

Abstract
The epidermis is a stratified epithelium which develops depending on the transcription factor p63, a member of the p53 family of transcription factors. p63 is strongly expressed in the innermost basal layer where highly proliferative epithelial cells reside. p63 functions as a molecular switch that initiates epithelial stratification or cell fate determination while regulating proliferation and differentiation of developmentally mature keratinocytes. p63 acts upstream of Dlx3 homeobox gene in a transcriptional regulatory pathway relevant to ectodermal dysplasia. Here we show that Dlx3 triggers p63 protein degradation by a proteasome-dependent pathway. Mutant ΔNp63α in which Threonine397 and Serine383 were replaced with Alanine as well as C-terminal truncated versions of ΔNp63α are resistant to Dlx3-mediated degradation. Transient expression of Dlx3 is associated with Raft phosphorylation. Dlx3 is unable to promote p63 degradation in Raft depleted MEF cells or upon pharmacological knockdown of Raft. Our data support a previously unrecognized role for Dlx3 in posttranslational regulation of ΔNp63α protein level, a mechanism that may contribute to reduce the abundance of ΔNp63α during differentiation of stratified epithelia.
Keywords: Dlx3, homeodomain protein, p53 homolog, keratinocyte differentiation, protein degradation, Raf1, protein phosphorylation
Introduction
The epidermis is a stratified epithelium which develops depending on the transcription factor p63, a member of the p53 family of transcription factors. p63 is strongly expressed in the innermost basal layer where highly proliferative epithelial cells reside. p63, is believed to function as a molecular switch that initiates epithelial stratification or cell fate determination in developing tissues, 1 while regulating proliferation and/or differentiation of developmentally mature keratinocytes.2 p63 is expressed as six distinct isoforms due to two alternative transcription start sites and splicing at the C-terminus. Transactivating (TA) isoforms contain an amino-terminal exon that encodes a p53-like transactivation domain whereas ΔNp63 isoforms lack this domain but contain the common DNA binding (DB) and oligomerization (00) domains. In addition, alternative splicing at the 3′ end generates TA and ΔNp63 proteins with different C-termini, denoted α, β and γ.3 Δp63α is the most abundant p63 isotype in the proliferating basal layer of many epithelial tissues. In mouse keratinocytes, multiple p63 isotypes (ΔN and TA) are expressed and differentially modulated during differentiation. In adult, ilNp63a is abundant in basal cells and decreases in the suprabasal layer of stratified epidermis while a smaller ΔN-form, reputed to be ΔNp63γ persists, suggesting unique functions for the two AN-forms.4 Forced expression of ΔNp63α in primary murine keratinocytes inhibits morphological differentiation induced by elevated extracellular Ca2+, abrogates Ca2+ -induced growth arrest and blocks expression of maturation-specific proteins keratin 10 and filaggrin.5
The Dlx3 homeodomain protein functions as a transcriptional activator expressed in placenta, bone, suprabasal layers of stratified epidermis and ectodermal appendages such as tooth and hair follicle.6 Targeted deletion of mouse Dlx3 gene is lethal because of placental defects.7 In cultured keratinocytes Dlx3 can be induced by treatment with 0.12 mM Ca2+.8 Transgenic misexpression of Dlx3 in the basal proliferative layer ofepidermis induces cell cycle arrest and leads to premature terminal differentiation.9
Increase in extracellular Ca2+ concentration is an important trigger of skin differentiation and it is involved in the formation of the spinous and granular layers. and epidermal barrier.10,11 Ca2+ addition to primary keratinocytes induces DIx3 gene transcription. Furthermore, we have demonstrated that TAp63α, but not ΔNp63α, is able to transactivate DIx3 gene promoter.12
It is well supported that ΔNp63α is required for the maintenance of the proliferative potential of basal keratinocytes and it is downregulated during keratinocyte differentiation.13 Accordingly, ΔNp63α overexpression in head and neck squamous carcinoma is associated with increased proliferation.14 The stability of p63 protein is regulated by post-translational modifications such as phosphorylation, ubiquitin and ubiquitin-like modifications.15,16,17 UV-B irradiation mediates phosphorylation-dependent degradation of ΔNp63α protein in mouse primary epidermal keratinocytes18 while TAp63α and γ proteins accumulate following genotoxic stress agents such as actinomycin D, bleomycin, etoposide and UV.16 In addition, it has been shown that p53 associates with ΔNp63α inducing its degradation through a caspase-dependent mechanism.19 Both ubiquitination and sumoylation have been reported to regulate p63 protein levels. The E3 ubiquitin ligases NEDD420 and ITCH/AIP417 can promote ubiquitination and degradation of p63. On the other hand, Ubc9 was found to associate with the C-terminal domain of p63α and catalyze SUMO-I conjugation at K637, impairing p63 stability and transcriptional activity. 15 Notwithstanding the progress in the knowledge of the mechanisms controlling p63 protein turnover, the association of these mechanisms with intrinsic signals governing coordinated changes in p63 protein levels during epidermal differentiation is largely unknown. In the present study, we demonstrate that the Dlx3 homeodomain protein can trigger proteasome-dependent p63 protein degradation. Because Dlx3 is unable to promote p63 degradation in Raf1 depleted MEF cells or upon pharmacological Raft knockdown, we suggest that Dlx3 can promote p63 protein degradation through a Raft-dependent pathway. This previously unrecognized mechanism may play a relevant role during differentiation of stratified epithelia.
