

The enzyme glutamine synthetase from M. truncatula has been expressed, purified and crystallized. The crystals belonged to the monoclinic space group P21 and diffracted to 2.35 Å resolution.
Keywords: nitrogen fixation, glutamine synthetase, Leguminosae, Medicago truncatula
Abstract
The condensation of ammonium and glutamate into glutamine catalyzed by glutamine synthetase (GS) is a fundamental step in nitrogen metabolism in all kingdoms of life. In plants, this is preceded by the reduction of inorganic nitrogen to an ammonium ion and therefore effectively articulates nitrogen fixation and metabolism. Although the three-dimensional structure of the dodecameric bacterial GS was determined quite some time ago, the quaternary architecture of the plant enzyme has long been assumed to be octameric, mostly on the basis of low-resolution electron-microscopy studies. Recently, the crystallographic structure of a monocotyledonous plant GS was reported that revealed a homodecameric organization. In order to unambiguously establish the quaternary architecture of GS from dicotyledonous plants, GS1a from the model legume Medicago truncatula was overexpressed, purified and crystallized. The collection of synchrotron diffraction data to 2.35 Å resolution allowed the determination of the three-dimensional structure of this enzyme by molecular replacement.
1. Introduction
Glutamine synthetase (GS; EC 6.3.1.2) is a central enzyme in nitrogen metabolism in all organisms; an in-depth understanding of the molecular details of its activity is therefore of crucial importance. GS catalyses the ATP-dependent synthesis of glutamine from ammonium ions and glutamate, which is the origin of essentially all nitrogenous compounds in the cell. In the case of plants, inorganic nitrogen is first reduced to ammonium ions before GS-catalysed incorporation into glutamine, thereby entering plant metabolism and becoming the origin of other organic forms of nitrogen; this ultimately represents the major route of entry of organic nitrogen for all animals. In plants, GS exists as a number of isoenzymes that are encoded by a small multigene family, generally with a single member encoding a plastid-located isoenzyme (GS2) and several genes coding for a number of cytosolic isoenzymes (GS1) ranging from two (GS1a and GS1b) in Medicago truncatula (Carvalho et al., 1997 ▶) to six in Zea mays (Li et al., 1993 ▶). The isoenzymes differ in catalytic properties and in subunit size, ranging from 39 kDa for the cytosolic forms to 42 kDa for the plastidic isoenzyme (Hirel & Lea, 2001 ▶), but very little is known about the structural determinants of the functional differences that are observed between the different isoforms of plant GS.
Three types of GS molecules, type I (GSI), type II (GSII) and type III (GSIII), have been described in living organisms based on molecular mass, quaternary structure, and gene sequence (Woods & Reid, 1993 ▶). GSIs are dodecameric prokaryotic enzymes with subunit molecular masses ranging from 44 to 60 kDa. GSIIs are found in eukaryotes and in a few soil-dwelling bacteria (belonging to the Rhizobiaceae, Frankiaceae and Streptomycetaceae families) and until recently were considered to be octameric enzymes composed of 35–50 kDa subunits (Eisenberg et al., 2000 ▶). Finally, GSIIIs have been identified in cyanobacteria (Reyes & Florencio, 1994 ▶) and two unrelated anaerobic bacteria (Goodman & Woods, 1993 ▶; Southern et al., 1986 ▶); although they were initially described as hexamers with 75–83 kDa subunits (Reyes & Florencio, 1994 ▶), a recent single-particle reconstruction of a type III glutamine synthetase revealed a dodecameric organization with many similarities to GSI (van Rooyen et al., 2006 ▶). While the three classes are structurally related, they differ significantly at the amino-acid sequence level, as well as in their mechanisms of regulation and sensibility to feedback inhibitors (Eisenberg et al., 2000 ▶).
