

The putative 4-amino-4-deoxychorismate lyase (TTHA0621) from T. thermophilus HB8 was cloned, overexpressed, purified and crystallized. Its crystal structure was determined by a combination of SAD and molecular-replacement methods and was refined to 1.93 Å resolution.
Keywords: 4-amino-4-deoxychorismate lyase, pyridoxal 5′-phosphate, folate biosynthesis, Thermus thermophilus HB8
Abstract
The pyridoxal 5′-phosphate-dependent enzyme 4-amino-4-deoxychorismate lyase converts 4-amino-4-deoxychorismate to p-aminobenzoate and pyruvate in one of the crucial steps in the folate-biosynthesis pathway. The primary structure of the hypothetical protein TTHA0621 from Thermus thermophilus HB8 suggests that TTHA0621 is a putative 4-amino-4-deoxychorismate lyase. Here, the crystal structure of TTHA0621 is reported at 1.93 Å resolution. The asymmetric unit contained four NCS molecules related by 222 noncrystallographic symmetry, in which the formation of intact dimers may be functionally important. The cofactor pyridoxal 5′-phosphate (PLP) binds to the protein in the large cleft formed by the N-terminal and C-terminal domains of TTHA0621. The high structural similarity and the conservation of the functional residues in the catalytic region compared with 4-amino-4-deoxychorismate lyase (PabC; EC 4.1.3.38) from Escherichia coli suggest that the TTHA0621 protein may also possess 4-amino-4-deoxychorismate lyase activity.
1. Introduction
Folate is an essential compound for animals, including humans. Folate deficiency in humans may lead to physiological disorders such as anaemia and neural tube defects in newborns (Lucock, 2000 ▶), while in older people it may cause mental disorders such as psychiatric syndromes and decreased cognitive performance (Calvaresi & Bryan, 2001 ▶; Hultherg et al., 2001 ▶). Recent studies have also suggested that folate has protective properties against cardiovascular diseases and some types of cancer (Boushey et al., 1995 ▶; Brattstrom & Wilcken, 2000 ▶). Food products with high folate content can be produced by the metabolic engineering of fermentative microbes.
p-Aminobenzoate, a component of folate, is derived from the aromatic precursor chorismate. Chorismate acts as a branch-point precursor for metabolites and is essential in the biosynthesis of many important aromatic products (Green & Nichols, 1991 ▶). These metabolites and their end products include anthranilate (a precursor of tryptophan), prephenate (a precursor of tyrosine and phenylalanine), p-aminobenzoate (a precursor of folic acid), isochorismate (a precursor of enterochelin and menaquinone) and p-hydroxybenzoate (a precursor of ubiquinone). In Escherichia coli, chorismate is converted into 4-amino-4-deoxychorismate (ADC) via chorismate synthetase components I and II (PabB and BabA; EC 6.3.5.8). Subsequently, 4-amino-4-deoxychorismate lyase (PabC; EC 4.1.3.38), which contains a tightly bound pyridoxal 5′-phosphate (PLP) cofactor, converts ADC into p-aminobenzoate (pABA) (Green & Nichols, 1991 ▶; Parsons et al., 2002 ▶).
Sequence analysis of the hypothetical TTHA0621 protein from Thermus thermophilus HB8 (hereafter referred to as TTHA0621) suggested that it possesses a putative 4-amino-4-deoxychorismate lyase activity. To our knowledge, the tertiary structure of the 4-amino-4-deoxychorismate lyase from E. coli (PabC) is the only structure of this enzyme solved to date (Nakai et al., 2000 ▶; Parsons et al., 2002 ▶). Here, we report the crystal structure at 1.93 Å resolution of the aforementioned TTHA0621 protein, a putative orthologue of E. coli PabC, complexed with the PLP cofactor.