Results
Dlx3 downregulates p63 protein level
Primary and immortalized keratinocytes express abundant ΔNp63α protein. We previously reported that Ca2+ addition to primary keratinocytes resulted in induction of Dlx3 gene expression.10 Surprisingly, transfection of Dlx3 in human immortalized HaCaT keratinocytes caused a remarkable decrease of endogenous ΔNp63α protein (Figure la). A similar effect was observed in PAM 212 keratinocytes, a naturally immortalized murine keratinocyte cell line, that express Dlx3 upon addition of Doxycycline (Figure lb), To probe the effect of DIx3 on ΔNp63α protein in a more physiological context, we developed siRNA duplexes that rapidly and efficiently downregulate Dlx3 in HaCaT cells. HaCaT cells seeded at 60% cell density and cultured in 0.45 mMCa2+ /Chelex-FBS do not express Dlx3. Induction of Dlx3 gene expression was evident upon addition of Ca2+ to a final concentration of 1.8 mM (Figure 1c, left panel). HaCaT cells, cultured in 1.8 mM Ca2+, were then transfected with scrambled or Dlx3 siRNA oligonucleotides. Figure 1c, right panel, clearly shows that reduction of endogenous Dlx3 protein resulted in a reproducible increase of ΔNp63α protein level, confirming that basal Dlx3 levels are important in controlling basal ΔNp63α levels.
Figure 1. DlKJ reduces endogenous ΔNp63α protein in keratinocytes.
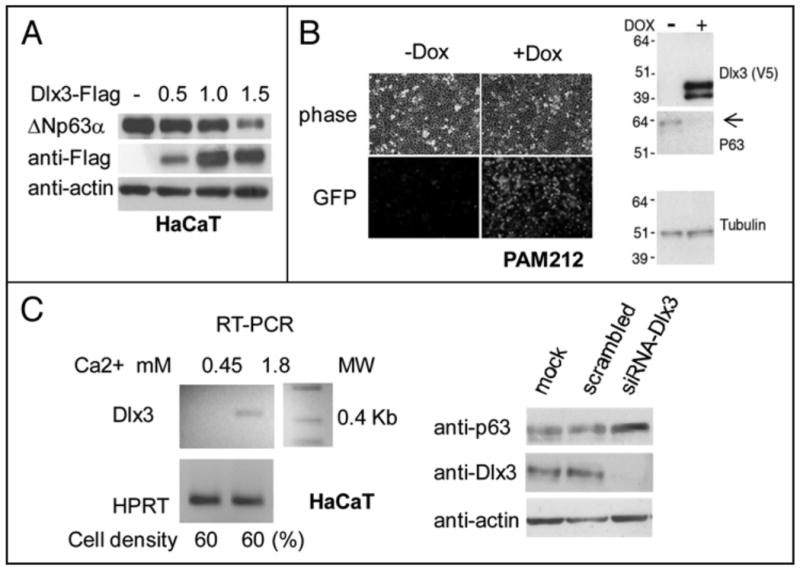
a) HaCaT cells transiently transfected with 0.5, 1.0 and 1.5 μg of Dlx3-Flag expression vector. Equal amounts of total protein (40 μg) were subjected to Western blot analysis for p63, Dlx3 (anti-Flag) and actin. b) Pictures showing OFP expression in PAM212-TetOn-V5Dlx3/GFP cells grown with (+ Dox) or without (- Dox) doxycycline for 24 hours. Phase views (upper panel) are shown to visualize cell density. Western blot of 20 μg of PAM212 nuclear extracts + and - Dox revealed with antibodies against Dlx3, p63 and tubulin. c) (Left panel) Total RNA was prepared from HaCaT cells seeded at 60% confluency (2.3 × 105) in 60 mM dishes and cultured for 16 hours in 0.45 or 1.8 mM Ca2+. Dlx3 mRNA levels were determined by RT-PCR with primers specific for human Dlx3 mRNA. Values were normalized for HPRT (Hypoxanthine-guanine phospporibosyl transferase) mRNA level. (Right panel) HaCaT cells seeded at 60% confluency in 1.8 mM Ca2+ were transfected with scrambled oligonucleotides or Dlx3 siRNA) NA oligonucleotides (10 nM). 24 hours later, cells extracts were analyzed by Western blot using antibodies against p63 and Dlx3. Actin was used as loading control. MW molecular weight marker.
