

The crystal structure of cold-shock protein E from S. typhimurium (StCspE) has been determined at 1.1 Å resolution.
Keywords: cold-shock proteins, Salmonella typhimurium
Abstract
In prokaryotic organisms, cold shock triggers the production of a small highly conserved family of cold-shock proteins (CSPs). CSPs have been well studied structurally and functionally in Escherichia coli and Bacillus subtilis, but Salmonella typhimurium CSPs remain relatively uncharacterized. In S. typhimurium, six homologous CSPs have been identified: StCspA–E and StCspH. The crystal structure of cold-shock protein E from S. typhimurium (StCspE) has been determined at 1.1 Å resolution and has an R factor of 0.203 after refinement. The three-dimensional structure is similar to those of previously determined CSPs and is composed of five antiparallel β-strands forming a classic OB fold/five-stranded β-barrel. This first structure of a CSP from S. typhimurium provides new insight into the cold-shock response of this bacterium.
1. Introduction
The cellular response to cold shock in prokaryotic organisms is defined by the production of a small set of cold-induced proteins (CIPs; Jones & Inouye, 1994 ▶, 1996 ▶). Most of these proteins are directly or indirectly involved in protein transcription and translation (reviewed in Thieringer et al., 1998 ▶). The most common of these are the small highly conserved family of cold-shock proteins (CSPs; Graumann & Marahiel, 1998 ▶). CSPs are a family of small acidic proteins with a molecular mass of approximately 7.4 kDa (Perl et al., 1998 ▶). CSPs have been shown to bind to stretches of 6–7 nucleotides (Lopez et al., 1999 ▶, 2001 ▶; Lopez & Makhatadze, 2000 ▶) of ssDNA with high affinity and a high degree of specificity (Graumann & Marahiel, 1994 ▶; Lopez et al., 1999 ▶; Max et al., 2006 ▶, 2007 ▶; Morgan et al., 2007 ▶). However, certain CSPs can bind in a nonspecific manner (Jiang et al., 1997 ▶). This ability to bind nucleic acids led to the hypothesis that CSPs could prevent mRNA secondary structures (Jiang et al., 1997 ▶) induced by low temperatures (Higgs, 2001 ▶) and as a result they are now commonly classified as RNA chaperones.
Two of the best characterized groups of bacterial CSPs are those from Bacillus subtilis and Escherichia coli. To date, nine CSPs have been identified in E. coli, CspA–I (Wang et al., 1999 ▶), and three in B. subtilis (Kunst et al., 1997 ▶), CspB, CspC and CspD. Three X-ray crystal structures of CSPs (B. subtilis CspB, B. caldolyticus Csp and E. coli CspA) have been determined (Schindelin et al., 1993 ▶, 1994 ▶; Delbruck et al., 2001 ▶), all of which comprise a single cold-shock domain (CSD) composed of five antiparallel β-strands forming a classic oligonucleotide/oligosaccharide (OB) fold/five-stranded β-barrel (Theobald et al., 2003 ▶), with the characteristic ribonucleotide protein 1 (RNP1) and RNP2 (Horn et al., 2007 ▶) motifs conserved on the DNA-binding surfaces (Fig. 1 ▶ a).
Figure 1.