Although glutamine synthetase was first purified and characterized from plants, the first GS structure to be determined was that of a prokaryotic GSI (PDB entry 2gls; Almassy et al., 1986 ▶; Yamashita et al., 1989 ▶) and many of the structural features of plant GS have been inferred from those of the bacterial enzyme (Eisenberg et al., 2000 ▶). The three-dimensional structure of GSI revealed a homododecamer, with 12 identical 52 kDa subunits organized in two stacked hexameric rings, an architecture that was also shared by the mycobacterial enzyme (PDB entry 1hto; Gill et al., 2002 ▶). The active sites of GS are formed at the interface between the N- and C-terminal domains of adjacent subunits (Almassy et al., 1986 ▶). Based on the sequence homology between the bacterial and eukaryotic enzymes, Eisenberg and coworkers suggested that the folding and packing of the subunits of the plant GS would be similar to those of the bacterial enzyme, proposing an octameric quaternary architecture with two tetrameric layers, each composed of two active-site-forming subunit pairs (Eisenberg et al., 1987 ▶). Early electron-microscopy studies of the cytosolic dicotyledonous GS enzymes from soybean (McParland et al., 1976 ▶) and lupin root nodules (Tsuprun et al., 1987 ▶) seemed to corroborate this hypothesis by reporting a cubic configuration for plant GS, with eight subunits arranged in two sets of planar tetramers. Recently, the structure of the cytosolic monocotyledonous GS1a isoenzyme from maize (PDB entry 2d3a) was determined by X-ray crystallography (Unno et al., 2006 ▶), revealing a decameric structure composed of two face-to-face pentameric rings with ten active sites formed at the interface between neighbouring subunits, a finding that was corroborated by the three-dimensional structure of mammalian GS (PDB entries 2ojw and 2uu7; Krajewski et al., 2008 ▶) and yeast GS (PDB entry 3fky; He et al., 2009 ▶). However, the report of an octameric organization for the cytosolic GS from Phaseolus vulgaris (Llorca et al., 2006 ▶) suggested that the quaternary organization of glutamine synthetases may differ between monocotyledonous and dicotyledonous plants.
In order to unambiguously assign the quaternary structure of glutamine synthetase from dicotyledonous plants, we engaged in determination of the three-dimensional structure of cytosolic GS isoform 1a from the model legume Medicago truncatula. Here, we report the overexpression, purification, crystallization and preliminary crystallographic analysis of GS1a from this organism.
2. Materials and methods
2.1. Purification of recombinant M. truncatula GS1a
The cDNA fragment coding for M. truncatula GS1a was removed from vector pTrc99A (Carvalho et al., 1997 ▶) using restriction enzymes NcoI and PstI and cloned into the (blunt) NheI site of pET28a (Novagen). The expression construct was confirmed by restriction analysis and DNA sequencing. The resulting pET28a-GS1a plasmid encoded an N-terminally His6-tagged fusion protein in which the sequence MGSSHHHHHHSSGLVPRGSHMAS precedes that of the full-length GS1a from M. truncatula. Expression in Escherichia coli BL21 (DE3) cells harbouring the pET28a-GS1a expression plasmid was induced with IPTG (final concentration 1 mM) at mid-exponential growth (OD600 = 0.5) and proceeded for 3–5 h at 310 K. The cells were harvested by centrifugation at 2800g, resuspended in 0.02 M potassium phosphate pH 7.4, 0.01 M magnesium sulfate, 0.005 M glutamate, 0.5 M NaCl, 0.02 M imidazole (buffer A), disrupted by sonication and centrifuged (60 min, 36 700g, 277 K) to remove cell debris. The crude protein extract was filtered through a 5 µm low-protein-binding filter and loaded onto a 5 ml Ni Sepharose column (GE Healthcare) equilibrated with buffer A. Elution of the bound fusion protein was achieved with buffer A supplemented with 0.23 M imidazole. GS1a-containing fractions were pooled and dialyzed against 0.02 M potassium phosphate pH 7.4, 0.005 M glutamate, 0.01 M magnesium sulfate and further purified by size-exclusion chromatography on a Sephacryl S-300 16/60 column (GE Healthcare) equilibrated in the same buffer. The fractions containing purified GS1a were pooled and concentrated to 8 mg ml−1 on a centrifugal concentration device with a 10 kDa molecular-weight cutoff membrane.
2.2. Crystallization, data collection and processing
2.2.1. GS1a crystallization
Initial crystallization conditions were screened at 293 K using the sitting-drop geometry with commercial sparse-matrix crystallization screens from Hampton Research and Molecular Dimensions. A condition that yielded microcrystals was identified [0.2 M triammonium citrate pH 7.0, 20%(w/v) polyethylene glycol 3350] and subjected to optimization by fine-grid screening. The best crystals were obtained at 287 K from sitting drops composed of 2 µl GS1a solution (8 mg ml−1 in 0.02 M potassium phosphate pH 7.4, 0.005 M glutamate, 0.01 M MgSO4) and 2 µl precipitant solution [0.12 M triammonium citrate pH 7.0, 9.2%(w/v) polyethylene glycol 3350] equilibrated against a 300 µl reservoir. The crystals were transferred sequentially to mother liquor with increasing concentrations [up to 25%(w/v)] of polyethylene glycol 3350 for a few seconds and flash-cooled by plunging them into liquid nitrogen.