To explore the mechanism behind the above observations, we used Saos2 cells, expressing no endogenous p63. As shown in Figure 2a, enforced expression of Dlx3 in Saos2 cells led to a marked reduction of transfected ΔNp63α protein level.
Figure 2. Dlx3 downregulates p63 at protein level.
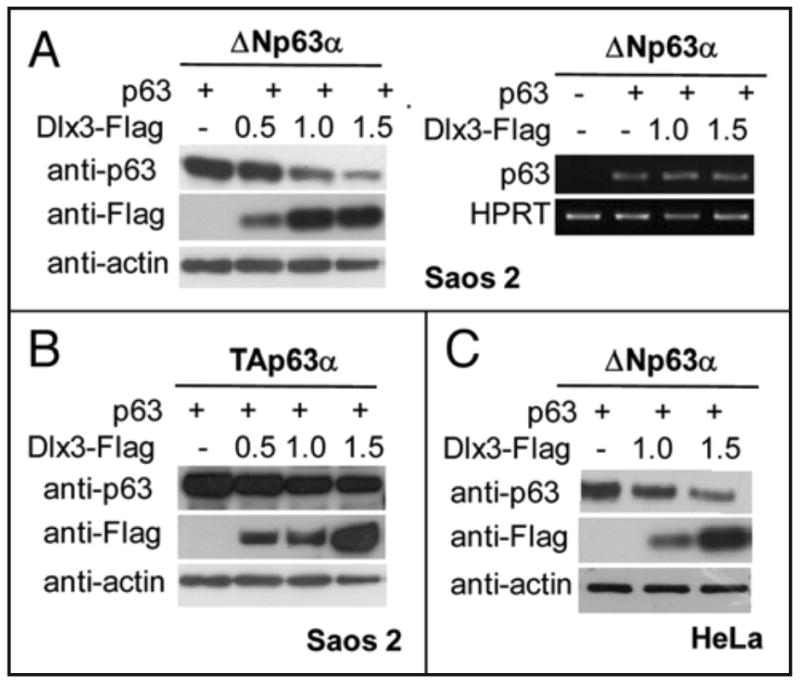
a) Equal amounts of mock transfected Saos2 cells and cells transfected with ΔNp63α alone or with Dlx3 (1.0 and 1.5 μg) were subjected to Western blot analysis for p63, Dlx3 (Flag) and actin (left panel). Equal aliquots of cells were used for total RNA extraction and subjected to RT-PCR analysis with p63 specific primers. Quantification of the p63 specific RT-PCR bands was performed using densitometry scanning and normalized respect to the HPRT internal control (right panel). b) Saos2 cells were transfected with TAp63α (0.2 μg) alone or with 0.5, 1.0 and 1.5 μg of Dlx3-Flag expression vector. Equal amounts of total protein (40 μg) were subjected to Western blot analysis and revealed for p63, Dlx3(Flag) and actin. c) HeLa cells were transfected with 1.0 and 1.5 μg of Dlx3-Flag expression vector. Equal amounts of total protein (40 μg) were subjected to Western blot analysis for p63, Dlx3 (Flag). Actin was used as a loading control.
Semiquantitative RT-PCR analysis on samples derived from cells that had been transfected with ΔNp63α alone or with Dlx3 vector showed that the abundance of ΔNp63α transcript was not altered by Dlx3 coexpression, suggesting that Dlx3 was affecting p63 protein stability (Figure 2b). Interestingly, TAp63α protein was much more resistant to Dlx3-mediated degradation (Figure 2c). Similar results were obtained in HeLa cells and mouse embryonic fibroblasts (MEFs) deleted for both copies of the Mdm2 and p53 genes (p53-/- mdm2-/-) (Figure 2d and data not shown), indicating that the mechanism responsible for the reduction of ∼p63a, following Dlx3 expression, was not keratinocyte-specific and p53/MDM2-independent.
