

The structure of d-lactate dehydrogenase from Aquifex aeolicus has been determined with each subunit of the homodimer in a ‘closed’ conformation and with the NAD+ cofactor and lactate (or pyruvate) bound at the inter-domain active-site cleft.
Keywords: d-lactate dehydrogenase, Aquifex aeolicus
Abstract
The crystal structure of d-lactate dehydrogenase from Aquifex aeolicus (aq_727) was determined to 2.12 Å resolution in space group P212121, with unit-cell parameters a = 90.94, b = 94.43, c = 188.85 Å. The structure was solved by molecular replacement using the coenzyme-binding domain of Lactobacillus helveticus d-lactate dehydrogenase and contained two homodimers in the asymmetric unit. Each subunit of the homodimer was found to be in a ‘closed’ conformation with the NADH cofactor bound to the coenzyme-binding domain and with a lactate (or pyruvate) molecule bound at the interdomain active-site cleft.
1. Introduction
Lactate dehydrogenase (LDH) catalyses the (reversible) reduction of pyruvate to lactic acid, concomitant with the oxidation of NADH to NAD+. During this process NADH transfers a hydride ion to pyruvate; in the reverse reaction NAD+ receives a hydride ion from lactate. Two evolutionarily distinct families of LDH enzymes (Kochhar et al., 1992 ▶) perform this step to yield a product that differs only in its chirality: l-lactate or d-lactate (Lamzin et al., 1995 ▶). The crystal structures of d-LDH from Lactobacillus bulgaricus (Razeto et al., 2002 ▶), L. helveticus (PDB code 2dld; C. R. Dunn & J. J. Holbrook, unpublished work) and L. pentosus (Shinoda et al., 2005 ▶) show that the enzyme consists of two βαβ domains, one of which binds NADH (the coenzyme domain) and the other of which binds the substrate (the catalytic domain), with the active site located at the interdomain cleft. The biologically significant unit in the d-LDH enzymes is probably a homodimer (Razeto et al., 2002 ▶).
As part of the RIKEN Structural Genomics Initiative, proteins from Aquifex aeolicus VF5 were targeted for high-throughput structure determination (Sugahara et al., 2008 ▶). Here, we report the crystal structure of a d-LDH in the fully closed conformation with lactate (or pyruvate) bound to the active site of each subunit of the functional dimer.
2. Materials and methods
2.1. Cloning, expression and purification
The gene encoding aq_727 protein (gi:15606122) was amplified via PCR using A. aeolicus VF5 genomic DNA and was cloned into the pET-21a expression vector (Merck Novagen, Darmstadt, Germany). The expression vector was introduced by heat shock into Escherichia coli Rosetta (DE3) strain (Merck Novagen, Darmstadt, Germany) and the recombinant strain was cultured in 3 l minimal medium containing 25 µg ml−1 selenomethionine, 30 µg ml−1 chloramphenicol and 50 µg ml−1 ampicillin. The cells were cultured for 3 h at 313 K after induction with 0.5 mg ml−1 EDTA when the OD at 600 nm reached 0.7. The harvested cells (6.8 g) were lysed by sonication in 20 mM Tris–HCl buffer pH 8.0 containing 50 mM NaCl on ice. The cell lysate was heat-treated at 363 K for 10 min and centrifuged at 200 000g for 60 min. The supernatant was applied onto a Resource Q column (GE Healthcare Biosciences) equilibrated with 50 mM Tris–HCl buffer pH 9.0 and eluted with a linear gradient of NaCl. The fractions that eluted in 0.06 M NaCl were further purified using a hydroxyapatite CHT10-I column (Bio-Rad Laboratories) equilibrated with 10 mM sodium phosphate buffer pH 7.0 and eluted with a linear gradient of sodium phosphate. The target sample, which eluted in the 130 mM sodium phosphate fraction, was collected and applied onto a HiLoad 16/60 Superdex 200 pg column (GE Healthcare Biosciences) equilibrated with 20 mM Tris–HCl buffer pH 8.0 containing 150 mM NaCl. The protein sample was analyzed by SDS–PAGE (Fig. 1 ▶ a) and the protein was confirmed by N-terminal amino-acid sequencing. After concentration to 20.1 mg ml−1 by ultrafiltration, the protein yield was 20.3 mg from 6.8 g of cells. Protein concentrations were measured by the Bradford method (Bradford, 1976 ▶).
Figure 1.

