
14.6% (± 4.3) |
35.4% (± 7.4) |
| C1.18.4 | 12.3* (± 2.1) |
22.7* (± 7.6) |
2.0* (± 0.2) |
12.5% (± 7.1) |
8.0* (± 4.6) |
8.0* (± 4.6) |
2.0* (± 0.4) |
21.2% (± 11.7) |
1.8% (± 1.5) |
0.0% | 4.1% (± 3.9) |
|
per hpf
All of the above lesions were markedly increased on day 30. Cellular infiltration around the vessels and bronchiole were increased to 93.1% and 81.8% respectively (Figure 1G and Table 1). More significant is our finding of an increase in fibrosis (35.4%) (Figure 1J) along with 14.6% lumen occlusion of the small airways (Figure 1J and Table 1). Lungs from animals treated with control C1.18.4 Ab or the anti-keratin Abs did not demonstrate any of the above changes (Figure 1H, 1K, and 1I, 1L). Similarly, administration of anti- H2Kb antibodies into the C57/BL6 mice or the W6/32 antibodies in the HLA-A2 transgenic C57/BL6 mice, resulted in epithelial hyperplasia, induction of fibrosis and occlusion of the small airways with a similar kinetics as observed in the anti- H2Kd administered BALB/c mice (data not shown). Moreover, histopathological analysis of the other organs, i.e., kidney, heart, liver and pancreas showed no significant morphological changes following administration of the anti-MHC class I Abs (data not shown).
Immunohistochemical staining of the cellular infiltration demonstrated a predominance of CD4+ cells (Figure 2A&G and Table 1) along with CD11b+ (Figure 2D&J and Table 1) macrophages around the bronchiole on days 15 and 30. In addition, a small percentage of CD8+ T cells were seen around the vessel but not around the bronchiole when compared to controls treated with C1.18.4 (2B, E, 2H, 2K) or anti-keratin ab administered mice (2C, 2F, 2I, 2L). Ab binding and complement deposition (C4d) on the lung parenchyma was noted (Figure 2M) in anti-H2Kd treated mice whereas no C4d deposition was observed in the controls (2N, 2O).
Figure 2. Administration of anti-MHC class I Abs induces significant increase in the CD4+ and CD11b+ cells in the lungs.
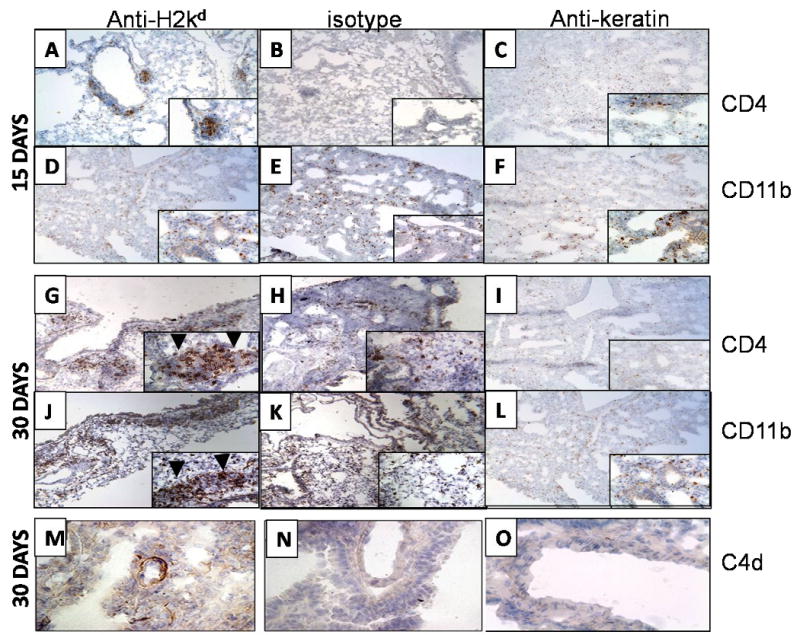
Anti-H2kd (A, D, G, J, M) or control (C1.18.4) Ab (B, E, H, K, N) or anti-keratin antibody (C, F, I, L, O) was administered endobronchially in BALB/c mice on days 1, 2, 3, 6 and weekly thereafter. The lungs were harvested on day 15 or 30 and analyzed for infiltration of CD4+ T cells, CD11b and C4d deposition by immunohistochemistry using mouse anti-CD4, anti-mouse CD11b Abs anti-mouse C4d antibodies. CD4 + cells infiltrated around the bronchiole (arrow head) and vessel at both 15 and 30 days (A, G) along with significant infiltration of CD11b + cells around the bronchiole (arrow head) and vessel at 15 and 30 days (D, J) in anti-H2kd administered mice whereas no cellular infiltration around the bronchiole and vessel was observed in isotype control (B,E, H and K) or anti-keratin antibody administered mice (C, F, I and L). A significant increase in the C4d deposition was observed in the lungs of anti-H2kd administered mice (M) when compared to control antibody administered mice (N, O). A significant increase in the levels of CD4+ T cells and CD11b+ cells was observed in the lungs of mice treated with anti-H2Kd Ab compared to mice treated with isotype control Ab.
Lung infiltrating lymphocytes secrete increased levels of IFN-γ and IL-4 upon stimulation with self antigens K-α1 tubulin or ColV
K-α1 tubulin and ColV specific T lymphocytes which infiltrate the lung following administration of anti-MHC class I Abs was enumerated. T cells were isolated from lung tissue at 15 days after anti-MHC class I Ab administration and stimulated with K-α1 tubulin or ColV in the presence of autologous splenocytes. As shown in Figure 3a & b, anti-MHC class I Ab administered animals had a significant increase in K-α1 tubulin and ColV reactive T cells in comparison to C1.18.4 administered animals. These autoreactive T cells include K-α1 tubulin reactive cells capable of secreting IFN-γ (80.0 vs. 43.3), IL-4 (33.9 vs. 0), and Col V reactive cells secreting IFN-γ (71.1 vs. 17.2), IL-4 (44.2 vs. 0). The response of T cells isolated from the lungs on day 30 both against K-α1 tubulin and ColV respectively were significantly elevated, IFN-γ (171.1 vs. 65.0), IL-4 (38.3 vs. 6.3), and IFN-γ (101.4 vs. 28.6), IL-4 (45.1 vs. 5.0). This observation indicates that administration of anti-MHC class I Abs increases the activation of autoimmune responses against the self antigens, ColV and K-α1 tubulin.
Figure 3. Administration of anti-MHC class I Ab increases the frequency of IFN-γ, IL-4 and IL-17 secreting T cells against K-α 1 tubulin and ColV in the lung.