2.2.2. Data collection and processing
X-ray diffraction data extending to 2.35 Å resolution were collected from a single crystal at 100 K using an ADSC Q4 detector on beamline ID14EH2 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France). Two data sets were collected in 1° oscillation steps over a range of 200° with a 350 mm sample-to-detector distance and 1 s exposure per frame for the low-resolution data set (extending to 3.5 Å) and in identical steps over a range of 360° with a 225 mm sample-to-detector distance and 6 s exposure per frame for the high-resolution data set (to the diffraction limit of the crystal). The recorded diffraction data were processed with MOSFLM (Leslie, 1991 ▶) and SCALA from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
2.3. Structure solution
The structure was solved by the molecular-replacement method with Phaser (McCoy et al., 2007 ▶) from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶), using the model of Z. mays GS1a (87% sequence identity; PDB entry 2d3a; Unno et al., 2006 ▶) with all nonglycine non-identical residues truncated to alanine as the search model.
3. Results and discussion
3.1. Protein expression and purification
Soluble and active M. truncatula glutamine synthetase (GS1a) was overexpressed in E. coli (data not shown) as an N-terminal fusion with a thrombin-cleavable hexahistidine tag. A simple two-step purification protocol allowed the recovery of 15% of the expressed GS1a, amounting to ∼30 mg of purified protein per litre of culture. This procedure yielded ∼96% pure recombinant protein, as judged by densitometric analysis of an SDS–PAGE separated sample (Fig. 1 ▶).
Figure 1.

Coomassie Blue-stained 12.5% SDS–PAGE of the purified recombinant GS1a from M. truncatula used for crystallization trials. Lane 1, molecular-mass markers. The apparent molecular mass (in kDa) of the marker proteins is given on the left. Lanes 2–5 contained 1, 2.5, 5 and 10 µg of recombinant GS1a, respectively.
3.2. Crystallization of GS1a
Crystals of recombinant M. truncatula GS1a grew to maximum dimensions of 0.4 × 0.15 × 0.1 mm (Fig. 2 ▶) and diffracted to 2.35 Å resolution on a synchrotron source. However, the GS1a crystals displayed a widely variable diffracting power and less than 2% of the large number of crystals screened yielded usable data.
Figure 2.

Single crystals of native GS1a from M. truncatula belonging to the monoclinic space group P21.
The data-collection and processing statistics are summarized in Table 1 ▶. The largest peaks (besides the origin) on a self-rotation function calculated with MOLREP (Vagin & Teplyakov, 1997 ▶) were found on both the κ = 72° and κ = 180° sections, indicating the presence of both fivefold and twofold local symmetry, which is consistent with the decameric arrangement of the enzyme assumed in calculating the value of the Matthews coefficient indicated in Table 1 ▶ (Matthews, 1968 ▶).
Table 1. Statistics of diffraction data collection.
Values in parentheses are for the outermost shell.
| X-ray source | ESRF ID14EH2 |
| Wavelength (Å) | 0.933 |
| Resolution range (Å) | 101–2.35 (2.48–2.35) |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 99.3, b = 101.7, c = 188.1, β = 103.7 |
| No. of observations (total/unique) | 935718/149655 |
| Multiplicity | 6.3 (5.6) |
| Rmerge† (%) | 7.7 (33.8) |
| Rp.i.m.‡ (%) | 3.1 (15.3) |
| Completeness (%) | 99.0 (96.8) |
| Mean I/σ(I) | 19.2 (4.0) |
| Mathews coefficient (Å3 Da−1) | 2.2 |
| Solvent content (%) | 44.7 |
| Wilson B factor (Å2) | 45.6 |
R
merge = 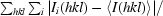
 , where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple observations of symmetry-related reflections.
, where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple observations of symmetry-related reflections.
R
p.i.m. = 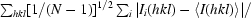 , where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple observations of symmetry-related reflections.
, where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity of multiple observations of symmetry-related reflections.
3.3. Structure solution
The molecular coordinates of the monomer of glutamine synthetase from Z. mays (PDB entry 2d3a; Unno et al., 2006 ▶) were used as a search model to solve the structure by the molecular-replacement method. The program Phaser (McCoy et al., 2007 ▶) located ten subunits of M. truncatula GS1a in the asymmetric unit (translation function Z score of 53.4 for the tenth monomer, with a log-likelihood gain of 10 188) arranged as two superposed (face-to-face) pentameric rings resembling the supramolecular arrangement of monocotyledonous GS (Unno et al., 2006 ▶). The three-dimensional model is currently under refinement.