Dlx3-induced ΔNp63 degradation requires specific residues located in the p63 α and β carboxyterminal tails
Although ΔNp63α is the most abundant p63 isoform in adult skin, we investigated whether Dlx3 could also reduce the level of the other ΔNp63α isoforms. Thus, we transfected ΔNp63α, β or γ in Saos2 cells, along with increasing amount of Dlx3. Interestingly, the levels of ΔNp63α and β, but not γ, were significantly reduced by Dlx3 (Figure 3a). These results indicated that aminoacid residues located in the β and α, but not γ tail, confer p63 protein sensitivity to the degradation mechanism dependent on Dlx3 expression. To identify these residues we used two constructs expressing carboxyterminal truncated ΔNp63 proteins, named ΔNp63Δ373 and Δ408 (a schematic representation is shown in Figure 3b). Each of these constructs was transfected with and without Dlx3 and compared with wild type ΔNp63α for sensitivity to Dlx3-mediated degradation. The ΔNp63αΔ373 protein level was almost unaffected by Dlx3, while ΔNp63αΔ408 was efficiently degraded (Figure 3c). Thus, we concentrated on the region encompassing aminoacid 373 to 408 of ΔNp63. By NetPhos 2.0 bioinformatic analysis we identified two potential phosphoacceptor site/s located in this region, a Serine at position 383 (score 0.87) and a Threonine at position 397 (score 0.74). Remarkably, Serine383 and Threonine397 belong to the Phospho Cluster IV, one of the four phopho-clusters identified on p63 by Finlan and Hupp employing the Group-based Phosphorylation prediction module (GpS).19 By site-directed mutagenesis, we generated the ΔNp63αT397A expression plasmid, bearing a Threonine to Alanine substitution at position 397 and the ΔNp63αS383A construct bearing a Serine to Alanine substitution at position 383. The substitution of Ser383 appeared to slightly affect Dlx3-induced ΔNp63α degradation while substitution of Thr397 leads to a lack of ΔNp63α sensitivity to Dlx3-driven degradation (Figure 3c). In addition, we generated the ΔNp63αS383AT397A double mutant. The ΔNp63αSer383Thr397A protein was almost completely resistant to Dlx3-mediated degradation (Figure 3c) suggesting that Dlx3-induced p63 degradation was dependent on these two specific residues. Immunofluorescence microscopy showed that ΔNp63αSer383Thr397A protein normally located in the nucleus, even when coexpressed with Dlx3 and was transcriptionally active, as tested on the p57Kip gene promoter (data not shown).21 As proteasome and lysosome pathways are both involved in the control of p63 protein degradation,19 we used the proteasome inhibitor MG132 and the lysosome inhibitors chloroquine and ammonium chloride. As shown in Figure 4a, MG132 led to significantly higher levels of p63 in presence of Dlx3. A similar result was observed in the presence of ALLNL while no effect was observed with the lysosome inhibitors (data not shown). These data suggest that Dlx3-mediated p63 degradation requires the proteasome activity. In order to verify what extent Dlx3 alters ΔNp63α protein stability we measured ΔNp63α protein half-life in Saos2 cells, with or without Dlx3, by cycloheximide chase. Transfected ΔNp63α protein was very stable with a half-life being greater than 9 hours. As shown in Figure 4b, the half-life of the ΔNp63αS383AT397A mutant protein was almost undistinguishable from that of wild type ΔNp63α. As expected, the expression of Dlx3 significantly reduced ΔNp63α protein level and the decay was faster with a half-life between 5 and 7 hours (Figure 4b). To confirm that S383 and T397 residues were involved in Dlx3-mediated p63 degradation we expressed the ΔNp63αS383AT397A double mutant in Saos2 cells, with Dlx3, and measured its protein half-life in cycloheximide-treated cells. As shown in Figure 4b, the half-life of ΔNp63α8383AT397A in presence of Dlx3 was only slightly affected suggesting that replacement of Serine383 and Threonine397 with Alanine resulted in a ΔNp63α protein resistant to Dlx3-mediated degradation.
FIGURE 3. Identification of the p63 residues involved in Dlx3-mediated p63 degradation.