(a) Alignment of the amino-acid sequences of cold-shock proteins for which X-ray crystal structures exist. (b) Alignment of the amino-acid sequences of cold-shock proteins from S. typhimurium with the major cold-shock protein from B. subtilis. Identical residues are shown in black, similar residues in grey and non-identical residues in white. Residues involved in DNA binding are marked with asterisks. Ribonucleotide protein motifs 1 and 2 are shown in red and blue, respectively. The abbreviations are as follows: S.T_CSP, cold-shock protein from S. typhimurium; E.C_CspA, cold-shock protein A from E. coli; B.S_CspB, cold-shock protein B from B. subtilis; B.C_Csp, cold-shock protein from B. caldolyticus. (c) Recombinant His6 StCspE and StCspE resolved on a HiLoad 26/60 Superdex-75 column. The column was calibrated with the molecular-weight standards catalase (232 kDa), albumin (67 kDa), ovalbumin (43 kDa), chymotrypsinogen A (25 kDa), ribonuclease A (13.7 kDa) and vitamin B (1.35 kDa) from Amersham Biosciences. The dashed lines on the trace correspond to the elution maxima of the standards. His6 StCspE (blue) eluted as a single peak with a retention volume of 13.79 ml, corresponding to a molecular weight of 12.5 kDa. StCspE (after His-tag cleavage; red) eluted as one major and one minor peak with retention volumes of 16.07 and 12.48 ml corresponding to molecular weights of 7.0 and 25 kDa, respectively. (d) SDS–PAGE analysis of 15 µg purified StCspE. (e) Electrophoretic mobility shift assay of StCspE and its complexes with ONc. 15–20 pmol 5′-end γ-32P-labelled ONc was mixed with increasing amounts of StCspE and incubated for 10 min at 277 K. Amounts of protein: lane 1, 500 pmol; lane 2, 250 pmol; lane 3, 125 pmol; lane 4, 62.5 pmol: lane 5, 31.25 pmol. Lane 6 was a control and contained 5′-end γ-32P-labelled ONc only. Protein–DNA complexes were separated on a 20% native polyacrylamide gel. (f) StCspE crystal after 24 h of growth. (g) StCspE crystal after 72 h of growth.
The CSD-containing proteins found in eukaryotic organisms (Y-box factors) share high sequence similarity with CSPs (Wolffe et al., 1992 ▶) and have similar structural architecture (Kloks et al., 2002 ▶), including the conserved DNA-binding motifs (RNP1 and RNP2). The homologous Y-box proteins regulate transcription and translation both positively and negatively (Matsumoto & Wolffe, 1998 ▶). It is clear that all CSPs and CSD-containing proteins belong to a large family of structurally related nucleic acid-binding proteins, suggesting similar functions.
In Salmonella typhimurium, six CSPs (StCspA–E and StCspH) have been identified (Fig. 1 ▶ b) and cold inducibility of StCspA, StCspB and StCspH has been reported (Craig et al., 1998 ▶; Jeffreys et al., 1998 ▶; Horton et al., 2000 ▶; Kim et al., 2001 ▶), although their functions have yet to be clearly elucidated. StCspA, StCspB, StCspC and StCspE exhibit the greatest sequence identity (Table 1 ▶) and all residues involved in ssDNA binding are highly conserved (Fig. 1 ▶ b). Although StCspE has not yet been shown to be cold-inducible, the high degree of sequence homology and the availability of large amounts of pure protein made it an ideal candidate for structural studies. Here, we present the purification, crystallization and X-ray structure of cold-shock protein E from S. typhimurium, providing new insight into the StCsp family.
Table 1. Sequence-identity (%) matrix for S. typhimurium CSPs.
| Sequence | CspA | CspB | CspC | CspD | CspE | CspH |
|---|---|---|---|---|---|---|
| CspA | 64.2 | 67.1 | 43.4 | 68.5 | 48.5 | |
| CspB | 64.2 | 68.5 | 46.0 | 70.0 | 45.7 | |
| CspC | 67.1 | 68.5 | 42.6 | 84.0 | 42.8 | |
| CspD | 43.4 | 46.0 | 42.6 | 42.6 | 26.3 | |
| CspE | 68.5 | 70.0 | 84.0 | 42.6 | 47.1 | |
| CspH | 48.5 | 45.7 | 42.8 | 26.3 | 47.1 |
2. Materials and methods
2.1. Construction of His6-StCspE expression vector
Forward (5′-dAGCCATATGTTAGAAGGTAAAGTAAAATAA-3′) and reverse (5′-dCGAAAGCTTTTACAGAGCAGTTACGTTTGCAGC-3′) primers were used in a PCR with S. typhimurium strain 1344 chromosomal DNA. Bases were added to the primers to introduce NdeI and HindIII restriction sites (bold). These sites were used to clone the PCR product into an NdeI–HindIII-digested pET28a vector (Novagen), resulting in the plasmid pET28a_StCspE. The pET expression vector also contains a cleavable hexahistidine (His6) fusion tag (GSHHHHHHSSGLVPRGSHM) that was added to the N-terminus of the StCspE gene to facilitate the purification of the recombinant protein.