2. Materials and methods
2.1. Protein expression and purification
The gene encoding the TTHA0621 protein (gi:55980590) was amplified via PCR using T. thermophilus HB8 genomic DNA and was cloned into the pET-11a expression vector (Merck Novagen). The expression vector was introduced into E. coli B834 (DE3) strain (Merck Novagen) and the recombinant strain was cultured in 6 l minimal medium containing 25 µg ml−1 selenomethionine and 50 µg ml−1 ampicillin. The harvested cells (43.1 g) were lysed on ice by sonication in 20 mM Tris–HCl buffer pH 8.0 containing 50 mM NaCl. The cell lysate was heat-treated at 343 K for 10 min and centrifuged at 200 000g for 60 min. The supernatant was applied onto a Resource ISO column (GE Healthcare Biosciences) equilibrated with 50 mM sodium phosphate buffer pH 7.0 containing 1.5 M ammonium sulfate and was eluted with a linear (1.5–0 M) gradient of ammonium sulfate. The target sample, which eluted in the 0.48 M ammonium sulfate fraction, was then applied onto a Resource Q column (GE Healthcare Biosciences) equilibrated with 20 mM Tris–HCl buffer pH 8.0 and was eluted with a linear gradient of NaCl. The target sample, which eluted in the 0.08 M NaCl fraction, was collected and applied onto a HiLoad 16/60 Superdex 75 pg column (GE Healthcare Biosciences) equilibrated with 20 mM Tris–HCl buffer pH 8.0 containing 150 mM NaCl. The protein sample was analyzed by SDS–PAGE and was confirmed by N-terminal amino-acid sequencing. After concentration to 11.7 mg ml−1 by ultrafiltration, the protein yield was 4.0 mg from 43.1 g of cells. The native protein sample was purified using the same method as used for the SeMet protein, except for the use of the E. coli BL21 (DE3) strain (Merck Novagen) and LB medium. After concentration to 14.2 mg ml−1 by ultrafiltration, the protein yield was 14.2 mg from 27.6 g of cells.
2.2. Crystallization and data collection
Crystals were grown by the sitting-drop vapour-diffusion method at 293 K. Each drop, consisting of 1.0 µl of a 11.7 mg ml−1 SeMet protein solution or a 14.2 mg ml−1 native protein solution and 1.0 µl reservoir solution [0.1 M HEPES buffer pH 8.0 containing 1.3 M Li2SO4 and 2%(v/v) PEG 200], was equilibrated against 100 µl reservoir solution. Crystals that were suitable for X-ray data collection appeared within two months and reached final dimensions of 0.3 × 0.22 × 0.22 mm (Fig. 1 ▶ a). The crystals were flash-cooled in a nitrogen-gas stream at 100 K using 25%(v/v) glycerol as a cryoprotectant.
Figure 1.

The structure of TTHA0621. (a) Crystals of the TTHA0621 protein. (b) Ribbon diagram of the tertiary structure of TTHA0621 (coloured in a rainbow ramp from blue at the N-terminus to red at the C-terminus). The PLP cofactor molecule is shown as a stick model. (c) Ribbon diagram of the quaternary structure of the TTHA0621 tetramer (shown in an arbitrary orientation) coloured by subunit. The N-terminal β-strands β1 of subunits A and D and those of their intact dimer subunits, B and C, respectively, form intermolecular antiparallel β-sheets in their respective dimers. Subunits A, B, C and D are coloured green, yellow, cyan and magenta, respectively. The PLP cofactor molecule is shown as a stick model. (d) Near the tetramer region. The weak tetramerization occurs through hydrophobic interactions between the four subunits (A–D) contributed by Phe154 and His156, which are shown as stick models. All figures were produced with PyMOL (DeLano, 2002 ▶) unless mentioned otherwise.
Anomalous diffraction data were collected on the PX10.1 beamline at the Daresbury Synchrotron Radiation Source (SRS, England) at three wavelengths, 0.97892 (peak), 0.979 (edge) and 0.92 Å (remote), for an Se-MAD experiment. The data sets were collected at 100 K using a MAR Mosaic 225 mm CCD detector. The crystal-to-detector distance was set to 150 mm. The diffraction data were integrated, reduced and scaled using the HKL-2000 software suite (Otwinowski & Minor, 1997 ▶). In addition, sulfur-SAD data for the native protein were collected at the in-house facility using a Rigaku FR-E SuperBright (Cr/Cu dual target) source at a wavelength of 2.29 Å (peak), corresponding to the S atom. The crystallographic data and refinement statistics are shown in Table 1 ▶. The asymmetric unit consisted of four molecules related by approximate 222 noncrystallographic symmetry (NCS), with a molecular mass of 27 kDa and 246 amino-acid residues per monomer and with a solvent content of about 57%.