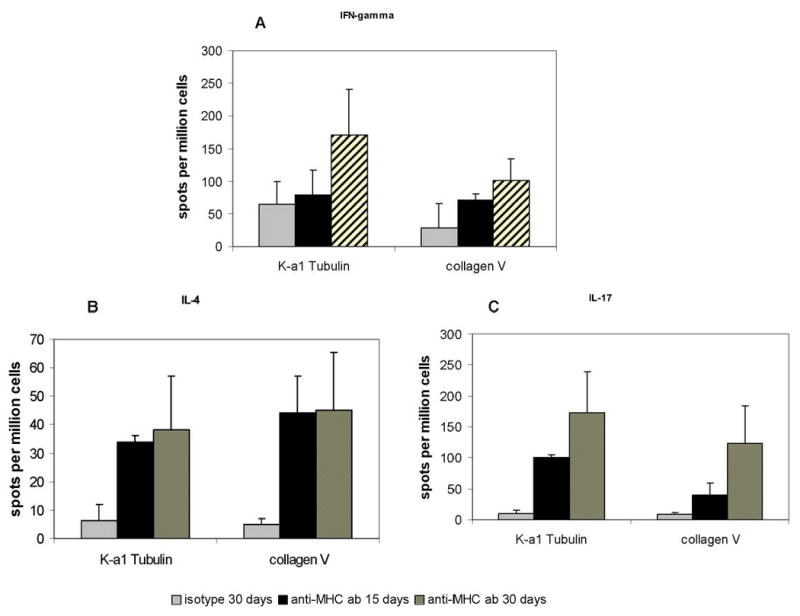
(A) Frequency of the IFN-γ secreting T cells against K-α 1 tubulin and ColV. (B) Frequency of the IL-4 secreting T cells against K-α 1 tubulin and ColV and C: Frequency of the IL-17 secreting T cells against K-α 1 tubulin and ColV. Anti-H2Kd or control Ab was administered endobronchially in BALB/c mice on days 1, 2, 3, 6 and weekly thereafter. The lungs were harvested on day 15 and 30. T cells infiltrating the lungs were harvested by collagenase digestion and the frequency of T cells secreting IFN-γ, IL-4 and IL-17 on stimulation with K-α 1 tubulin and collagen were analyzed by ELISPOT. The values are represented as means ± SD using data obtained from 5 ELISPOT assays. The grey bars represent the isotype control, the solid bars represent the T cells isolated from lungs on day 15 and the black bars represent the T cells isolated on day 30. A significant increase in the frequency of IFN-γ, IL-4 and IL-17 secreting T cells against K-α 1 tubulin and ColV was observed in mice administered anti-H2kd Ab when compared to controls (*=P<0.05, control vs. anti-MHC class I Ab).
Increased ColV specific IL-17 production by the lung infiltrating T lymphocytes
T cells, isolated from lung tissue at 15 and 30 days, after anti-MHC class I Ab administration and stimulated with K-α1 tubulin or ColV in the presence of autologous splenocytes, were analyzed by ELISPOT to enumerate the frequency of IL-17 secreting T cells. As shown in Figure 3c, anti-MHC class I Ab administered animals had a significant increase in K-α1 tubulin/ColV reactive T cells in comparison to C1.18.4 administered animals indicating that autoreactive T cells include cells capable of secreting IL-17 at 15 days (101.1 vs. 13.6 for K-α1 tubulin and 39.2 vs. 0 for Col V in anti-MHC class I Ab compared to isotype control respectively) and at 30 days (173.0 vs. 10.3 for K-α1 tubulin and123.7 vs. 8.2 for ColV).
Specific binding of MHC class I molecules with Abs result in up regulation of chemokines and its receptors on the lung parenchyma
Since marked cellular infiltration and epithelial hyperplasia was seen following the administration of anti-MHC class I Abs into the lung, we analyzed for upregulation of the genes for chemokines and their receptors which may be involved in promoting cellular infiltration into the lung and causing epithelial hyperplasia. As shown in Figure 4a, lungs harvested on day 4 following treatment with anti-MHC class I Ab showed a several fold increase in relation to C1.18.4 in the expression of results in an increased expression of CCL9 (9.2 fold, p<0.01), CCR2 (10.6 fold, p<0.01), CXCL1 (6.35 fold, p<0.01) and CXCL12 (8.0 fold, p<0.01). Other genes analyzed in this panel which includes genes for CC, CXC motif ligands and their receptors did not demonstrate any significant change in comparison to C1.18.4 treated animals.
Figure 4. Administration of anti-MHC class I Abs increases the expression of chemokines, cytokines and growth factor in the lungs.
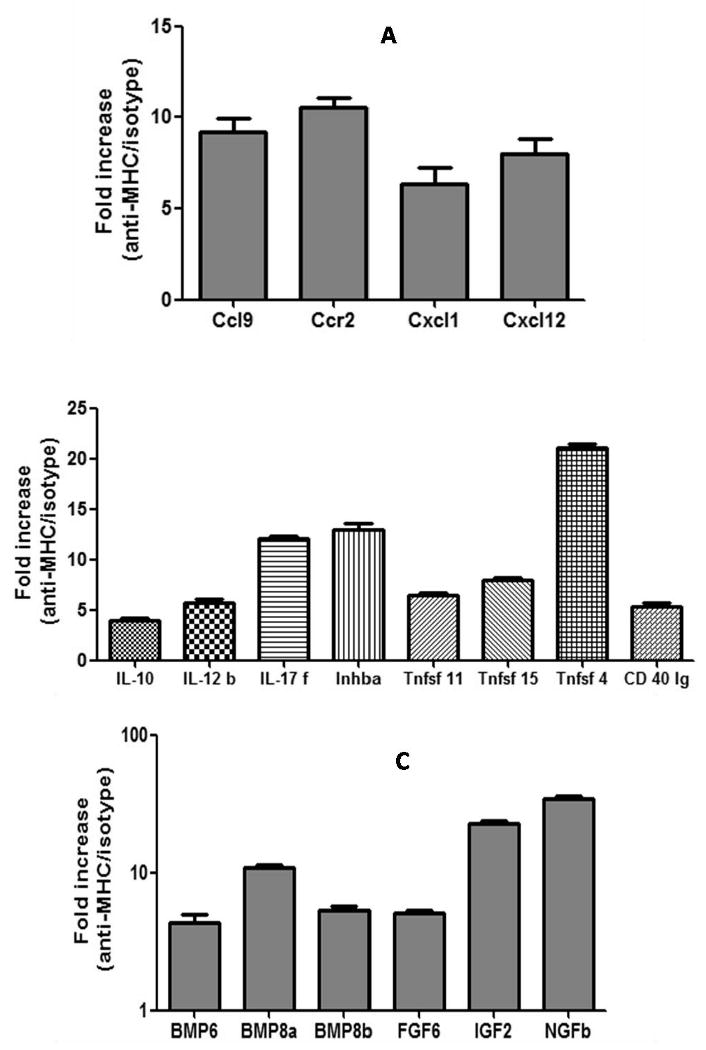
(A) Expression levels of chemokines and their receptors. (B) Expression levels of the cytokines. (C) Expression levels of the growth factor. Anti-H2Kd or control Ab was administered endobronchially in BALB/c mice on days 1, 2, 3, 6 and weekly thereafter. The lungs were harvested on day 4 (chemokines) and 15 (cytokines and growth factor) and analyzed using a quantitative real-time PCR array. RNA was extracted from the lung tissue and 1μg of the RNA was reverse transcribed and used as template for the quantitative real time PCR array (chemokines and receptor PCR array, common cytokines PCR array and growth factor PCR array, Superarray Inc, Frederick, MD). CT values obtained from the quantitative real-time PCR were analyzed by using the PCR array analysis software from Superarray Inc. The samples were normalized using the expression levels of the house keeping genes GAPDH, and HPRT1. The data represents the average values obtained from 3 different experiments. A significant (p>0.05) increase in the expression of chemokines (CCL9: 9.2, CCR2: 10.6, CXCL1: 6.35 and CXCL12: 8.0), cytokines (IL-10: 4.0, IL-12b: 5.66, IL-17f: 12.17, Inhba: 13.0, Tnfsf11: 6.5, Tnfsf15: 8.0, Tnfsf 4: 21.11, CD40 lg: 5.28) and growth factors (Bmp6: 4.30, Bmp8a: 10.88, Bmp8b: 5.28, Fgf6: 5.07, Fgf2: 37.46 and IL-4: 290.82) was observed in mice administered anti-H2Kd compared to controls.