2.2. Expression, purification and characterization of recombinant StCspE
Competent Rosetta BL21 (DE3) E. coli cells (Novagen) were transformed with pET28a_StCspE. Single colonies of transformed E. coli harbouring the StCspE gene construct were picked from Luria–Bertani (LB) kanamycin plates (50 µg ml−1) and used to inoculate 50 ml LB medium (containing 50 µg ml−1 kanamycin). Cultures were grown overnight at 310 K with agitation. 20 ml of overnight culture was used to inoculate 1 l LB medium containing kanamycin (50 µg ml−1). 1 l cultures were grown to an OD600 of 0.5–0.7 and expression was induced at 310 K by adding IPTG (isopropyl β-d-1-thiogalactopyranoside) to a final concentration of 1 mM. The incubation was then continued for 4 h and the cells were harvested at 8000 rev min−1 in a JLA-9.1000 rotor for 12 min at 283 K. Pellets were then frozen in liquid nitrogen and stored at 193 K. Cell pellets were resuspended in lysis buffer [20 mM Tris–HCl pH 8.0, 500 mM NaCl, 0.1 mM phenylmethylsulfonyl fluoride (PMSF) and 1 mM EDTA-free protease inhibitors] to a final volume of 30 ml per 5 g of cells and lysed by pulsed sonication on ice for 5 min. The lysate was centrifuged at 23 000 rev min−1 at 283 K for 45 min. The supernatant was loaded onto a 12 ml Ni–NTA agarose column at 1 ml min−1. The column was washed with eight column volumes of buffer A (50 mM NaH2PO4, 300 mM NaCl and 20 mM imidazole pH 8.0) and the protein was eluted over an eight column-volume elution gradient with buffer B (50 mM NaH2PO4, 300 mM NaCl and 250 mM imidazole pH 8.0). Eluted fractions were pooled and concentrated using a Vivaspin column (molecular-weight cutoff 3.5 kDa). The protein purity was excellent, but DNA contamination was high (as determined from A 280:A 260). To remove bound nucleic acids, 1 ml His6 StCspE (20 mg ml−1) was added to 9 ml buffer C (8 M urea, 300 mM NaCl and 50 mM Tris pH 8.0) and incubated at room temperature (RT) overnight. The sample was loaded onto a self-packed 2 ml Ni–NTA gravity column at RT and washed with ten column volumes of buffer C. The protein was eluted with buffer D (8 M urea, 300 mM NaCl and 50 mM Tris pH 5.0; made immediately prior to use) by manually collecting 2 ml fractions. Fractions containing high concentrations of His6 StCspE were either refolded immediately or could be stored at RT (free from any source of light) for up to two months prior to refolding. The protein was refolded by loading 200 µl His6 StCspE (∼30 mg ml−1) onto a HiLoad 26/60 Superdex 75 column (Amersham Biosciences) pre-equilibrated in buffer E (20 mM HEPES, 150 mM NaCl and 3 mM EDTA). The column flow rate was 0.5 ml min−1 for the first 2 ml (to quickly dilute the urea) and was then reduced to 0.2 ml min−1 (to allow the protein to refold). This method was based upon that used in a previous paper (Werner et al., 1994 ▶). To obtain adequate amounts of protein for crystallization, the refolding procedure was repeated 10–15 times. Protein samples were concentrated and buffer-exchanged into the required buffer (PD-10 column; Amersham Biosciences) using standard protocols. For cleavage of the histidine tag, the protein was buffer-exchanged (PD-10 column) into buffer E (20 mM Tris pH 7.5). 1.5 units of thrombin protease were added per milligram of His6 StCspE and incubated at RT for 4 h with gentle agitation. Thrombin was removed by loading the protein sample onto a self-packed 0.5 ml benzamidine gravity-flow column pre-equilibrated in buffer E. The protein was eluted by adding 2 ml buffer E to the column. To remove the free histidine tag, the protein was concentrated to 200 µl (20 mg ml−1) and loaded onto a HiLoad 26/60 Superdex 75 column pre-equilibrated with buffer E. StCspE was eluted using buffer E and quantified using both the Bradford method and the theoretical extinction coefficient ∊280nm = 5500 M −1 cm−1. Electrophoretic mobility shift assays (EMSA) were performed as detailed previously (Morgan et al., 2007 ▶).