Table 1. Summary of data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| SeMet | Native | |
|---|---|---|
| Data collection | ||
| Source | SRS, PX10.1 | Rigaku FR-E SuperBright (Cr/Cu dual target) |
| Wavelength (Å) | 0.9790 | 2.29 |
| Space group | P212121 | P212121 |
| Unit-cell parameters (Å) | a = 65.61, b = 133.58, c = 141.98 | a = 66.33, b = 135.81, c = 142.20 |
| Resolution (Å) | 50.00–1.93 (2.00–1.93) | 45.64–2.47 (2.56–2.47) |
| Completeness (%) | 96.8 (72.0) | 93.9 (57.4) |
| Redundancy | 13.1 (5.0) | 14.0 (8.6) |
| Average I/σ(I) | 26.5 (1.9) | 53.3 (10.1) |
| Rmerge† (%) | 6.3 (57.8) | 5.4 (21.8) |
| Refinement statistics | ||
| No. of molecules in ASU | 4 | |
| Resolution limits (Å) | 20–1.93 | |
| R factor/Rfree‡ (%) | 19.3/24.9 | |
| B factors (Å2) | ||
| Mean | 33.7 | |
| PLP cofactor | 26.1 | |
| PEG/PGE | 58.1 | |
| Water | 44.8 | |
| No. of residues | 982 | |
| No. of PLP cofactors | 4 | |
| No. of PEG molecules | 2 | |
| No. of PGE molecules | 2 | |
| No. of water molecules | 1318 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.013 | |
| Bond angles (°) | 1.54 | |
R
merge = 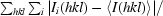
 .
.
R = 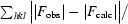
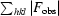 . R
free was calculated with 5% of data omitted from refinement.
. R
free was calculated with 5% of data omitted from refinement.
2.3. Structure determination and refinement
We initially attempted to determine the structure by the MAD method using SOLVE/RESOLVE (Terwilliger & Berendzen, 1999 ▶). The MAD procedure yielded four distinct peaks corresponding to the four NCS-related Se atoms, with an overall mean figure of merit of 0.25, despite the fact that the protein chain contained only one methionine residue at the N-terminus (Met1). Density modification using the program RESOLVE (Terwilliger, 2000 ▶) resulted in an increase in the figure of merit to 0.67 and generated a partially interpretable map. In parallel, we also obtained sulfur anomalous diffraction data using the in-house Cr-target facility. The structure corresponding to the Cr-target data was solved by the SAD procedure using SHELXC/SHELXD (Sheldrick, 2008 ▶). Four sites corresponding to anomalous S atoms of the protein and nine sites corresponding to anomalous SO4 2− molecules from the Li2SO4 used as a crystallization reagent were obtained from the SAD procedure. These sites were refined by SOLVE/RESOLVE and the SAD phases after density modification yielded about 70% of the protein chains corresponding to the four NCS-related molecules (N. Watanabe et al., unpublished data). We then used this partial structure as a model for molecular replacement using MOLREP (Vagin & Teplyakov, 1997 ▶) as incorporated in the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶) and the SeMAD data. The correctly positioned tetramer (consisting of chains A, B, C and D), which possesses approximate 222 NCS, was subjected to refinement with CNS (Brünger et al., 1998 ▶) and subsequently to warpNtrace refinement in ARP/wARP (Morris et al., 2003 ▶). A total of 848 of the 984 residues corresponding to the TTHA0621 tetramer were identified from the ARP/wARP tracing. Moreover, several rounds of manual fitting and refitting were performed using the program Coot (Emsley & Cowtan, 2004 ▶) in combination with refinement using REFMAC5 (Murshudov et al., 1997 ▶). Long stretches of residual electron density were observed after refinement of the protein structure and the PLP cofactor molecules. This residual density was modelled by short and long fragments of polyethylene glycol, diethylene glycol (PEG) and triethylene glycol (PGE), respectively. The source of the PEG/PGE molecules could be the polydisperse PEG 200 molecule used in crystallization. The refined model of the tetramer in the asymmetric unit contains 982 residues, four PLP molecules, four SO4 2− molecules, two PEG molecules, two PGE molecules and 1318 water molecules, with a final R of 19.3% and an R free of 24.9% at 1.93 Å resolution. The Thr245 and Glu246 residues in chain A, Glu246 in chain B and Glu246 in chain C are missing from the final structure. The stereochemistry of the complex structure was good, as assessed using MolProbity (Lovell et al., 2003 ▶). Ramachandran plot analysis (Laskowski et al., 1993 ▶) of this structure revealed that 99.2% of all residues were in the allowed region and 0.8% were in the disallowed region. The outlier residues are Leu11 and Arg226 in chain A, Leu11 and Arg226 in chain B, Gly171 and Arg226 in chain C and Glu170 and Arg226 in chain D. The electron density was clear within these regions. The refinement statistics are summarized in Table 1 ▶.