(a) Acrylamide SDS–PAGE analysis of d-lactate dehydrogenase from A. aeolicus overproduced in E. coli. (b) Crystals of A. aeolicus aq_727 prior to flash-cooling in glycerol cryoprotectant.
2.2. Crystallization
Crystallization was performed by the sitting-drop vapour-diffusion method at 293 K. Each drop consisted of 1.0 µl 20.3 mg ml−1 protein solution and 1.0 µl reservoir solution. In a preliminary screening, small crystals appeared using a crystallization reagent consisting of 0.1 M MES buffer pH 6.0 containing 30%(w/v) PEG 200 and 5%(w/v) PEG 3000 (Cryo I condition No. 13, Emerald BioSystems). After optimization, large crystals were obtained using a crystallization reagent consisting of 0.1 M MES buffer pH 6.0 containing 25%(w/v) PEG 200. Crystals suitable for X-ray data collection appeared within 10 d and reached final dimensions of 0.2 × 0.05 × 0.01 mm. The crystals (Fig. 1 ▶ b) were flash-cooled in a nitrogen-gas stream at 100 K using 20%(v/v) glycerol as a cryoprotectant.
2.3. Data collection and processing
Experiments were performed at the Daresbury Synchrotron Radiation Source (SRS) using the combined crystallography/X-ray absorption beamline 10.1, employing a Si(111) sagittally focused monochromator tuned to a wavelength of 0.98 Å. Diffraction data were recorded at 100 K. Diffraction resolution was improved by annealing the crystal (Ellis et al., 2002 ▶), a procedure that also reduced the mosaic spread to 0.38°. Images were recorded using a MAR Mosaic 225 CCD detector and were processed (indexed, integrated and scaled) using HKL-2000 (Otwinowski & Minor, 1997 ▶). The crystal was found to belong to space group P212121, with unit-cell parameters a = 90.94, b = 94.43, c = 188.85 Å. The estimated solvent content was 54% for four molecules in the asymmetric unit.
2.4. Structure solution and refinement
Diffraction data were collected to 2.12 Å resolution from a single SeMet-derivative crystal. However, the anomalous signal was found to be poor for this crystal and structure solution was instead accomplished by molecular replacement with MOLREP (Vagin & Teplyakov, 1997 ▶), using as a search model the structure of d-lactate dehydrogenase from L. helveticus (PDB code 2dld; C. R. Dunn & J. J. Holbrook, unpublished work) obtained from the Protein Data Bank (Abola et al., 1987 ▶), which has 36% identity and 59% similarity to the target sequence; the model consisted of one subunit of 2dld. The whole 2dld subunit was unsuccessful as a search model; it was therefore modified by removing the catalytic domain and using only the coenzyme-binding domain as the search model. This resulted in a score of 0.24 and an R factor of 57.7% for four copies in the asymmetric unit. This model fragment was first improved by applying rigid-body and restrained refinement using REFMAC (Murshudov et al., 1997 ▶) and was then used in a second run of MOLREP, this time with the catalytic domain intact. This resulted in a score of 0.29 and an R factor of 55.8%. Several cycles of refinement using REFMAC and rebuilding with Coot (Emsley & Cowtan, 2004 ▶) were then carried out. During the structure-solution and refinement procedures and as the electron-density maps improved, the sequence for A. aeolicus was gradually introduced into the model to replace the sequence of L. helveticus. Water molecules were added to the structure using Coot with a 1σ 2F c − F o cutoff. The stereochemistry was checked using PROCHECK (Laskowski et al., 1993 ▶) and MolProbity (Davis et al., 2007 ▶). The Ramachandran plot reported 90.3% of residues to be in the core region and 9.7% to be in additionally allowed regions. The R factor and R free of the final model were 16.8% and 21.6%, respectively. Table 1 ▶ summarizes the data-collection and refinement parameters. The structure was deposited in the PDB under accession code 3kb6.
Table 1. Data-collection and refinement parameters.
Values in parentheses are for the highest resolution shell (2.20–2.12 Å).
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 90.94, b = 94.43, c = 188.85 |
| Resolution (Å) | 38.0–2.12 |
| Unique reflections | 84352 |
| Completeness (%) | 93.3 (70.0) |
| Redundancy | 8.8 (3.4) |
| Rmerge† (%) | 8.3 (28.0) |
| I/σ(I) | 24.3 (5.1) |
| R factor‡ (%) | 16.8 |
| Rfree‡ (%) | 21.6 |
| B factors (Å2) | |
| Wilson plot | 24.4 |
| Protein | 27.6 |
| Water | 38.6 |
| NAD+ cofactor | 21.5 |
| Lactate/pyruvate | 21.5 |
| Propanoic acid | 45.8 |
| PEG | 56.6 |
| Glycerol | 64.2 |
| R.m.s. deviations | |
| Bond distances (Å) | 0.014 |
| Bond angles (°) | 1.41 |
| ESU§ (Å) | 0.12 |
R
merge = 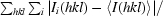
 , where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple symmetry-related observations.