2.3. Crystallization and data collection
Purified StCspE was concentrated (using a Vivaspin column, molecular-weight cutoff 3.5 kDa) to 10 mg ml−1 in a buffer containing 20 mM Tris pH 7.5. Crystals were obtained at 290 K by vapour diffusion using the hanging-drop technique (Blundell & Johnson, 1976 ▶). The drops were formed by mixing 1.5 µl protein solution with 1.5 µl of a well solution composed of 28% PEG 20 000, 0.05 M 3-[(1,1-dimethyl-2-hydroxyethyl)amino]-2-hydroxypropanesulfonic acid (AMPSO) pH 9 and 1% glycerol. Crystals appeared after 24 h (Fig. 1 ▶ f) and grew to maximum dimensions of 1.3 × 0.2 × 0.05 mm after 72 h (Fig. 1 ▶ g). Intensity data were collected to a resolution of 1.1 Å from a single crystal on beamline 10.1 at the Synchrotron Radiation Source (SRS), Daresbury, United Kingdom. Data were then processed with MOSFLM (Potterton et al., 2003 ▶) and scaled with SCALA (Evans, 2006 ▶). Data-collection statistics are given in Table 2 ▶.
Table 2. Data-collection and refinement statistics for StCspE.
Values in parentheses are for the outermost resolution shell.
| Data statistics | |
| Space group | P21 |
| Unit-cell parameters | |
| a () | 29.40 |
| b () | 46.80 |
| c () | 46.72 |
| () | 90.0 |
| () | 103.4 |
| () | 90.0 |
| Resolution range () | 45.361.10 (1.161.10) |
| Molecules per ASU | 2 |
| Observed reflections | 260904 (27528) |
| Unique reflections | 47339 (7261) |
| I/(I) | 15.7 (2.5) |
| Completeness (%) | 99.82 (100) |
| Multiplicity | 5.2 (3.8) |
| R merge † | 4.7 (38.9) |
| Refinement statistics | |
| R factor† (%) | 20.33 |
| R free ‡ (%) | 23.42 |
| No. of protein atoms | 1073 |
| No. of water molecules | 100 |
| Mean B factor for protein (2) | 18.86 |
| R.m.s. deviations from ideal geometry | |
| Bond distance () | 0.02 |
| Bond angle () | 1.94 |
| Ramachandran statistics | |
| Most favoured regions (%) | 99.25 [138/140] |
| Additional allowed regions (%) | 0.75 [2/140] |
| Residues in disallowed regions (%) | 0 |
R
merge = 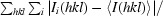
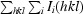 .
.
R factor and R
free = 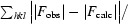
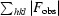 .
.
2.4. Structure determination
Phasing was performed with ACORN (Cowtan & Zhang, 1999 ▶) using the positions of ten amino acids (12–21) from the homologous structure of E. coli CspA previously solved at 2.0 Å resolution (Schindelin et al., 1994 ▶). Using the phases from ACORN, an initial structure was built automatically using ARP/wARP (Perrakis et al., 1999 ▶) to give 112 residues out of a total of 144. Model building was manually completed using the program Coot (Emsley & Cowtan, 2004 ▶). The model was refined in REFMAC (Murshudov et al., 1997 ▶), including automatic water-molecule placement using ARP/wARP and construction of alternative side-chain conformations. Anisotropic B factors were then refined and additional water molecules were added. Water molecules were accepted based on the following criteria: a peak of height not less than 3.2σ in the difference maps, hydrogen-bonding distances to protein atoms of between 2.0 and 3.5 Å and a B factor of less than 45 Å2. Areas of disorder were carefully modelled into F o − F c electron density and the changes in R/R free values were used to assess the final model quality. This resulted in a final model of 138 residues, composed of two StCspE molecules (molecule 1/chain A containing Ser−11 to Leu69 and molecule 2/chain B containing Lys3–Leu69) and 100 water molecules. Further addition of water molecules using a lower σ cutoff led to an increase in the R/R free values. The R/R free values converged for 20 cycles of REFMAC at 0.20 and 0.23, respectively (Table 1 ▶). Coordinates for the 1.1 Å structure have been deposited in the Protein Data Bank with code 3i2z. The geometry of the model was assessed using MolProbity (Davis et al., 2007 ▶). Two residues fall outside the most favoured region of the Ramachandran plot, both of which are found in flexible regions [the N-terminus (Lys3, chain B) and loop 4 (Asn56, chain A)].