Acknowledgments
We acknowledge the ESRF for the provision of synchrotron-radiation facilities and the ESRF staff for assistance in using beamline ID14EH2. This work was funded in part by Fundação para a Ciência e a Tecnologia, Portugal, through grants POCI/AGR/61025/2004 and REEQ/564/BIO/2005 (EU-FEDER and POCI 2010).
References
- Almassy, R. J., Janson, C. A., Hamlin, R., Xuong, N.-H. & Eisenberg, D. (1986). Nature (London), 323, 304–309. [DOI] [PubMed]
- Carvalho, H., Sunkel, C., Salema, R. & Cullimore, J. V. (1997). Plant Mol. Biol.35, 623–632. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Eisenberg, D., Almassy, R. J., Janson, C. A., Chapman, M. S., Suh, S. W., Cascio, D. & Smith, W. W. (1987). Cold Spring Harb. Symp. Quant. Biol.52, 483–490. [DOI] [PubMed]
- Eisenberg, D., Gill, H. S., Pfluegl, G. M. & Rotstein, S. H. (2000). Biochim. Biophys. Acta, 1477, 122–145. [DOI] [PubMed]
- Gill, H. S., Pfluegl, G. M. & Eisenberg, D. (2002). Biochemistry, 41, 9863–9872. [DOI] [PubMed]
- Goodman, H. J. & Woods, D. R. (1993). J. Gen. Microbiol.139, 1487–1493. [DOI] [PubMed]
- He, Y. X., Gui, L., Liu, Y. Z., Du, Y., Zhou, Y., Li, P. & Zhou, C. Z. (2009). Proteins, 76, 249–254. [DOI] [PubMed]
- Hirel, B. & Lea, P. J. (2001). Plant Nitrogen, edited by P. J. Lea & J.-F. Morot-Gaudry, pp. 79–99. Berlin: Springer-Verlag.
- Krajewski, W. W., Collins, R., Holmberg-Schiavone, L., Jones, T. A., Karlberg, T. & Mowbray, S. L. (2008). J. Mol. Biol.375, 217–228. [DOI] [PubMed]
- Leslie, A. G. W. (1991). Crystallographic Computing 5: From Chemistry to Biology, edited by D. Moras, A. D. Podjarny & J. C. Thierry, pp. 27–38. Oxford University Press.
- Li, M. G., Villemur, R., Hussey, P. J., Silflow, C. D., Gantt, J. S. & Snustad, D. P. (1993). Plant Mol. Biol.23, 401–407. [DOI] [PubMed]
- Llorca, O., Betti, M., Gonzalez, J. M., Valencia, A., Marquez, A. J. & Valpuesta, J. M. (2006). J. Struct. Biol.156, 469–479. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst.40, 658–674. [DOI] [PMC free article] [PubMed]
- McParland, R. H., Guevara, J. G., Becker, R. R. & Evans, H. J. (1976). Biochem. J.153, 597–606. [DOI] [PMC free article] [PubMed]
- Reyes, J. C. & Florencio, F. J. (1994). J. Bacteriol.176, 1260–1267. [DOI] [PMC free article] [PubMed]
- Rooyen, J. M. van, Abratt, V. R. & Sewell, B. T. (2006). J. Mol. Biol.361, 796–810. [DOI] [PubMed]
- Southern, J. A., Parker, J. R. & Woods, D. R. (1986). J. Gen. Microbiol.132, 2827–2835. [DOI] [PubMed]
- Tsuprun, V. L., Mesyanzhinova, I. V., Milgrom, Y. M. & Kalashnikova, T. (1987). Biochim. Biophys. Acta, 892, 130–137. [DOI] [PubMed]
- Unno, H., Uchida, T., Sugawara, H., Kurisu, G., Sugiyama, T., Yamaya, T., Sakakibara, H., Hase, T. & Kusunoki, M. (2006). J. Biol. Chem.281, 29287–29296. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- Woods, D. R. & Reid, S. J. (1993). FEMS Microbiol. Rev.11, 273–283. [DOI] [PubMed]
- Yamashita, M. M., Almassy, R. J., Janson, C. A., Cascio, D. & Eisenberg, D. (1989). J. Biol. Chem.264, 17681–17690. [DOI] [PubMed]