

A new crystal form of N. europaea hydroxylamine oxidoreductase (space group P21212) diffracted to 2.25 Å resolution at a third-generation synchrotron X-ray source.
Keywords: hydroxylamine oxidoreductase, heme P460
Abstract
Hydroxylamine oxidoreductase (HAO) from Nitrosomonas europaea is a homotrimeric protein that catalyzes the oxidation of hydroxylamine to nitrite. Each monomer, with a molecular weight of 67.1 kDa, contains seven c-type hemes and one heme P460, the porphyrin ring of which is covalently linked to a tyrosine residue from an adjacent subunit. HAO was first crystallized and structurally characterized at a resolution of 2.8 Å in 1997. The structure was solved in space group P63 and suffered from merohedral twinning. Here, a crystallization procedure is presented that yielded untwinned crystals belonging to space group P21212, which diffracted to 2.25 Å resolution and contained one trimer in the asymmetric unit. The unit-cell parameters were a = 140.7, b = 142.6, c = 107.4 Å.
1. Introduction
Hydroxylamine oxidoreductase (HAO) from the autotrophic bacterium Nitrosomonas europaea catalyzes the four-electron oxidation of hydroxylamine (NH2OH) to nitrite (NO2 −) (Hooper & Nason, 1965 ▶). The conversion of ammonia to nitrite via hydroxylamine is rate-limiting in nitrification in most environments and HAO is thus important in the global nitrogen cycling of eutrophic ecosystems (Kowalchuk & Stephen, 2001 ▶). HAO exists as a homotrimer and each monomer of molecular weight 67.1 kDa contains seven c-type hemes as well as a modified c-type heme known as P460 that is the site of catalysis (Hooper, 1978 ▶; Hooper & Terry, 1977 ▶; Terry & Hooper, 1981 ▶). Heme P460 was first characterized from bacterial extracts as a soluble CO-binding heme-like pigment that was primarily associated with HAO and had a ferrous Soret band at 465 nm (Hooper, 1978 ▶; Hooper & Terry, 1977 ▶; Rees & Nason, 1965 ▶; Terry & Hooper, 1981 ▶). The HAO-catalyzed reaction is unusual as it involves the withdrawal of electrons from an iron-coordinated substrate; the only heme in biology known to do this. Using proteolysis/mass spectrometry and NMR, its unusual spectral and redox properties were proposed to be the result of a cross-link between a meso carbon of the porphyrin and an aromatic ring carbon of a tyrosine residue (Arciero et al., 1993 ▶). This cross-link was subsequently observed in the only HAO crystal structure described to date, which was solved in space group P63 to 2.8 Å resolution (Igarashi et al., 1997 ▶). The structure also showed that the porphyrin–tyrosine bond was between adjacent subunits, giving a covalent homotrimeric architecture. The crystals used in the earlier study took 2–3 months to grow and suffered from merohedral twinning. The medium resolution of the data limited the ability of the authors to describe the P460 heme cross-link in detail and did not enable modelling of water molecules. In addition, the 47 C-terminal residues of each subunit (proposed to contain a membrane anchor) were not clearly visible in the electron density of the HAO crystal structure.
Recently, the crystal structure of a small homodimeric protein from N. europea was reported to 1.8 Å resolution (Pearson et al., 2007 ▶). This protein, which can oxidize hydroxylamine, is termed cytochrome P460 and is the only other protein known to contain heme P460 (Erickson & Hooper, 1972 ▶; Miller et al., 1984 ▶; Numata et al., 1990 ▶; Zahn et al., 1994 ▶). In this case the cross-link with a meso carbon of the porphyrin is from the side-chain amine group of a conserved lysine rather than a tyrosine ring carbon as proposed for HAO (Arciero & Hooper, 1997 ▶; Pearson et al., 2007 ▶; Arciero et al., 1993 ▶). However, the different properties of a nitrogen (in cytochrome P460) compared with an aromatic ring carbon (in HAO) led to the suggestion that the HAO cross-link might be chemically more equivalent if it was between the HAO tyrosine hydroxyl and a porphyrin meso carbon. This idea was supported by a serious steric clash in the HAO structural model between the cross-linked tyrosine hydroxyl and atoms of the porphyrin ring (Igarashi et al., 1997 ▶). However, the experimental evidence that the cross-link was with the tyrosine ring carbon was very strong. In particular, NMR analysis of a proteolyzed cross-link-containing fragment could only account for three of the aromatic ring protons of the tyrosine (Arciero et al., 1993 ▶). Owing to the medium resolution of the X-ray diffraction data, the placement of the cross-link in the HAO crystal structure model must have relied heavily on these NMR results. Unfortunately, with only two heme P460-containing proteins known, we can only hope to gain an understanding of the unusual properties of heme P460 if the true nature of the cross-link in both proteins is unequivocally defined. To resolve these issues, a different more highly diffracting crystal form of HAO was needed.