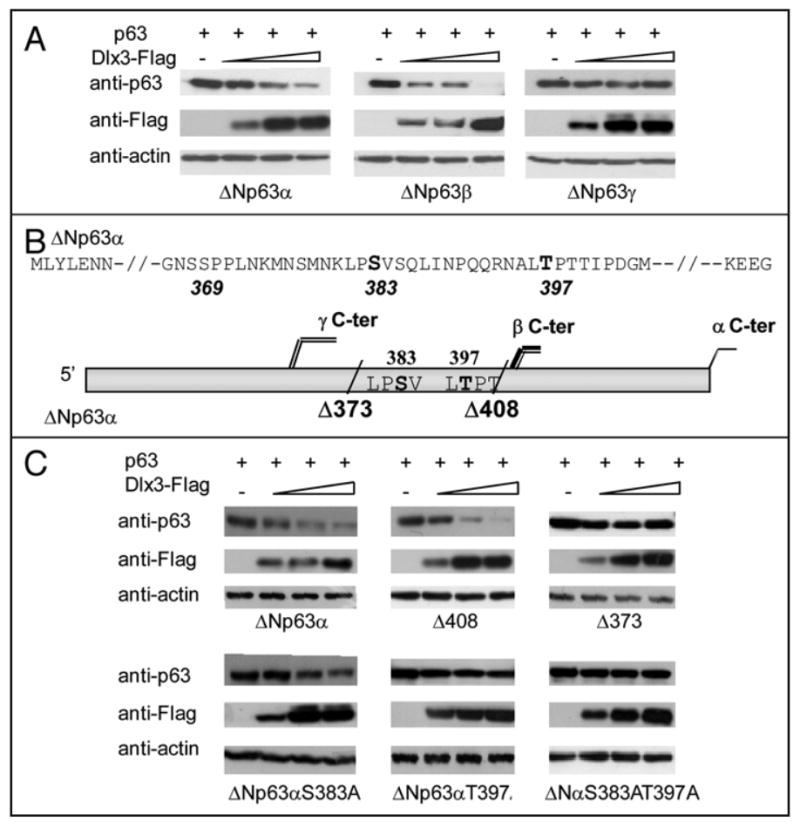
a) Representative experiments showing the effect of Dlx3 on transiently expressed ΔNp63α, ΔNp63αβ and ΔNp63γ. Each p63 isoform (0.2 μg) was transiently expressed in Saos2 cells alone or with 0.5, 1.0 and 1.5 μg of Dlx3-Flag expression vector. At 24 hrs after transfection cell extracts were subjected to Western blot analysis with anti-p63 and anti-Flag antibodies. Actin was used as loading control. b) Schematic representation showing the position of stop codons inserted in the ΔNp63α c-DNA sequence to generate the Δ373 and Δ408 truncated versions of ΔNp63α. Serine and Threonine at position 383 and 397 are also indicated. c) Saos2 cells transfected with 0.2 μg of expression plasmids encoding ΔNp63α, Δ408, Δ373, ΔNp63αS383A, ΔNp63αT397A or ΔNp63αS383AT397A with 0.5, 1.0 and 1.5 μg of Dlx3-Flag expression vector. At 24 hrs after transfection cell extracts were subjected to Western blot analysis with anti-p63 and anti-Flag antibodies. Actin was used as loading control.
FIGURE 4. Dlx3 reduces p63 protein half-life and induces proteasome-mediated p63 degradation.
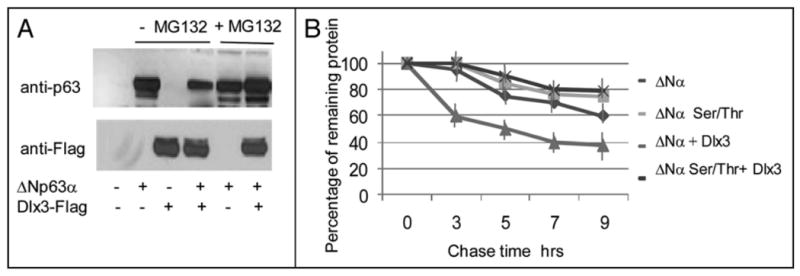
a) Extracts from Saos2 cells transfected with the indicated plasmids were treated or not with 10 μM MG132 (Z-Ieu-leu-leu-CHO). 48 hours after transfection cell extracts were prepared and subjected to Western blot analysis. p63 and Dlx3 were revealed with anti-p63 and anti-Flag antibodies, respectively. b) Saos2 cells transfected with ΔNp63α or ΔNp63αS383AT397A alone or with 1 μ.g of Dlx3-Flag vector were treated with cyclohexamide (CHX) for the indicated times. Equal amounts of total proteins were subjected to Western blot analysis for p63 and Dlx3. The plot represents the quantification of the p63 half life by densitometry scanning expressed as percentage of the remaining protein. Each profile represents the mean of three independent experiments. Standard Deviations are also shown.