3. Results and discussion
3.1. Overall structure of the TTHA0621 protein
The overall tertiary structure of TTHA0621 (amino acids 1–246) comprises a long N-terminal extended segment (residues 1–23), a small N-terminal domain (residues 24–96) and a large C-terminal domain (residues 103–246) (Fig. 1 ▶ b). The small N-terminal domain is formed by three β-strands (β2–β4) and two α-helices (α1 and α2). The β-strands (β2–β4) are arranged in an antiparallel manner. The two helices, α1 and α2, are located on the same side of the β-sheet.
The C-terminal domain possesses an α+β fold and is formed by eight β-strands (β5–β12) and three α-helices (α3–α5). The α-helices face outwards and are exposed to the solvent region. A six-amino-acid fragment (Tyr97–Leu102) connects β4 of the small domain and a 310-helix (Ser103–Arg106) of the large domain. The arrangement of these two domains in the overall structure produces a large cleft in which the PLP cofactor is deeply buried (Fig. 1 ▶ b). The flexible C-terminal region is stabilized by the formation of an intramolecular disulfide bridge between Cys78 and Cys243 (not shown).
3.2. Oligomeric structure
The asymmetric unit contains four subunits, which form a tetramer composed of a dimer of dimers (Fig. 1 ▶ c). The dimers are formed between subunits A and B and between subunits C and D. The dimer of TTHA0621 comprising subunits A and B is used for description unless stated otherwise. The N-terminal β-strand β1 (Met1–Leu4) of subunit A forms an intersubunit antiparallel β-sheet with β4 (Val89–Pro96) of subunit B (Fig. 1 ▶ c). In addition to this intermolecular interaction, the long flexible region comprising Ala12–Ser23 in the N-terminal extended segment, the hairpin connecting β3 and β4 and the long flexible segment connecting β5 and α3 in each monomer contribute substantial intersubunit interactions to form a stable dimer. The buried surface area per monomer within the intact dimers is about 2300 Å2 (∼18% buried surface area), as calculated by the program SURFACE in the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
The loop regions comprising Ala153–His156 from each monomer are close to each other in the tetramer arrangement (Fig. 1 ▶ d). The stacking of the Phe154 residues of the four subunits contributes to the intermolecular hydrophobic interactions between them. Moreover, the His156 residues of subunits A and B form hydrophobic interactions with the Phe154 residues of subunits D and C, respectively. The dimer interfaces between subunits A and D and between subunits B and C each bury a total surface area per monomer of about 500 Å2 (∼4% buried surface area). In this weak dimer arrangement, only five electrostatic interactions are observed (not shown). Hence, we speculate from the above analyses that the tetrameric arrangement of the subunits in the asymmetric unit may arise from crystal packing.
3.3. The cofactor PLP-binding site
The long extended loop connecting β5 and β6 and the flexible segment connecting β6 and α3 create a large cavity between the small and large domains of TTHA0621 (Figs. 1 ▶ and 2 ▶). The cofactor pyridoxal 5′-phosphate (PLP) is deeply buried and tightly bound to the protein in the cavity region. The PLP molecule mostly interacts with the large C-terminal domain of the TTHA0621 protein. The pyridine ring of PLP is sandwiched between a short loop connecting β8 and β9 on one side and the loop connecting β10 and α4 on the opposite side of the pyridine ring. The top of the pyridine ring is capped by the loop connecting β6 and α3. In addition, the phosphate moiety of PLP sits on the surface formed by the N-terminal region of helix α4 and the C-terminal end of β-strand β12.
Figure 2.

The PLP cofactor-binding region. A stereoview of the interaction of the cofactor PLP with TTHA0621 is shown. The interacting residues and PLP are shown as stick models. A water molecule, shown as red sphere, bridges PLP and Tyr130. Electrostatic interactions are depicted by dashed lines.
Within the PLP-binding site region, ten electrostatic interactions are observed between TTHA0621 and the PLP cofactor (Table 2 ▶). The side-chain NZ atom of the catalytic residue Lys126 forms a Schiff base bond to the C4 atom of PLP. Asp159 is hydrogen bonded to N1 of the pyridine ring of PLP (Fig. 2 ▶). A water molecule bridges Tyr130 and O3′ of PLP. Furthermore, the phosphate group of PLP interacts with Arg44, Ile183, Thr184 and Ser218 as well as with three water molecules.