Specific binding of MHC class I molecules of the lung parenchyma with Abs result in up regulation of several cytokines
To analyze the genes which are activated following anti-MHC class I Ab treatment, the expression of 96 cytokines relation gene between lymphocyte purified from lung tissues treated with anti-MHC class I Ab and C1.18.4 at 15 days after treatment were determined. As shown in Figure 4b, lymphocyte purified from lung tissues treated with anti-MHC class I Ab showed a significant increase in the expression of IL-10: 4.0 fold, IL-12b: 5.66 fold, IL-17f: 12.17 fold, Inhba: 13.0 fold, Tnfsf11: 6.5 fold, Tnfsf15: 8.0 fold, Tnfsf 4: 21.11 fold, CD40 lg: 5.28 fold.
Specific binding of MHC class I molecules of the lung parenchyma with Abs result in up regulation of growth factors on the lung parenchyma
Lungs harvested on day 30 post administration of anti-MHC class I Abs demonstrated significant fibrosis and luminal occlusion of the bronchioles. To determine the growth factors which may be involved in this process, we analyzed expression of 96 well known growth factor genes in lungs harvested after 15 days of administering either anti-MHC class I or control Ab C1.18.4. As presented in Figure 4c, lungs treated with H2kd demonstrated significant fold increase in the expression of many growth factors including Bmp6: 4.30 fold, Bmp8a: 10.88 fold, Bmp8b: 5.28 fold, Fgf6: 5.07 fold, Igf2: 37.46 fold and Ngfb: 290.82 fold.
Production of auto-Abs against self antigens ColV and K-α1 tubulin following administration of anti-MHC class I Abs
Development of auto-Abs against ColV and K-α1 tubulin has been demonstrated in LTx recipients with chronic rejection as well as in animal models of lung allograft rejection (25). To determine whether anti-MHC class I Ab administration into the lung can result in auto-Ab formation, we analyzed serial serum samples from the animals that developed lesions similar to chronic rejection following human LTx. As shown in Figure 5a, 30 days post anti-MHC class I Ab administered mice reacted with tubulin. In addition, sera obtained at 15, 30 and 60 days post anti-MHC class I Ab administration also reacted to another self antigen ColV (Figure 5b). These results concur with earlier findings from both human and animal LTx studies indicating that auto-Abs to self-antigens may play a role in the pathogenesis of chronic rejection. Further, our results indicate that anti-MHC class I Abs can indeed expose otherwise cryptic antigens and immune responses to these antigens can be elicited following the development of anti-donor MHC Abs.
Figure 5. Administration of Anti-MHC class I Abs leads to development of autoAbs against self antigens K-α1 tubulin and ColV.
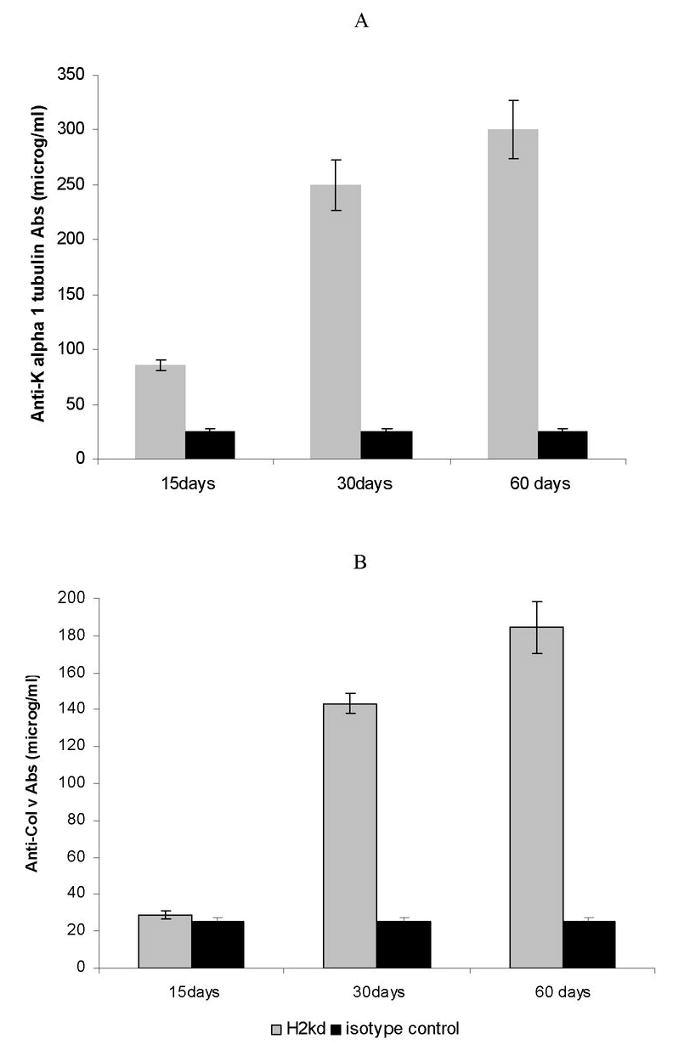
Anti-H2Kd or control Ab was administered endobronchially in BALB/c mice (n=5 each) on days 1, 2, 3, 6 and weekly thereafter. Sera were collected from the mice on day 15, 30 and 60. ELISA was performed for Abs to self antigens in the serum of mice treated with anti-MHC Abs or control Abs using purified K-α1 tubulin or ColV and as antigen. Administration of anti-H2Kd Ab results in the increased concentrations of anti-K-α 1 tubulin (A) and ColV (B) specific Abs on days 15, 30 and 60 compared to control mice treated with C1.18.4 Abs.