2. Experimental
2.1. Production of hydroxylamine oxidoreductase
HAO was purified from N. europaea according to the procedure described in Arciero et al. (1991 ▶). In brief, suspended N. europaea cells were disrupted by three rounds of freeze–thaw (from 263 to 277 K). The resulting homogenate was centrifuged at 20 000g for 20 min and the supernatant was fractionated with ammonium sulfate. The 60–80% ammonium sulfate precipitate was resuspended in 50 mM potassium phosphate buffer pH 7.5 and then dialyzed against 10 mM potassium phosphate buffer pH 7.5. This was then subjected to two sequential gel-filtration chromatography steps, a Sephadex G-100 column followed by a Sepharose CL-6B column, both of which were equilibrated with 0.2 M KCl, 50 mM potassium phosphate buffer pH 7.5, with the HAO fraction from the first column being concentrated by ultracentrifugation before loading onto the second column. Following this, homogeneous HAO was obtained by ion-exchange chromatography on a DEAE-Sephacel column equilibrated with 20 mM Tris–HCl pH 8.1 and developed with a 0.0–0.3 M NaCl gradient. The protein sample was stored in 1.5 ml microcentrifugation tubes at 193 K for 13 years (the storage buffer is unknown). Prior to crystallization, the protein was buffer-exchanged into 20 mM HEPES–NaOH pH 7.0 using an Amicon Ultra Centrifugal Device (Millipore) with a cutoff of 100 kDa. After the buffer exchange the protein was concentrated to 28 mg ml−1 as determined using Bradford reagent (Sigma).
2.2. Crystallization
High-throughput crystallization trials were performed through the Hauptman–Woodward Medical Research Institute Inc., Buffalo, New York, USA. The initial screen tested 1536 unique conditions using the microbatch-under-oil crystallization method with equal amounts (0.2 µl) of protein and crystallization cocktail at room temperature. The crystallization condition that was chosen for subsequent optimization contained 0.1 M potassium nitrate, 0.1 M MES pH 6 and 40%(v/v) PEG 400. Optimization of the pH and the PEG 400 concentration around this condition was performed at 293 K in a 72-Well Microbatch Plate (Hampton Research) with equal amounts (2 µl) of protein and crystallization cocktail under 10 µl paraffin oil (Hampton Research). A single crystal was washed in two successive 8 µl drops of the crystallization solution and then flash-frozen in liquid nitrogen in a 0.2–0.3 mm CryoLoop mounted on a CrystalCap Magnetic base (both from Hampton Research). No additional cryoprotectant was needed.
2.3. X-ray diffraction data collection, processing and analysis
X-ray diffraction data collection was carried out on the Structural Biology Center (SBC) beamline 19-ID-D at the Advanced Photon Source (APS), Argonne National Laboratory, Argonne, Illinois, USA. 120° of data were collected from a single crystal using an ADSC CCD detector and a 100 × 100 µm beam. Data collection was performed at cryogenic temperatures (100 K). The data were processed with HKL-2000 (Otwinowski & Minor, 1997 ▶). Analysis of the diffraction data was carried out using the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results
3.1. Crystallization
The final crystallization condition that produced the crystals used for X-ray diffraction data collection contained 0.1 M potassium nitrate, 0.1 M MES pH 7.5 and 46%(v/v) PEG 400. Small plate-shaped crystals appeared within a matter of days and grew to a final length of ∼200 µm within two weeks (Fig. 1 ▶).
Figure 1.

Crystals of HAO grown by the microbatch-under-oil method.
3.2. Preliminary X-ray diffraction studies
The HAO crystal diffracted to 2.25 Å resolution at the APS. A diffraction image is shown in Fig. 2 ▶. The data were indexed as orthorhombic and the systematic absences present in the diffraction pattern indicated that the crystal belonged to space group P21212. The unit-cell parameters were refined to a = 140.7, b = 142.6, c = 107.4 Å. The X-ray data-collection and processing statistics are summarized in Table 1 ▶.
Figure 2.