3. Results and discussion
In the refolding gel-filtration step, His6 StCspE eluted as single peak (Fig. 1 ▶ c) with a retention volume of 13.79 ml, corresponding to a molecular weight of 12.5 kDa and signifying that His6 StCspE exists as a monomer in solution. After removal of the hexahistidine tag, StCspE eluted as two distinct species with retention volumes of 12.48 and 16.07 ml (Fig. 1 ▶ c). On comparison with molecular-weight standards, the 16.07 ml peak corresponded to a molecular weight of 7 kDa or a single StCspE molecule (the actual molecular weight of StCspE is 7.7 kDa). The 12.48 ml peak corresponded to the molecular-weight standard chymotrypsinogen (25 kDa) or a hydrodynamic radii of 20.9 Å, suggesting higher order oligomeric species. An StCspE monomer has a hydrodynamic radius of 15.5 Å (Fig. 2 ▶ d) and a dimer has a radius of 22 Å (similar to chymotrypsinogen), suggesting that the 12.48 ml retention peak is equivalent to a StCspE dimer. Only StCspE from the major peak (16.07 ml) was used in crystallization trials. StCspE was more than 95% pure, as judged by SDS–PAGE (Fig. 1 ▶ d), with an experimentally determined molecular weight of 7.7 kDa (data not shown).
Figure 2.

The X-ray crystal structure of StCspE. (a) A three-dimensional ribbon diagram of StCspE, forming a classic five-stranded β-barrel structure. The five β-strands have been coloured to highlight their positions: β-1 (green), β-2 (blue), β-3 (yellow), β-4 (red) and β-5 (magenta). (b) An alternative view (rotated 90° along the y axis) of the same StCspE structure. (c) The amino acids (shown in yellow) which form potential hydrogen bonds (dashed red lines) between the two StCspE monomers, as calculated using the PISA program (Krissinel & Henrick, 2005 ▶). Residues which hydrogen bond to one another are (1) Ala58–Lys15, (2) Asp28–Trp10, (3) Lys9–Asp28, (4) Trp10–Pro61, (5) Asn12–Gly60, (6) Glu46–Lys27 and (7) Glu13–Lys59. (d) Three-dimensional ribbon diagram of StCspE (purple) superimposed onto the EcCspA (green) structure. Loops between β-strands are numbered L1–L4. Loop 4 of StCspE follows a different direction to that in all other CSP structures. The dashed lines show the maximum diameter of a StCspE monomer (measured from the Cα of Gly40 in chain A to the Cα of Ala58 in chain A) and dimer (measured from the Cα of Gly40 in chain A to the Cα of Asn39 in chain B). (e) The electrostatic surface of StCspE (calculated using PyMOL) showing the highly electropositive DNA-binding face.
EMSA were conducted to verify that refolded StCspE had retained DNA-binding activity. A single-stranded 25-mer (ONc; 5′-dATCCTACTGATTGGCCAAGGTGCTG-3′) substrate containing the mammalian Y-box binding motif (bold; Graumann & Marahiel, 1994 ▶), labelled at the 5′-end with γ-32P, was used to examine the DNA-binding activity of refolded StCspE. Fig. 1 ▶(e) shows a gel-shift experiment performed with ONc in the presence of increasing amounts of StCspE. StCspE bound the ssDNA oligonucleotide, indicating that StCspE had correctly refolded during the purification process. The high molar ratio of protein to ssDNA required for complex formation is indicative that StCspE binds relatively weakly to the ATTGG sequence; indeed, this has also been observed for B. subtilis CspB (K d = 5.3 µM at 288 K; Zeeb et al., 2006 ▶).