Neutralization of IL-17 results in decreased auto-Ab production and fibrosis in the lung
IL-17 is a pleiotropic cytokine implicated in autoimmune disorders and in development of auto-Abs. To determine the role of IL-17 in the mechanism by which anti-MHC class I Abs induce autoimmunity we administered neutralizing anti-IL17 Abs (eBioscience, San Diego, CA) or control Abs (50 μg) intravenously on day 1, 3, 5 and weekly thereafter into BALB/c mice administered anti-H2Kd Abs endobronchially. Analysis of the sera from these mice collected on day 30 showed a significant reduction in the development of auto-Abs against K-α1 tubulin and ColV by ELISA similar to the levels observed in the control Ab treated mice (Figure 6a). More significantly, histopathological analysis of the lung showed a significant reduction in the levels of cellular infiltration, epithelial hyperplasia and fibrosis (Figure 6b) compared to those animals treated with anti-MHC class I Ab alone. These results indicate that IL-17 play a crucial role in the induction of autoimmunity following administration of anti-MHC class I Abs into the lung.
Figure 6. Neutralization of IL-17 blocks the induction of autoimmune responses and development of bronchial occlusion.
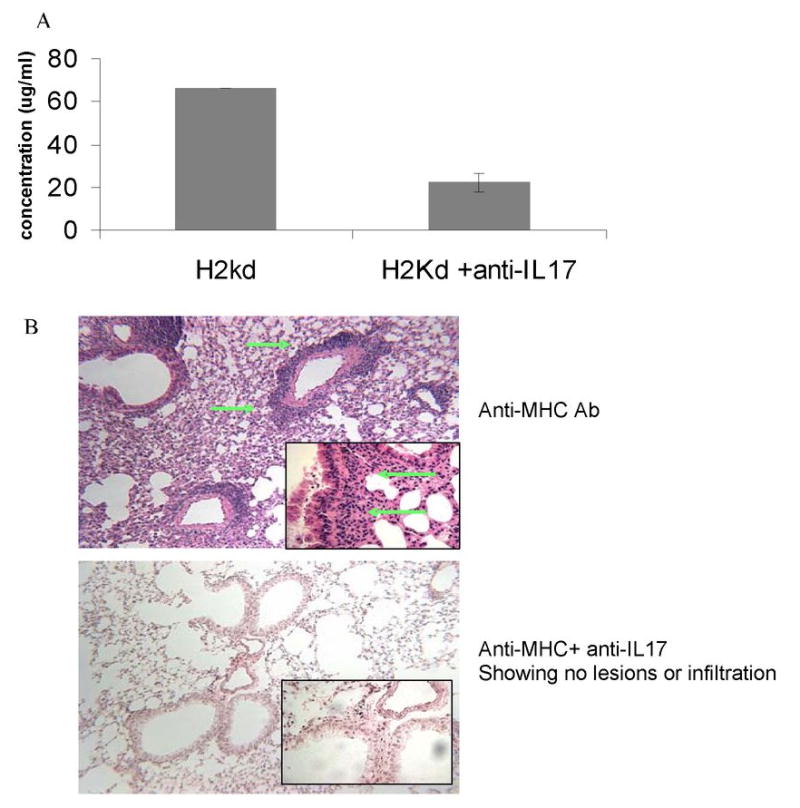
Anti-H2Kd or control Ab was administered endobronchially in BALB/c mice on days 1, 2, 3, 6 and weekly thereafter. 50 μg of neutralizing anti-IL17 or control Abs were administered intravenously on day 1, 3, 5 and weekly thereafter (n=5 each). Serum and lungs of these mice were harvested on day 30 and analyzed for the development of auto-Abs and histopathological analysis. (A) Quantitation of the anti-tubulin Abs by ELISA showed a 3 fold reduction in the concentrations of anti-tubulin Abs in IL-17 neutralized mice compared to controls. (B) H& E staining of the lungs showed a significant reduction in T cell infiltration and bronchial occlusion similar to that observed in control mice treated with isotype control Ab.
Discussion
Understanding the mechanisms for the immunopathogenesis of chronic allograft rejection remains a major goal towards improving the long term success of transplanted organs. There is considerable evidence that alloimmune response against the mismatched MHC antigens plays a pivotal role in the pathogenesis of chronic rejection (26). In the past, it was presumed that mostly T cell immune responses including direct cytotoxicity of the organ (27) or indirect antigen presentation of the mismatched allopeptides (28) resulting in both cytokine mediated (23) and allo-Ab complement mediated damage of the organ (29) played a significant role in the pathogenesis of chronic rejection. There is emerging evidence for an important role for allo-Abs in the pathogenesis of chronic rejection of kidneys (6), heart (7) and lung (8). Previous studies from our laboratory have also shown not only a significant correlation between the development of Abs during the post-transplant period but also the emergence of detectable anti-HLA Abs months prior to the onset of BOS following LTx (30). It has been demonstrated that anti-MHC class I Ab binding to the endothelial lining or to the epithelial surface can activate signaling cascades resulting in growth factor production leading to fibrosis and occlusion, a cardinal feature of all chronic rejection (31, 32). However, persistence of detectable anti-MHC allo-Abs is not a general feature in allograft recipients undergoing chronic rejection (33) and there is controversy that the presence of Abs during the post-transplant period is merely a marker for other immunological events occurring. In this communication we present evidence that allo-Ab binding leads to auto-Ab production which is responsible for the pathogenesis of chronic rejection. We provide evidence for the first time that anti-MHC class I Ab binding to the lung parenchyma results in cellular infiltration of the lung resulting in endotheliitis and epithelial hyperplasia leading to auto-Ab production to two epithelial self antigens, i.e., ColV and K-α1 tubulin.
In an allograft, all of the Abs directed to the mismatched MHC antigens will have the capacity to bind to the allograft. To mimic this, we directly administered anti-MHC class I Ab to the lung via endobronchial route. Therefore, we believe that most, if not all, of the Abs will have a chance to bind and saturate the lung MHC molecules prior to getting into the circulation. This assumption turned to be valid since we found lesions in the lung and not in other organs. Specificity of the MHC Abs to cause the lesion was confirmed since isotype matched C1.18.4 Abs failed to cause the lesions (Figure 1B, 1E, 1H and 1K). In addition, we also did not see any lesions when Abs to keratin which binds to the epithelial cell surface was administered endobronchially (Figure 1C, 1F, 1I and 1L). Therefore, Ab binding alone was not sufficient to induce the lesion and specificity to MHC class I was required. Moreover, similar lesions observed in the C57/BL6 mice and the HLA-A2 transgenic C57/BL6 mice following administration of anti- H2Kb and W6/32 antibody respectively, with a similar kinetics demonstrating that the observation is independent of strain and antibody. Therefore, the data presented here along with the earlier findings (32) indicates that anti-donor MHC Abs developed during the post-transplant period will bind specifically to the allograft resulting in the lesions seen in chronic rejection.