Table 2. Potential electrostatic interactions between PLP and TTHA0621.
| PLP | TTHA0621 | Distance (Å) |
|---|---|---|
| OP1 | Arg44 NH1 | 2.71 |
| Arg44 NH2 | 2.99 | |
| Ile183 N | 2.90 | |
| OP2 | Thr184 N | 2.93 |
| Thr184 OG1 | 2.90 | |
| OP3 | Ser218 OG | 2.72 |
| Ser218 N | 3.04 | |
| N1 | Asp159 OD1 | 2.91 |
| C4† | Lys126 NZ | 2.89 |
| O3‡ | Tyr130 | — |
C4 of PLP forms a Schiff-base bond to Lys126 NZ.
A water molecule bridges O3 of PLP and Tyr130.
3.4. Comparison with E. coli PabC protein
The structure of PabC from E. coli has previously been determined in unbound (PDB code 1et0; Nakai et al., 2000 ▶) and PLP-bound forms (PDB code 1i2l and 1i2k; Parsons et al., 2002 ▶). E. coli PabC forms a functionally important dimer. As the dimer arrangements are similar in E. coli PabC and TTHA0621, we speculate that the TTHA0621 dimer may also be functionally important. The DaliLite program (Holm & Park, 2000 ▶) was employed to perform sequence and superposition analyses. Superposition of the TTHA0621 structure (subunit A) onto the PabC structure (subunit A) yielded an r.m.s.d. value of 2.0 Å for 225 Cα atoms. Although the sequence similarity between TTHA0621 and E. coli PabC is low (21% identity; Fig. 3 ▶), the overall structures of these two proteins are similar (Fig. 4 ▶). However, superposition of the TTHA0621 dimer onto the E. coli PabC dimer revealed significant structural deviations between TTHA0621 and E. coli PabC (Fig. 4 ▶ a). Intriguingly, the intersubunit antiparallel β-sheet observed in the TTHA0621 dimer is absent in the E. coli PabC dimer. Another major structural difference exists in the loop region connecting β3 and β4. The E. coli PabC structure has a long flexible 12-amino-acid loop (Gly87–Ala98, PabC numbering), whereas the corresponding region in TTHA0621 contains a short hairpin loop. In the E. coli PabC dimer structure this flexible loop from one subunit protrudes into the substrate-binding region of the other subunit of the dimer and plays a critical role in the interaction with PLP (see details below). Another striking feature is also observed in the segment connecting β12 and α5: a three-amino-acid insertion (Gly248–Val250) in E. coli PabC forms an extended flexible loop, whereas the corresponding region in TTHA0621 possesses a relatively shorter loop conformation.
Figure 3.

Sequence alignment of the putative 4-amino-4-deoxychorismate lyase (TTHA0621) from T. thermophilus (Q5SKM2_THET8) and the 4-amino-4-deoxychorismate lyase (PabC) from E. coli (PABC_ECOLI). The secondary-structural features of TTHA0621 are indicated above the alignment. The residues that potentially interact with the PLP cofactor are indicated by blue circles. The conserved catalytic residue Lys161, which forms a Schiff base with PLP, is indicated by a red triangle. The colours reflect the similarity (conserved residues are shown as white characters on a red background, residues that are similar within a group are shown as red characters and residues that are similar across groups are shown in blue frames). 310-Helices are denoted by the symbol η. The sequence was aligned and displayed using ClustalW (Thompson et al., 1994 ▶) and ESPript (Gouet et al., 1999 ▶), respectively.
Figure 4.

Comparison of TTHA0621 with E. coli PabC. (a) Superposition of TTHA0621 (light orange) on E. coli PabC (light green) for the Cα atoms corresponding to one subunit (top) of the dimer (PDB code 1i2k; Parsons et al., 2002 ▶). In the E. coli PabC dimer a long flexible loop (pink) from one monomer, which is inserted between strands β3 and β4, protrudes into the catalytic site of the other monomer. (b) Near the cofactor-binding site region. The interacting residues and the PLP cofactors of the THA0621 and E. coli PabC complexes are shown in green and slate, respectively. The interacting residues and the inhibitor DCS in the E. coli PabC–DCS complex are shown in yellow. (c) Near the O3′ atom of PLP, conservation of Tyr is observed between the T. thermophilus and E. coli enzymes. In the E. coli PabC structure Tyr92, which lies in the flexible region (pink) of the neighbouring subunit, forms intermolecular interactions with the PLP cofactor bound to the other subunit, whereas in TTHA0621 a water molecule bridges Tyr130 and PLP within the monomer. The nonconserved residues in E. coli PabC are labelled with asterisks.