StCspE crystallized in space group P21 and was refined using data to 1.1 Å resolution (Table 2 ▶). The final model was composed of two molecules of StCspE (Fig. 2 ▶ a; chains A and B). All residues had clearly defined electron density except for the first four N-terminal residues of chain B. The three crystal structures of CSPs (Schindelin et al., 1993 ▶, 1994 ▶; Delbruck et al., 2001 ▶) and the complexes formed with dT6 (Max et al., 2006 ▶, 2007 ▶) superimpose well with StCspE, with average r.m.s. deviation values of 0.63 and 0.67 Å, respectively, for 64 of a possible 67 residues (main-chain atoms). Loop 4 of StCspE was excluded as it adopted an unusual conformation compared with all other CSPs (Fig. 2 ▶ d). Two StCspE molecules in the asymmetric unit pack with their DNA-binding pockets (Zeeb & Balbach, 2003 ▶) face to face, held together by six hydrogen bonds (Fig. 2 ▶ c). Gel-filtration analysis shows that StCspE exists as both a monomer and a higher oligomeric species (likely to be a dimer) in solution (Fig. 1 ▶ c). The homologous CspE from E. coli, which differs in only four amino acids, forms dimers in solution (Johnston et al., 2006 ▶). Therefore, one could presume that the StCspE dimer observed in the asymmetric unit may be equivalent to oligomeric species formed in solution, although this requires further analysis. Interestingly, CspB from B. subtilis (BsCspB) also exists as a crystallographic dimer but with a different intermolecular interface formed through six hydrogen bonds connecting β-strand 4 of each chain (Schindelin et al., 1993 ▶). BsCspB is also a monomer and dimer in solution depending on the concentration of phosphate ions (Makhatadze & Marahiel, 1994 ▶). Variation of the concentration of phosphate ions did not affect the formation of the StCspE dimer in solution (data not shown). Formation of the StCspE dimer via the electropositive DNA-binding face (Fig. 2 ▶ e) is interesting, although further study is required to analyse the oligomerization and ssDNA interaction.
Supplementary Material
PDB reference: cold-shock protein E, 3i2z, r3i2zsf
Footnotes
Recombinant StCspE was numbered as follows: Ser−1, His0, Met1 (start) to Leu69.
References
- Blundell, T. & Johnson, L. (1976). Protein Crystallography. New York: Academic Press.
- Cowtan, K. D. & Zhang, K. Y. (1999). Prog. Biophys. Mol. Biol. 72, 245–270. [DOI] [PubMed]
- Craig, J. E., Boyle, D., Francis, K. P. & Gallagher, M. P. (1998). Microbiology, 144, 697–704. [DOI] [PubMed]
- Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B. III, Snoeyink, J., Richardson, J. S. & Richardson, D. C. (2007). Nucleic Acids Res. 35, W375–W383. [DOI] [PMC free article] [PubMed]
- Delbruck, H., Mueller, U., Perl, D., Schmid, F. X. & Heinemann, U. (2001). J. Mol. Biol. 313, 359–369. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Graumann, P. & Marahiel, M. A. (1994). FEBS Lett. 338, 157–160. [DOI] [PubMed]
- Graumann, P. L. & Marahiel, M. A. (1998). Trends Biochem. Sci. 23, 286–290. [DOI] [PubMed]
- Higgs, P. G. (2001). Q. Rev. Biophys. 33, 199–253. [DOI] [PubMed]
- Horn, G., Hofweber, R., Kremer, W. & Kalbitzer, H. R. (2007). Cell. Mol. Life Sci. 64, 1457–1470. [DOI] [PMC free article] [PubMed]
- Horton, A. J., Hak, K. M., Steffan, R. J., Foster, J. W. & Bej, A. K. (2000). Antonie Van Leeuwenhoek, 77, 13–20. [DOI] [PubMed]
- Jeffreys, A. G., Hak, K. M., Steffan, R. J., Foster, J. W. & Bej, A. K. (1998). Curr. Microbiol. 36, 29–35. [DOI] [PubMed]
- Jiang, W., Hou, Y. & Inouye, M. (1997). J. Biol. Chem. 272, 196–202. [DOI] [PubMed]
- Johnston, D., Tavano, C., Wickner, S. & Trun, N. (2006). J. Biol. Chem. 281, 40208–40215. [DOI] [PubMed]
- Jones, P. G. & Inouye, M. (1994). Mol. Microbiol. 11, 811–818. [DOI] [PubMed]
- Jones, P. G. & Inouye, M. (1996). Mol. Microbiol. 21, 1207–1218. [DOI] [PubMed]
- Kim, B. H., Bang, I. S., Lee, S. Y., Hong, S. K., Bang, S. H., Lee, I. S. & Park, Y. K. (2001). J. Bacteriol. 183, 5580–5588. [DOI] [PMC free article] [PubMed]
- Kloks, C. P., Spronk, C. A., Lasonder, E., Hoffmann, A., Vuister, G. W., Grzesiek, S. & Hilbers, C. W. (2002). J. Mol. Biol. 316, 317–326. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2005). Computational Life Sciences, edited by M. R. Berthold, R. Glen, K. Diederichs, O. Kohlbacher & I. Fischer, pp. 163–174. Berlin/Heidelberg: Springer-Verlag.