Part of an X-ray diffraction image from the HAO crystal used for data collection. Red arcs indicate diffraction resolution.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Space group | P21212 |
| Unit-cell parameters (Å) | |
| a | 140.7 |
| b | 142.6 |
| c | 107.4 |
| Wavelength (Å) | 0.97948 |
| Resolution range (Å) | 50–2.25 (2.33–2.25) |
| No. of unique reflections | 92971 (7287) |
| Completeness (%) | 90.8 (72.0) |
| Redundancy | 4.8 (4.6) |
| Rmerge† | 0.115 (0.406) |
| Mean I/σ(I) | 10.5 (3.9) |
R
merge = 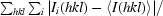
 , where Ii(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity of reflection hkl.
, where Ii(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity of reflection hkl.
Cell-content analysis indicated that three monomers (one HAO homotrimer) were present in the asymmetric unit, with a solvent content of 54% (Matthews, 1968 ▶). Unlike the previous HAO data in space group P63, no merohedral twinning was detected in the P21212 crystal form. The estimated mosaicity of the scaled data was 0.43° and the average B factor derived from a Wilson plot was 36.8 Å2 (French & Wilson, 1978 ▶). Efforts are now under way to solve the P21212 structure of HAO by molecular replacement using the P63 HAO structural model (PDB code 1fgj; Igarashi et al., 1997 ▶). This should enable resolution of the conundrum regarding the true structure of the heme P460–tyrosine cross-link in HAO.
Acknowledgments
X-ray data were collected at the Argonne National Laboratory Structural Biology Center at the Advanced Photon Source (APS). Argonne is operated by UChicago Argonne LLC for the US Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. We thank Stephan L. Ginell PhD and the staff of SBC-CAT beamline 19-ID for valuable assistance during X-ray data collection and processing. In-house X-ray data were collected at the Kahlert Structural Biology Laboratory at The University of Minnesota, and we thank Ed Hoeffner for his support. Computer resources were provided by the Basic Sciences Computing Laboratory of the University of Minnesota Supercomputing Institute and we thank Can Ergenekan for his support. This research was supported by an Office of the Dean of the Graduate School of the University of Minnesota grant to ABH and a Minnesota Partnership for Biotechnology and Medical Genomics Grant SPAP-05-0013-P-FY06 to CMW.
References
- Arciero, D. M., Balny, C. & Hooper, A. B. (1991). Biochemistry, 30, 11466–11472. [DOI] [PubMed]
- Arciero, D. M. & Hooper, A. B. (1997). FEBS Lett.410, 457–460. [DOI] [PubMed]
- Arciero, D. M., Hooper, A. B., Cai, M. & Timkovich, R. (1993). Biochemistry, 32, 9370–9378. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Erickson, R. H. & Hooper, A. B. (1972). Biochim. Biophys. Acta, 275, 231–244. [DOI] [PubMed]
- French, S. & Wilson, K. (1978). Acta Cryst. A34, 517–525.
- Hooper, A. B. (1978). Microbiology – 1978, edited by D. Schlessinger, pp. 299–304. Washington DC: American Society for Microbiology.
- Hooper, A. B. & Nason, A. (1965). J. Biol. Chem.240, 4044–4057. [PubMed]
- Hooper, A. B. & Terry, K. R. (1977). Biochemistry, 16, 455–459. [DOI] [PubMed]
- Igarashi, N., Moriyama, H., Fujiwara, T., Fukumori, Y. & Tanaka, N. (1997). Nature Struct. Biol.4, 276–284. [DOI] [PubMed]
- Kowalchuk, G. A. & Stephen, J. R. (2001). Annu. Rev. Microbiol.55, 485–529. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Miller, D. J., Wood, P. M. & Nicholas, D. J. D. (1984). J. Gen. Microbiol.130, 3049–3054.
- Numata, M., Saito, T., Yamazaki, T., Fukumori, Y. & Yamanaka, T. (1990). J. Biochem.108, 1016–1021. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pearson, A. R., Elmore, B. O., Yang, C., Ferrara, J. D., Hooper, A. B. & Wilmot, C. M. (2007). Biochemistry, 46, 8340–8349. [DOI] [PMC free article] [PubMed]
- Rees, M. & Nason, A. (1965). Biochem. Biophys. Res. Commun.21, 248–256. [DOI] [PubMed]
- Terry, K. R. & Hooper, A. B. (1981). Biochemistry, 20, 7026–7032. [DOI] [PubMed]
- Zahn, J. A., Duncan, C. & DiSpirito, A. A. (1994). J. Bacteriol.176, 5879–5887. [DOI] [PMC free article] [PubMed]