, where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple symmetry-related observations.
R = 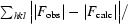
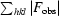 . R
free is the same but calculated for a test set not used in structural refinement.
. R
free is the same but calculated for a test set not used in structural refinement.
Estimated standard uncertainty based on maximum likelihood as implemented in REFMAC.
3. Results and discussion
The asymmetric unit comprises two homodimers containing four NAD+ molecules, with lactate (or pyruvate) molecules bound at each of the four active sites, and five propanoic acid molecules. Neither the cofactor nor ligands were added and they presumably come from the expression and protein-purification stages. In addition, 18 PEG molecules from the crystallization medium and 11 glycerol molecules from the cryoprotectant solution were found bound to the protein surfaces. A total of 858 water molecules were present in the final model. Each subunit comprises 334 amino acids, with the catalytic domain comprising residues 1–102 and 300–334 and the coenzyme-binding domain comprising residues 103–299. The average B factor of the catalytic domain is larger than that of the coenzyme-binding domain, signifying greater flexibility of this segment, which is consistent with its role in domain closure during catalysis (Lamzin et al., 1994 ▶). In both molecules of the dimer the catalytic domain has adopted a fully closed conformation (Fig. 2 ▶). Movement of the catalytic domain from the open to the closed conformation arises from a rotation about the ‘hinge’ region around residues Ser97 and Thr299 and results in the creation of hydrogen bonds between residues Lys34, Tyr51 and Asp76 of the catalytic domain and residues Glu270, Glu260 and Arg231, respectively, of the coenzyme-binding domain (Fig. 2 ▶ d). The equivalent residues in the open conformation (Fig. 2 ▶ c) are separated by more than 15 Å. The aromatic ring of Tyr297 is oriented perpendicular to the substrate and may play a role in ligand discrimination in concert with several hydrophobic residues that are packed around the active site (Razeto et al., 2002 ▶).
Figure 2.

Functional homodimers of three members of the d-lactate dehydrogenase family. (a) A. aeolicus d-LDH is in the closed conformation, with a pyruvate/lactate ligand (black) bound at the NAD+-binding site (magenta) in both subunits of the dimer. (b) The asymmetric structure from L. bulgaricus (PDB code 1j49; Razeto et al., 2002 ▶) is shown, with a sulfate ion (black) in place of the substrate in one partially closed subunit and with the second (apo) subunit in the open conformation. (c) The fully open apo structure from L. helveticus (PDB code 2dld). The coenzyme-binding domain of 2dld was used as the molecular-replacement search model. (d) Superposition of the coenzyme-binding domains of A. aeolicus (green) and L. helveticus (brown) monomers, shown as ribbons in two perpendicular orientations with the NAD+ cofactor omitted for clarity. The figure illustrates the following: (i) the rotation of the catalytic domain about the ‘hinge’ axis, (ii) the locations of the residues involved in hydrogen bonding in the closed conformation, with residues from the catalytic and coenzyme-binding domains shown as yellow and black sticks, respectively, and (iii) the B-factor distribution in the monomer, with larger thermal disorder of the catalytic domain indicated by increased thickness of the ribbons. The r.m.s. deviations of the superposition of the coenzyme-binding domains of L. bulgaricus and L. helveticus with A. aeolicus are 1.0 and 0.93 Å, respectively.
The enzyme was expressed and crystallized with a substrate molecule bound to the active site (Fig. 3 ▶) with water molecules excluded. The electron density for this molecule is consistent with it being either pyruvate or lactate. Thus, either pyruvate is present at the active site but turnover has not occurred, leaving the enzyme in its closed conformation, or lactate has been formed but is trapped at the active site because domain opening has not been triggered. The substrate is oriented so that the carboxylate groups are in a position to interact with the backbone N atoms of Val73 and Gly74. One carboxylate group is also able to interact with the Tyr96 side chain. These three residues are all supplied by the catalytic domain. From the coenzyme domain, substrate binding is mediated by the Arg231 and His294 side chains, which are both available to make contact with the carbonyl group of the substrate, while Arg231 is oriented so that it also interacts with the second carboxylate group. The nicotinamide ring of the coenzyme (shown in the background in Fig. 3 ▶) is oriented face-on and directly adjacent to the substrate molecule, where it is ideally positioned for transfer of a hydride ion during catalysis. The relative orientations of the substrate and cofactor moieties and the dual interaction of the Arg231 with carboxylate and carbonyl groups of the substrate confirms the ligand-binding model originally proposed by Stoll et al. (1996 ▶).