Dlx3-mediated p63 degradation is regulated by Raf signalling
While characterizing ΔNp63α levels in cells exposed to ALLNL by SDS-PAGE and immunoblotting, in addition to there being a band corresponding to ΔNp63α, we also detected an additional slowly migrating form of the protein in extracts from cells expressing Dlx3 (Figure Sa, black arrow). To investigate whether the more slowly migrating form of ΔNp63α was due to a change in its phosphorylation status we carried out a λ-PPase assay followed by high resolution SDS-PAGE. Under these electrophoretic conditions any electrophoretic mobility shifts becomes more evident. As .6.Np63a is a phosphoprotein,14 treatment with A-PPase results in a shift of p63 electrophoretic mobility (Figure 5b lane 2). However, when .6.Np63a was cotransfected with Dlx3 the p63 band appeared to be slightly retarded and this effect was almost completely reversed by λ-PPase treatment (Figure 5b lane 3 and 4). This result implies that Dlx3 can change the phosphorylation status of p63. Dlx3 is expressed in the suprabasal layers of stratified epidermis.6 Differentiation of stratified epidermis largely depends upon protein kinase C (PKC) activation. 13 Several PKC isoforms are expressed in the epidermis (α, δ, ε, μ and ζ) and they can signal to downstream effectors through Raf1. Thus, we examined the possible contribution of Raf signaling in Dlx3-mediated p63 degradation by using GW5074, a specific pharmacological Raf inhibitor. Saos2 cells, transfected with a fixed amount of p63 and Dlx3 expression vectors, were exposed for 3, 6 and 8 hours to 10μM GW5074. As shown in Figure 6a, inhibition of Raf resulted in a substantial recovery of p63 protein level (Figure 6a, lanes from 4 to 6). On the other hand, pharmacological inhibition of PI3K and AKT kinase activity with Ly294002 or Wortmannin had no effect on Dlx3-mediated p63 degradation (data not shown). To more rigorously address the involvement of Raf1 kinase in DIx3-mediated p63 degradation, we used mouse embryonic fibroblasts deleted for both copies of Raf1 gene (Raf1-/- MEFs). Remarkably, in absence of Raf1, ΔNp63α was completely unaffected by DIx3 (Figure 6a, lanes 2, 3 and 4). Significantly, the introduction of RafBxB, an activated form of Raf1 into Raf1 null cells restored the ability of Dlx3 to induce p63 degradation (Figure 6b, lanes 6 to 8). Furthermore, we observed that RafBxB enhanced the expression level of transfected Dlx3 (Figure 6b, lanes from 5 to 8) and this effect was independent from p63 as it was observed even in absence of p63 (Figure 6c). Next, we investigated the effect of activated Raf1 on ΔNp63α using Saos2 cells transfected with wild type ΔNp63α or ΔNp63αS383AT397A and increasing amount of RafBxB plasmid. The expression levels of both wild type and double mutant p63 proteins were slightly increased upon RafBxB overexpression (Figure 7a and 7b, lanes from 5 to 7). This was in agreement with the observation that treatment of cells with GW5074 reduces p63 protein level (see Figure 6a lanes 1 and 2). Nonetheless, RafBxB overexpression appeared to cooperate with Dlx3, to induce a more pronounced i1Np63a protein degradation (Figure 7a, compare lanes 2, 3, 4 with lanes 8, 9 and 10). This effect was not observed with the ΔNp63αS383AT397A protein (Figure 7b, lanes 8 to 10), further confirming that S383 and T397 are absolutely required to allow p63 degradation induced by Dlx3.
FIGURE 5. Dlx3 induces ΔNp63α phosphorylation.