A comparison of the PLP-binding site regions between TTHA0621 and E. coli PabC revealed that the binding site and the interacting residues are well conserved (Fig. 4 ▶ b). However, Tyr92 in E. coli PabC, which is located in the extended loop region between β3 and β4 of subunit A, forms a hydrogen-bond interaction with the O3′ atom of PLP bound to the other subunit B of the dimer (Fig. 4 ▶ c). Although this type of interaction is absent in the TTHA0621 structure, Tyr130 (Arg144 in E. coli), which lies between the β5 strand and the α3 helix, forms a similar electrostatic interaction with PLP through a water molecule within the subunit complex. The side chains of Thr184 and Ser218 in TTHA0621 are hydrogen bonded to PLP, whereas these residues are replaced by Met201 and Ala237, respectively, in E. coli PabC. Since the overall tertiary structure, the dimerization arrangements and the binding mode of the PLP interaction in TTHA0621 are similar to those in E. coli PabC, we speculate that TTHA0621 may indeed possess 4-amino-4-deoxychorismate lyase activity and that its catalytic mechanism may be similar to that of the 4-amino-4-deoxychorismate lyase from E. coli. However, further structural analyses of the TTHA0621 complex and biochemical studies are essential to confirm the proposed hypothesis.
Supplementary Material
PDB reference: TTHA0621, 2zgi, r2zgisf
Acknowledgments
We thank Mr Yoshihiro Agari, Mr Takeshi Ishii, Mr Hitoshi Iino and Dr Akeo Shinkai for their assistance with sample preparation. We are grateful to Ms Tomoko Nakayama and Ms Azusa Ishii for clerical assistance. This work was supported in part by the RIKEN Structural Genomics/Proteomics Initiative (RSGI), the National Project on Protein Structural and Functional Analyses, Ministry of Education, Culture, Sports, Science and Technology of Japan. This work was supported by the Science and Technology Facilities Council, Daresbury Laboratory UK and beamline 10.1 at the Synchrotron Radiation Source, which was supported by Biotechnology and Biological Sciences Research Council Grant BB/E001971 (to SSH and RWS).
References
- Boushey, C. J., Beresford, G. A., Omenn, G. S. & Motulsky, A. G. (1995). JAMA, 274, 1049–1057. [DOI] [PubMed]
- Brattstrom, J. & Wilcken, D. E. (2000). Am. J. Clin. Nutr.72, 315–323. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Calvaresi, E. & Bryan, J. (2001). J. Gerontol. B, 56, 327–339. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DeLano, W. L. (2002). PyMOL Molecular Viewer. DeLano Scientific, San Carlos, California, USA. http://www.pymol.org.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Métoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Green, J. M. & Nichols, B. P. (1991). J. Biol. Chem.266, 12971–12975. [PubMed]
- Holm, L. & Park, J. (2000). Bioinformatics, 16, 566–567. [DOI] [PubMed]
- Hultherg, B., Isaksson, A., Nilsson, K. & Gustafson, L. (2001). Psychiatry, 16, 873–878. [DOI] [PubMed]
- Laskowski, R. A., Moss, D. S. & Thornton, J. M. (1993). J. Mol. Biol.231, 1049–1067. [DOI] [PubMed]
- Lovell, S. C., Davis, I. W., Arendall, W. B. III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2003). Proteins, 50, 437–450. [DOI] [PubMed]
- Lucock, M. (2000). Mol. Genet. Metab.71, 121–138. [DOI] [PubMed]
- Morris, R. J., Perrakis, A. & Lamzin, V. S. (2003). Methods Enzymol.374, 229–244. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nakai, T., Mizutani, H., Miyahara, I., Hirotsu, K., Takeda, S., Jhee, K.-H., Yoshimura, T. & Esaki, N. (2000). J. Biochem.128, 29–38. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Parsons, J. F., Jensen, P. Y., Pachikara, A. S., Howard, A. J., Eisenstein, E. & Ladner, J. E. (2002). Biochemistry, 41, 2198–2208. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Terwilliger, T. C. (2000). Acta Cryst. D56, 965–972. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res.22, 4673–4680. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: TTHA0621, 2zgi, r2zgisf
PDB reference: TTHA0621, 2zgi, r2zgisf