Figure 3.

(a) The active site of subunit A of A. aeolicus d-LDH (subunits B, C and D in the asymmetric unit were identical). (a) The orientation of the substrate bound in the cleft between the two domains, with the intermolecular contacts to the protein residues (length 2.7–2.9 Å) indicated by dashed lines. The catalytic domain is distinguished from the coenzyme-binding domain by having its C atoms coloured yellow. (b) The 2F o − F c electron-density map at the active site, contoured at the 1.2σ level. The electron density on the coenzyme has been omitted for clarity. (c) The F o − F c OMIT map at the active site showing electron density at the 3σ level with the substrate omitted from the model. (d) The quality of the 2F o − F c electron-density map for the NAD+ cofactor contoured at 1.2σ. The molecular-graphics figures were all obtained using PyMOL (DeLano, 2008 ▶).
Supplementary Material
PDB reference: d-lactate dehydrogenase, 3kb6, r3kb6sf
Acknowledgments
We thank Mr Yoshihiro Agari and Dr Akeo Shinkai for their help in sample preparation. This work was supported in part by the RIKEN Structural Genomics/Proteomics Initiative (RSGI), the National Project on Protein Structural and Functional Analyses, Ministry of Education, Culture, Sports, Science and Technology of Japan. This work was supported by the Synchrotron Radiation Department at the Science and Technology Facilities Council, Daresbury Laboratory UK and X-ray data were collected on beamline 10.1 at the Synchrotron Radiation Source, which was supported by Biotechnology and Biological Sciences Research Council Grant BB/E001971 (to SSH and RWS).
References
- Abola, A., Bernstein, F. C., Bryant, S. H., Koetzle, T. F. & Weng, J. (1987). Crystallographic Databases – Information Content, Software Systems, Scientific Applications, edited by F. H. Allen, G. Bergerhoff & R. Sievers, pp. 107–132. Bonn/Cambridge/Chester: Data Commission of the International Union of Crystallography.
- Bradford, M. M. (1976). Anal. Biochem.72, 248–254. [DOI] [PubMed]
- Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B. III, Snoeyink, J., Richardson, J. S. & Richardson, D. C. (2007). Nucleic Acids Res.35, W375–W383. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2008). PyMOL Molecular Viewer. DeLano Scientific, Palo Alto, California, USA. http://www.pymol.org.
- Ellis, M. J., Antonyuk, S. & Hasnain, S. S. (2002). Acta Cryst. D58, 456–458. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Kochhar, S., Hunziker, P. E., Leong-Morgenthaler, P. & Hottinger, H. (1992). Biochem. Biophys. Res. Commun.184, 60–66. [DOI] [PubMed]
- Lamzin, V. S., Dauter, Z., Popov, V. O., Harutyunyan, E. H. & Wilson, K. S. (1994). J. Mol. Biol.236, 759–785. [DOI] [PubMed]
- Lamzin, V. S., Dauter, Z. & Wilson, K. S. (1995). Curr. Opin. Struct. Biol.5, 830–836. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Razeto, A., Kochhar, S., Hottinger, H., Dauter, M., Wilson, K. S. & Lamzin, V. S. (2002). J. Mol. Biol.318, 109–119. [DOI] [PubMed]
- Shinoda, T., Arai, K., Shigematsu-Iida, M., Ishikura, Y., Tanaka, S., Yamada, T., Kimber, M. S., Pai, E. F., Fushinobu, S. & Taguchi, H. (2005). J. Biol. Chem.280, 17068–17075. [DOI] [PubMed]
- Stoll, V. S., Kimber, M. S. & Pai, E. F. (1996). Structure, 4, 437–447. [DOI] [PubMed]
- Sugahara, M. et al. (2008). J. Struct. Funct. Genomics, 9, 21–28. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: d-lactate dehydrogenase, 3kb6, r3kb6sf
PDB reference: d-lactate dehydrogenase, 3kb6, r3kb6sf