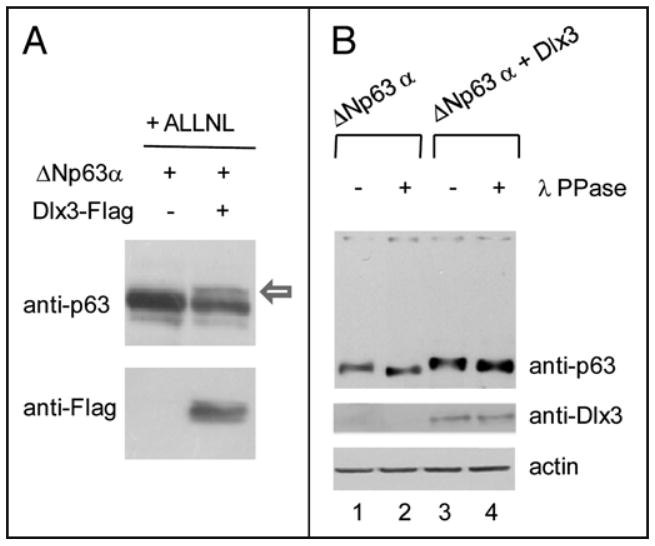
a) Saos2 cells, treated with 10 μM ALLNL (N-acetyl leucyl-leucyl norlucinal) were transfected with ΔNp63α alone or along with Dlx3-Flag vector (1.0 μg), as indicated. At 24 hours after transfection cell lysates were prepared and analysed by SOS-PAGE (b) Saos2 cells, treated with 10 μM ALLNL were transfected with ΔNp63α alone or along with Dlx3-Flag vector (1.0 μg), as indicated. At 24 hours after transfection cell lysates were treated (lanes 2 and 4) or not (lanes 1 and 3) with λ protein phosphatase (λ PPase), analyzed by high resolving SDS-PAGE (see materials and methods) and blotted with antibodies to p63 and actin.
FIGURE 6. Raf kinase activity is involved in Dlx3-induced p63 degradation.
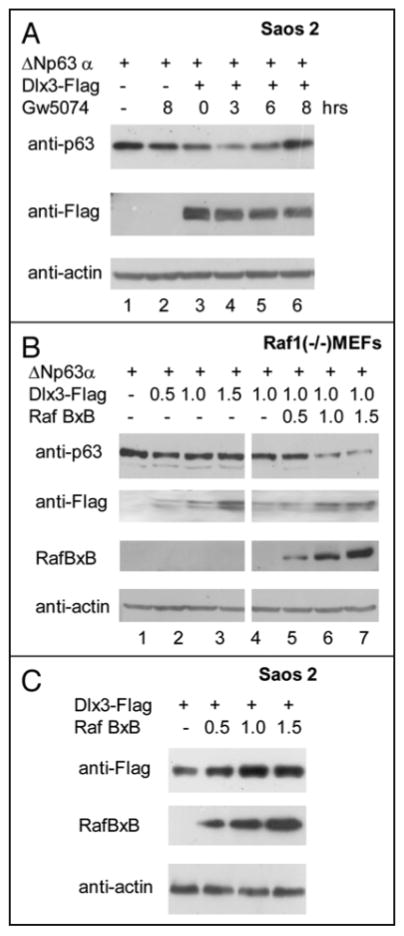
a) Saos2 cells transfected with 0.2 μg of ΔNp63α plasmid alone (lane 1 and 2) or with 1.5 μg of Dlx3-Flag plasmid (lanes from 3 to 6) were treated with 10 μM GW5074 as indicated. At 24 hours after transfection cell lysates were blotted with antibodies to p63, Flag and actin. b) Raf1 null Mouse Embryo Fibroblasts (MEFs) were transfected with ΔNp63α plasmid (0.2 μg) alone or with the indicated amounts of Dlx3-Flag and RafBxB plasmids. At 24 hrs after transfection cell Iysates were blotted with antibodies to p63, Flag, Raf1 and actin. c) Dlx3-Flag plasmid (0.5 ug) was transfected in Saos2 cells alone or with the indicated amounts of RafBxB expression vector. 24 hours after transfection cell lysates were blotted with antibodies to Flag. Antibodies against total Raf1 were used to check for the level of RafBxB while antibodies to actin were used as loading control.
FIGURE 7. Raf1 cooperates with Dlx3 to induce p63 protein degradation.