Cellular infiltration into the lung 15 days following treatment with anti-MHC class I Ab was impressive. Initial infiltration was noted around the small vessels leading to endotheliitis (Figure 1A). This is consistent with the reports demonstrating that endotheliitis precedes clinical signs of chronic rejection in human LTx recipients (34). Analysis of the chemokine expression in the lungs by quantitative real time PCR demonstrated a significant increase in the expression of several chemokines (CCL9 9.2 fold, CCR2 10.6 fold, CXCL1 6.3 fold and CXCL12 8.0 fold). CCL9 is a member of the CC motif chemokine ligand that is expressed by the epithelial cells and plays a critical role in mucosal immunity, tissue homeostasis (35) and recruitment of dendritic cells (36). CCL9 through its receptor CCR1 has been shown to play a role in induction of autoimmunity as well as in transplant rejection (37). CCR2, a monocyte chemoattractant, has also been shown to be expressed in the lung epithelial cells and is thought to be involved in IL-13 mediated pathogenesis as well as in lung re-epithelialization following injury (38). Studies in experimental animal model have also shown that CCR2 plays a crucial role in recruitment of the leucocytes in the lung and contribute to rejection and development of BOS (39). CXCL12 has been shown to be critically involved in the germinal center formation especially in response to self antigens (22). Based on our findings of up regulation of several chemokines and their receptors (Figure 4a) following administration of anti-MHC class I, it is reasonable to assume that anti-MHC class I binding induces the signals for infiltration of inflammatory cells into the organ which begins at the endothelial parenchymal junction.
Alloimmune response to mismatched HLA antigens have been considered to be the primary insult leading to chronic rejection following human LTx (40). Frequency of indirect antigen presentation of the allo-MHC peptides have been shown to be increased in patients undergoing chronic rejection (26). In addition, Abs to mismatched HLA antigens have been demonstrated in patients with BOS (5) and more significantly precedes the development of clinical evidence of BOS (8, 30). Surprisingly, many patients with BOS have no detectable Abs to mismatched HLA antigens giving rise to the possibility that immune responses to non-HLA antigens may play a significant role in the pathogenesis of BOS. Our results clearly demonstrate that anti-MHC class I Abs developed during the post-transplant period upon binding to the organ expose otherwise cryptic determinants on self-antigens leading to immune responses against them. Data presented here (Figure 4c) also indicate that these auto-Abs can bind to the epithelial cells and other cells, activate them to produce BMP, FGF family of growth factors which may play an important role in the pathogenesis of BOS. These results are consistent with the notion that auto-Abs play a significant role in the pathogenesis of chronic rejection (10) as reported for a role for cardiac myosin and vimentin in the pathogenesis of coronary artery diseases following heart transplantation (13, 41). There is also strong evidence for autoreactivity to ColV in the pathogenesis of chronic rejection following human LTx (19) as well as animal models of lung allograft rejection (15). Both direct and indirect allorecognition pathways have been shown to contribute to rejection following transplantation (42). Studies analyzing the immune responses to self antigens following transplantation has clearly demonstrated that the indirect allorecognition pathway plays a crucial role (15, 43). Moreover studies in the chronic lung disease models have shown that there is an increase in the frequency of the B cells in the lung following chronic injury and the B cells can provide help to the T cells by acting as antigen presenting cells (44). Our preliminary analysis of the cells infiltrating the lung indicates that they are activated and secrete IFN-γ, IL-4 as well as IL-17 (Figure 3). In vitro studies demonstrate that this population consists of cells responding to K-α1 tubulin as well as ColV. It is of interest that the auto antigen ColV has been shown to participate in human and animal models of lung allograft rejection (16, 43, 45, 46). This was followed by the development of Abs to both of these auto antigens. It is of interest that cells infiltrating the lung produced large amounts of IL-17 following stimulation with both K-α1 tubulin and ColV (Figure 3). IL-17 has been shown to be a potent pro-inflammatory cytokine that acts on epithelial cells, ECs, fibrocytes and stromal cells leading to a pro-inflammatory signal which results in enhanced pro-inflammatory cytokine and chemokine secretion (17). IL-17 has also been shown to play a crucial role in the induction of humoral autoimmune responses (18). Analysis of the T cell responses to ColV in LTx recipients has been shown to be dependent on CD4 T cells and monocytes (19). This response was found to be dependent on both IL-17, TNF-α and IL-1β. The risk of BOS development was observed to correlate with the severity of the ColV specific T cell responses (19). Further, in this study, it was also shown that the interaction between IL-17 and monocytes is crucial in induction of the ColV specific responses. IL-17 has been also shown to play a crucial role in the formation of germinal centers (GCs) and regulate the movement of the B cells in the GCs through chemokine CXCL12 and CXCL13 (22). Studies have also demonstrated that a deficiency in the IL-17 or blocking of IL-17 results in a decrease in the production of auto-Abs (20, 21). Therefore, our finding of IL-17 producing T cells following stimulation with self antigens indicate that these T cells provide signals towards activation of B cells resulting in Abs to self antigens such as K- α 1 tubulin and ColV. Moreover, as shown in Figure 6a, neutralization of IL-17 resulted in a significant reduction in the levels of Abs to the self antigen K-α1 tubulin. More significantly, neutralization of IL-17 also resulted in abrogation of all of the lesions induced by anti-MHC class I Abs, i.e., a decreased cellular infiltration, epithelial hyperplasia and fibrosis in the lungs following administration of anti-MHC class I Abs. These observations are in agreement with the report that in IL-17 deficient mice that lack IL-17 signaling significantly reduce the induction of autoimmunity and down modulate the formation of germinal centers leading to a reduction in the titer of auto-Abs (22). Hence we propose that IL-17 is a critical mediator in the induction of autoimmunity following ligation of the anti-MHC class I Abs resulting in the development of chronic rejection following LTx.
By day 30 post Ab administration, we also noted marked epithelial hyperplasia (Figure 1) which is similar to that seen in BOS. Increased growth factor production in the lung milieu was also noted at this period. These changes eventually lead to increased fibrosis and luminal occlusion of the small airway (Figure 1J). Analysis of the profile of the growth factor produced in the lungs showed a significant increase in the expression of FGF and BMP family of proteins (Figure 4c). Earlier in vitro studies from our lab as well as others have demonstrated that binding of the anti-MHC class I Abs to the epithelial cells as well as the ECs produce significantly high quantities of several growth factors including epithelial growth factor (EGF), bFGF, PDGF, TGF-β (32,47-50), and granulocyte-monocyte colony-stimulating factor (GM-CSF) (51). Studies in the literature have also shown that the BMP family of proteins act upstream of FGF to regulate T cell development and epithelial remodeling in the lungs. BMP has been shown to up regulate adhesion molecule expression in the ECs and a critical inflammatory signal in atherogenic lesions. These changes eventually lead to increased fibrosis and luminal occlusion (Figure 1J). All of these changes are consistent with the hypothesis that anti-MHC class I can induce autoimmune response which lead to auto-Ab production to self antigens. We postulate that not only Abs to donor MHC but also cellular immune responses to mismatched donor MHC as well as viral infections of the respiratory tract can result in damage to the airway epithelial cells leading to exposure of otherwise cryptic determinants of self antigens including K-α1 tubulin and ColV. This can result in Ab production to these auto-antigens which then can induce chronic stimuli of the endothelium and epithelium of the lung parenchyma resulting in various growth factor production which are necessary for inducing epithelial hyperplasia, fibrosis of the lung parenchyma leading to luminal occlusion. This fits well with our current dogma that the number of acute cellular rejection episodes (52, 53) and number of upper respiratory viral infections (54) correlates with incidence of chronic rejection following human LTx.