- Kunst, F. et al. (1997). Nature (London), 390, 249–256.
- Lopez, M. M. & Makhatadze, G. I. (2000). Biochim. Biophys. Acta, 1479, 196–202. [DOI] [PubMed]
- Lopez, M. M., Yutani, K. & Makhatadze, G. I. (1999). J. Biol. Chem. 274, 33601–33608. [DOI] [PubMed]
- Lopez, M. M., Yutani, K. & Makhatadze, G. I. (2001). J. Biol. Chem. 276, 15511–15518. [DOI] [PubMed]
- Makhatadze, G. I. & Marahiel, M. A. (1994). Protein Sci. 3, 2144–2147. [DOI] [PMC free article] [PubMed]
- Matsumoto, K. & Wolffe, A. P. (1998). Trends Cell Biol. 8, 318–323. [DOI] [PubMed]
- Max, K. E., Zeeb, M., Bienert, R., Balbach, J. & Heinemann, U. (2006). J. Mol. Biol. 360, 702–714. [DOI] [PubMed]
- Max, K. E., Zeeb, M., Bienert, R., Balbach, J. & Heinemann, U. (2007). FEBS J. 274, 1265–1279. [DOI] [PubMed]
- Morgan, H. P., Estibeiro, P., Wear, M. A., Max, K. E., Heinemann, U., Cubeddu, L., Gallagher, M. P., Sadler, P. J. & Walkinshaw, M. D. (2007). Nucleic Acids Res. 35, e75. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Perl, D., Welker, C., Schindler, T., Schroder, K., Marahiel, M. A., Jaenicke, R. & Schmid, F. X. (1998). Nature Struct. Biol. 5, 229–235. [DOI] [PubMed]
- Perrakis, A., Morris, R. & Lamzin, V. S. (1999). Nature Struct. Biol. 6, 458–463. [DOI] [PubMed]
- Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. (2003). Acta Cryst. D59, 1131–1137. [DOI] [PubMed]
- Schindelin, H., Jiang, W., Inouye, M. & Heinemann, U. (1994). Proc. Natl Acad. Sci. USA, 91, 5119–5123. [DOI] [PMC free article] [PubMed]
- Schindelin, H., Marahiel, M. A. & Heinemann, U. (1993). Nature (London), 364, 164–168. [DOI] [PubMed]
- Theobald, D. L., Mitton-Fry, R. M. & Wuttke, D. S. (2003). Annu. Rev. Biophys. Biomol. Struct. 32, 115–133. [DOI] [PMC free article] [PubMed]
- Thieringer, H. A., Jones, P. G. & Inouye, M. (1998). Bioessays, 20, 49–57. [DOI] [PubMed]
- Wang, N., Yamanaka, K. & Inouye, M. (1999). J. Bacteriol. 181, 1603–1609. [DOI] [PMC free article] [PubMed]
- Werner, M. H., Clore, G. M., Gronenborn, A. M., Kondoh, A. & Fisher, R. J. (1994). FEBS Lett. 345, 125–130. [DOI] [PubMed]
- Wolffe, A. P., Tafuri, S., Ranjan, M. & Familari, M. (1992). New Biol. 4, 290–298. [PubMed]
- Zeeb, M. & Balbach, J. (2003). Protein Sci. 12, 112–123. [DOI] [PMC free article] [PubMed]
- Zeeb, M., Max, K. E., Weininger, U., Low, C., Sticht, H. & Balbach, J. (2006). Nucleic Acids Res. 34, 4561–4571. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: cold-shock protein E, 3i2z, r3i2zsf