Based on our observations we propose that ligation of the anti-MHC class I Ab to MHC molecules on epithelial and ECs results in the increased expression of CXCL12, CCL9, that signal through CCR2 and CXCR4 resulting in increased leucocyte infiltration in the lungs and exposure of cryptic determinants of self antigens leading to an increased frequency of autoreactive Th17 T cells. The TH17 T cells in combination with CXCL12 lead to increase in response to self antigens leading to auto-Ab production. Therefore, neutralization of IL-17 can be a viable therapeutic option that could prevent the induction of autoimmunity and development of chronic rejection. Direct activation of the epithelial and ECs by both Abs to MHC antigens as well as self antigens can also result in the increased expression of fibrogenic growth factors and proliferation, the hallmarks of chronic allograft rejection. Therefore, we provide evidence for the first time that autoimmunity which is induced by Ab ligation of MHC class I molecule plays an important role in the pathogenesis of chronic rejection following LTx. We are currently testing the hypothesis that prevention of development of autoimmunity by early recognition and control of alloimmune responses or viral infections have the potential to prevent development of chronic rejection following human lung transplantation.
Acknowledgments
We thank Billie Glasscock for her assistance in preparing this manuscript.
“The project described was supported by Grant Number NIH HL66452 (TM) from NHLBI” and “the contents are solely the responsibility of the authors and do not necessarily represent the official views of the NHLBI.”
References
- 1.Trulock EP, Edwards LB, Taylor DO, Boucek MM, Mohacsi PJ, Keck BM, Hertz MI. The Registry of the International Society for Heart and Lung Transplantation: Twentieth Official adult lung and heart-lung transplant report--2003. J Heart Lung Transplant. 2003;22:625–635. doi: 10.1016/s1053-2498(03)00182-7. [DOI] [PubMed] [Google Scholar]
- 2.Griffith BP, Paradis IL, Zeevi A, Rabinowich H, Yousem SA, Duquesnoy RJ, Dauber JH, Hardesty RL. Immunologically mediated disease of the airways after pulmonary transplantation. Ann Surg. 1988;208:371–378. doi: 10.1097/00000658-198809000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boehler A, Estenne M. Obliterative bronchiolitis after lung transplantation. Curr Opin Pulm Med. 2000;6:133–139. doi: 10.1097/00063198-200003000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Suciu-Foca N, Reed E, D'Agati VD, Ho E, Cohen DJ, Benvenisty AI, McCabe R, Brensilver JM, King DW, Hardy MA. Soluble HLA antigens, anti-HLA antibodies, and antiidiotypic antibodies in the circulation of renal transplant recipients. Transplantation. 1991;51:593–601. doi: 10.1097/00007890-199103000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Smith MA, Sundaresan S, Mohanakumar T, Trulock EP, Lynch JP, Phelan DL, Cooper JD, Patterson GA. Effect of development of antibodies to HLA and cytomegalovirus mismatch on lung transplantation survival and development of bronchiolitis obliterans syndrome. J Thorac Cardiovasc Surg. 1998;116:812–820. doi: 10.1016/S0022-5223(98)00444-9. [DOI] [PubMed] [Google Scholar]
- 6.Mizutani K. Serial ten-year follow-up of HLA and MICA antibody production prior to kidney graft failure. Am J Transplant. 2005;5:1–8. doi: 10.1111/j.1600-6143.2005.01016.x. [DOI] [PubMed] [Google Scholar]
- 7.Reed EF, Hong B, Ho E, Harris PE, Weinberger J, Suciu-Foca N. Monitoring of soluble HLA alloantigens and anti-HLA antibodies identifies heart allograft recipients at risk of transplant-associated coronary artery disease. Transplantation. 1996;61:566–572. doi: 10.1097/00007890-199602270-00009. [DOI] [PubMed] [Google Scholar]
- 8.Sundaresan S, Mohanakumar T, Smith MA, Trulock EP, Lynch J, Phelan D, Cooper JD, Patterson GA. HLA-A locus mismatches and development of antibodies to HLA after lung transplantation correlate with the development of bronchiolitis obliterans syndrome. Transplantation. 1998;65:648–653. doi: 10.1097/00007890-199803150-00008. [DOI] [PubMed] [Google Scholar]
- 9.Fedoseyeva EV, Zhang F, Orr PL, Levin D, Buncke HJ, Benichou G. De novo autoimmunity to cardiac myosin after heart transplantation and its contribution to the rejection process. J Immunol. 1999;162:6836–6842. [PubMed] [Google Scholar]
- 10.Benichou G, Alessandrini A, Charrad RS, Wilkes DS. Induction of autoimmunity after allotransplantation. Front Biosci. 2007;12:4362–4369. doi: 10.2741/2393. [DOI] [PubMed] [Google Scholar]
- 11.Barber LD, Whitelegg A, Madrigal JA, Banner NR, Rose ML. Detection of vimentin-specific autoreactive CD8+ T cells in cardiac transplant patients. Transplantation. 2004;77:1604–1609. doi: 10.1097/01.tp.0000129068.03900.25. [DOI] [PubMed] [Google Scholar]
- 12.Caldas C, Luna E, Spadafora-Ferreira M, Porto G, Iwai LK, Oshiro SE, Monteiro SM, Fonseca JA, Lemos F, Hammer J, Ho PL, Kalil J, Coelho V. Cellular autoreactivity against heat shock protein 60 in renal transplant patients: peripheral and graft-infiltrating responses. Clin Exp Immunol. 2006;146:66–75. doi: 10.1111/j.1365-2249.2006.03195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka M, Zwierzchoniewska M, Mokhtari GK, Terry RD, Balsam LB, Robbins RC, Fedoseyeva EV. Progression of alloresponse and tissue-specific immunity during graft coronary artery disease. Am J Transplant. 2005;5:1286–1296. doi: 10.1111/j.1600-6143.2005.00880.x. [DOI] [PubMed] [Google Scholar]
- 14.Rolls HK, Kishimoto K, Dong VM, Illigens BM, Sho M, Sayegh MH, Benichou G, Fedoseyeva EV. T-cell response to cardiac myosin persists in the absence of an alloimmune response in recipients with chronic cardiac allograft rejection. Transplantation. 2002;74:1053–1057. doi: 10.1097/00007890-200210150-00028. [DOI] [PubMed] [Google Scholar]
- 15.Haque MA, Mizobuchi T, Yasufuku K, Fujisawa T, Brutkiewicz RR, Zheng Y, Woods K, Smith GN, Cummings OW, Heidler KM, Blum JS, Wilkes DS. Evidence for immune responses to a self-antigen in lung transplantation: role of type V collagen-specific T cells in the pathogenesis of lung allograft rejection. J Immunol. 2002;169:1542–1549. doi: 10.4049/jimmunol.169.3.1542. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida S, Haque A, Mizobuchi T, Iwata T, Chiyo M, Webb TJ, Baldridge LA, Heidler KM, Cummings OW, Fujisawa T, Blum JS, Brand DD, Wilkes DS. Anti-type V collagen lymphocytes that express IL-17 and IL-23 induce rejection pathology in fresh and well-healed lung transplants. Am J Transplant. 2006;6:724–735. doi: 10.1111/j.1600-6143.2006.01236.x. [DOI] [PubMed] [Google Scholar]
- 17.Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998;160:3513–3521. [PubMed] [Google Scholar]
- 18.Irmler IM, Gajda M, Brauer R. Exacerbation of antigen-induced arthritis in IFN-gamma-deficient mice as a result of unrestricted IL-17 response. J Immunol. 2007;179:6228–6236. doi: 10.4049/jimmunol.179.9.6228. [DOI] [PubMed] [Google Scholar]
- 19.Burlingham WJ, Love RB, Jankowska-Gan E, Haynes LD, Xu Q, Bobadilla JL, Meyer KC, Hayney MS, Braun RK, Greenspan DS, Gopalakrishnan B, Cai J, Brand DD, Yoshida S, Cummings OW, Wilkes DS. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest. 2007;117:3498–3506. doi: 10.1172/JCI28031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang JL, Subbotin VM, Antonysamy MA, Troutt AB, Rao AS, Thomson AW. Interleukin-17 antagonism inhibits acute but not chronic vascular rejection. Transplantation. 2001;72:348–350. doi: 10.1097/00007890-200107270-00035. [DOI] [PubMed] [Google Scholar]
- 21.Sonderegger I, Rohn TA, Kurrer MO, Iezzi G, Zou Y, Kastelein RA, Bachmann MF, Kopf M. Neutralization of IL-17 by active vaccination inhibits IL-23-dependent autoimmune myocarditis. Eur J Immunol. 2006;36:2849–2856. doi: 10.1002/eji.200636484. [DOI] [PubMed] [Google Scholar]
- 22.Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, Le TV, Lorenz RG, Xu H, Kolls JK, Carter RH, Chaplin DD, Williams RW, Mountz JD. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 23.Bharat A, Narayanan K, Street T, Fields RC, Steward N, Aloush A, Meyers B, Schuessler R, Trulock EP, Patterson GA, Mohanakumar T. Early posttransplant inflammation promotes the development of alloimmunity and chronic human lung allograft rejection. Transplantation. 2007;83:150–158. doi: 10.1097/01.tp.0000250579.08042.b6. [DOI] [PubMed] [Google Scholar]
- 24.Yamaguchi Y, Fujio K, Shoda H, Okamoto A, Tsuno NH, Takahashi K, Yamamoto K. IL-17B and IL-17C are associated with TNF-alpha production and contribute to the exacerbation of inflammatory arthritis. J Immunol. 2007;179:7128–7136. doi: 10.4049/jimmunol.179.10.7128. [DOI] [PubMed] [Google Scholar]
- 25.Goers TA, Ramachandran S, Aloush A, Trulock E, Patterson GA, Mohanakumar T. De novo production of K-alpha1 tubulin-specific antibodies: role in chronic lung allograft rejection. J Immunol. 2008;180:4487–4494. doi: 10.4049/jimmunol.180.7.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuo E, Maruyama T, Fernandez F, Mohanakumar T. Molecular mechanisms of chronic rejection following transplantation. Immunol Res. 2005;32:179–185. doi: 10.1385/IR:32:1-3:179. [DOI] [PubMed] [Google Scholar]
- 27.Hornick PI, Mason PD, Yacoub MH, Rose ML, Batchelor R, Lechler RI. Assessment of the contribution that direct allorecognition makes to the progression of chronic cardiac transplant rejection in humans. Circulation. 1998;97:1257–1263. doi: 10.1161/01.cir.97.13.1257. [DOI] [PubMed] [Google Scholar]
- 28.Chen W, Murphy B, Waaga AM, Willett TA, Russell ME, Khoury SJ, Sayegh MH. Mechanisms of indirect allorecognition in graft rejection: class II MHC allopeptide-specific T cell clones transfer delayed-type hypersensitivity responses in vivo. Transplantation. 1996;62:705–710. doi: 10.1097/00007890-199609270-00001. [DOI] [PubMed] [Google Scholar]
- 29.Smith JD, Hamour IM, Banner NR, Rose ML. C4d fixing, luminex binding antibodies - a new tool for prediction of graft failure after heart transplantation. Am J Transplant. 2007;7:2809–2815. doi: 10.1111/j.1600-6143.2007.01991.x. [DOI] [PubMed] [Google Scholar]
- 30.Jaramillo A, Smith MA, Phelan D, Sundaresan S, Trulock EP, Lynch JP, Cooper JD, Patterson GA, Mohanakumar T. Development of ELISA-detected anti-HLA antibodies precedes the development of bronchiolitis obliterans syndrome and correlates with progressive decline in pulmonary function after lung transplantation. Transplantation. 1999;67:1155–1161. doi: 10.1097/00007890-199904270-00012. [DOI] [PubMed] [Google Scholar]
- 31.Harris PE, Bian H, Reed EF. Induction of high affinity fibroblast growth factor receptor expression and proliferation in human endothelial cells by anti-HLA antibodies: A possible mechanism for transplant atherosclerosis. J Immunol. 1997;159:5697–5704. [PubMed] [Google Scholar]
- 32.Jaramillo A, Smith CR, Maruyama T, Zhang L, Patterson GA, Mohanakumar T. Anti-HLA class I antibody binding to airway epithelial cells induces production of fibrogenic growth factors and apoptotic cell death: a possible mechanism for bronchiolitis obliterans syndrome. Hum Immunol. 2003;64:521–529. doi: 10.1016/s0198-8859(03)00038-7. [DOI] [PubMed] [Google Scholar]
- 33.McKenna RM, Takemoto SK, Terasaki PI. Anti-HLA antibodies after solid organ transplantation. Transplantation. 2000;69:319–326. doi: 10.1097/00007890-200002150-00001. [DOI] [PubMed] [Google Scholar]
- 34.Luckraz H, Goddard M, McNeil K, Atkinson C, Charman SC, Stewart S, Wallwork J. Microvascular changes in small airways predispose to obliterative bronchiolitis after lung transplantation. J Heart Lung Transplant. 2004;23:527–531. doi: 10.1016/j.healun.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 35.Shaykhiev R, Bals R. Interactions between epithelial cells and leukocytes in immunity and tissue homeostasis. J Leukoc Biol. 2007;82:1–15. doi: 10.1189/jlb.0207096. [DOI] [PubMed] [Google Scholar]
- 36.Zhao X, Sato A, Dela Cruz CS, Linehan M, Luegering A, Kucharzik T, Shirakawa AK, Marquez G, Farber JM, Williams I, Iwasaki A. CCL9 is secreted by the follicle-associated epithelium and recruits dome region Peyer's patch CD11b+ dendritic cells. J Immunol. 2003;171:2797–2803. doi: 10.4049/jimmunol.171.6.2797. [DOI] [PubMed] [Google Scholar]
- 37.Horuk R. BX471: a CCR1 antagonist with anti-inflammatory activity in man. Mini Rev Med Chem. 2005;5:791–804. doi: 10.2174/1389557054867057. [DOI] [PubMed] [Google Scholar]
- 38.Zhu Z, Ma B, Zheng T, Homer RJ, Lee CG, Charo IF, Noble P, Elias JA. IL-13-induced chemokine responses in the lung: role of CCR2 in the pathogenesis of IL-13-induced inflammation and remodeling. J Immunol. 2002;168:2953–2962. doi: 10.4049/jimmunol.168.6.2953. [DOI] [PubMed] [Google Scholar]
- 39.Belperio JA, Keane MP, Burdick MD, Lynch JP, 3rd, Xue YY, Berlin A, Ross DJ, Kunkel SL, Charo IF, Strieter RM. Critical role for the chemokine MCP-1/CCR2 in the pathogenesis of bronchiolitis obliterans syndrome. J Clin Invest. 2001;108:547–556. doi: 10.1172/JCI12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finn OJ, Debruyne LA, Bishop DK. T cell receptor (TCR) repertoire in alloimmune responses. Int Rev Immunol. 1996;13:187–207. doi: 10.3109/08830189609061747. [DOI] [PubMed] [Google Scholar]
- 41.Azimzadeh AM, Pfeiffer S, Wu GS, Schroder C, Zhou H, Zorn GL, 3rd, Kehry M, Miller GG, Rose ML, Pierson RN., 3rd Humoral immunity to vimentin is associated with cardiac allograft injury in nonhuman primates. Am J Transplant. 2005;5:2349–2359. doi: 10.1111/j.1600-6143.2005.01022.x. [DOI] [PubMed] [Google Scholar]
- 42.Waanders MM, Heidt S, Koekkoek KM, Zoet YM, Doxiadis II, Amir A, Heemskerk MH, Mulder A, Brand A, Roelen DL, Claas FH. Monitoring of indirect allorecognition: wishful thinking or solid data? Tissue Antigens. 2008;71:1–15. doi: 10.1111/j.1399-0039.2007.00979.x. [DOI] [PubMed] [Google Scholar]
- 43.Sumpter TL, Wilkes DS. Role of autoimmunity in organ allograft rejection: a focus on immunity to type V collagen in the pathogenesis of lung transplant rejection. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1129–1139. doi: 10.1152/ajplung.00330.2003. [DOI] [PubMed] [Google Scholar]
- 44.Lindell DM, Berlin AA, Schaller MA, Lukacs NW. B cell antigen presentation promotes Th2 responses and immunopathology during chronic allergic lung disease. PLoS ONE. 2008;3:e3129. doi: 10.1371/journal.pone.0003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bharat A, Fields RC, Steward N, Trulock EP, Patterson GA, Mohanakumar T. CD4+25+ regulatory T cells limit Th1-autoimmunity by inducing IL-10 producing T cells following human lung transplantation. Am J Transplant. 2006;6:1799–1808. doi: 10.1111/j.1600-6143.2006.01383.x. [DOI] [PubMed] [Google Scholar]
- 46.Wilkes DS. The role of autoimmunity in the pathogenesis of lung allograft rejection. Arch Immunol Ther Exp (Warsz) 2003;51:227–230. [PubMed] [Google Scholar]
- 47.Mauck KA, Hosenpud JD. The bronchial epithelium: a potential allogeneic target for chronic rejection after lung transplantation. J Heart Lung Transplant. 1996;15:709–714. [PubMed] [Google Scholar]
- 48.Zhao J, Sime PJ, Bringas P, Gauldie J, Warburton D. Epithelium-specific adenoviral transfer of a dominant-negative mutant TGF-beta type II receptor stimulates embryonic lung branching morphogenesis in culture and potentiates EGF and PDGF-AA. Mech Dev. 1998;72:89–100. doi: 10.1016/s0925-4773(98)00019-7. [DOI] [PubMed] [Google Scholar]
- 49.Wagner CR, Morris TE, Shipley GD, Hosenpud JD. Regulation of human aortic endothelial cell-derived mesenchymal growth factors by allogeneic lymphocytes in vitro. A potential mechanism for cardiac allograft vasculopathy. Journal of Clinical Investigation. 1993;92:1269–1277. doi: 10.1172/JCI116699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Brien AD, Standiford TJ, Christensen PJ, Wilcoxen SE, Paine R. Chemotaxis of alveolar macrophages in response to signals derived from alveolar epithelial cells. J Lab Clin Med. 1998;131:417–424. doi: 10.1016/s0022-2143(98)90142-1. [DOI] [PubMed] [Google Scholar]
- 51.Marini M, Soloperto M, Mezzetti M, Fasoli A, Mattoli S. Interleukin-1 binds to specific receptors on human bronchial epithelial cells and upregulates granulocyte/macrophage colony-stimulating factor synthesis and release. Am J Respir Cell Mol Biol. 1991;4:519–524. doi: 10.1165/ajrcmb/4.6.519. [DOI] [PubMed] [Google Scholar]
- 52.Hachem RR, Khalifah AP, Chakinala MM, Yusen RD, Aloush AA, Mohanakumar T, Patterson GA, Trulock EP, Walter MJ. The significance of a single episode of minimal acute rejection after lung transplantation. Transplantation. 2005;80:1406–1413. doi: 10.1097/01.tp.0000181161.60638.fa. [DOI] [PubMed] [Google Scholar]
- 53.Khalifah AP, Hachem RR, Chakinala MM, Yusen RD, Aloush A, Patterson GA, Mohanakumar T, Trulock EP, Walter MJ. Minimal acute rejection after lung transplantation: a risk for bronchiolitis obliterans syndrome. Am J Transplant. 2005;5:2022–2030. doi: 10.1111/j.1600-6143.2005.00953.x. [DOI] [PubMed] [Google Scholar]
- 54.Khalifah AP, Hachem RR, Chakinala MM, Schechtman KB, Patterson GA, Schuster DP, Mohanakumar T, Trulock EP, Walter MJ. Respiratory viral infections are a distinct risk for bronchiolitis obliterans syndrome and death. Am J Resp Crit Care Med. 2004;170:181–187. doi: 10.1164/rccm.200310-1359OC. [DOI] [PubMed] [Google Scholar]