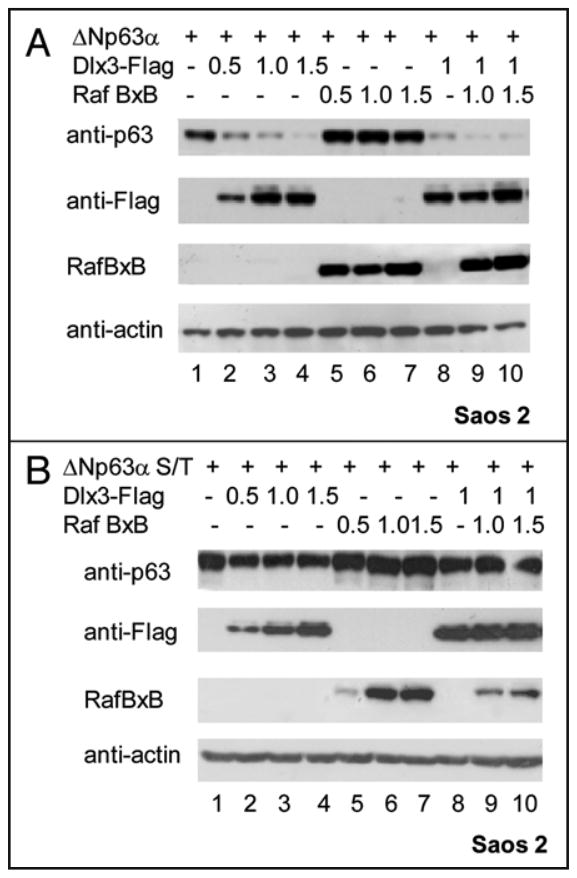
a) ΔNp63α plasmid was transiently transfected in Saos2 cells alone (lane 1), or with the indicated amounts of Dlx3-Flag and RafBXB expression vectors. 24 hours after transfection cell Iysates were blotted with antibodies to p63, Flag and actin. Antibodies against Raft were used to check for the level of RafBXB. b) ΔNp63αS383AT397A plasmid was transiently transfected in Saos2 cells alone or with the indicated amounts of Dlx3 and RafBxB expression vectors. 24 hours after transfection cell lysates were blotted with antibodies to p63, Flag and actin. Antibodies against Raf1 were used to check for the level of RafBXB.
Dlx3 interacts with and induces Raf1 phosphorylation
Given the dependence of Dlx3-mediated p63 degradation on Raf1, we investigated whether Dlx3 had a direct effect on Raf1. First, we carried out Real Time PCR analyses on samples derived from mock or Dlx3 transfected HaCaT cells. We found that Dlx3 does not significantly change the amount of Raf1 mRNA (Figure 8a). Consistent with this finding, we found that the total amount of Raf1 protein was unaffected by Dlx3 expression (Figure 8b and c). Conversely, by using antibodies that specifically recognize Raf1 phosphorylated at Serine 338, an activated form of Raf1, we found that phosphorylated Raf1 markedly increased in response to Dlx3 expression, both in Saos2 cells and HaCaT keratinocytes. (Figure 8b and c). Taken together, these data indicate that Dlx3 expression induces Raft kinase activity, which mediates p63 phosphorylation, thus targeting p63 for proteasomal degradation. To address the potential interaction between Dlx3 and Raf1, we performed immunoprecipitation in HaCaT cells. Extracts from HaCaT cells transfected with V5-tagged Dlx3 were immunoprecipitated with V5 antibodies. Western blot analysis of the immunoprecipitation products revealed that endogenous Raf1 was pulled-down with Dlx3 (Figure 9a). Then, we speculated that Raft might also interact with p63. Indeed, ΔNp63α was coimmunoprecipitated in Raf1 immunocomplexes but was completely absent in anti-GF′P immunoprecipitated (Figure 9b). The reciprocal experiment confirmed that p63 and Raf1 are able to form a complex (Figure 9c). A direct interaction between Raf1 and p63 as well as between Raf1 and Dlx3 was also confirmed in Saos2 cells using either endogenous Raf1 or transfected RafBxB (data not shown). In parallel experiments, we tested whether p63 could physically interact with Dlx3. Coimmunoprecipitation assays were performed using extracts from Saos2 cells co-transfected with Flag-Dlx3 and ΔNp63α expression plasmids. Whole extracts were immunoprecipitated with anti-p63 (4A4) antibodies and subjected to Western blot analysis with anti-Flag antibodies. Reciprocal experiments were done and in both cases, interaction between p63 and Dlx3 was not detected (Figure 9d and data not shown). Finally, to explore the possibility that interaction between p63 and Raft could mediate p63 protein phosphorylation, we performed a kinase assay. Saos2 cells were transfected with ΔNp63α, Dlx3 or ΔNp63α and Dlx3. We then immunoprecipitated p63 and Raf1, as indicated in Figure 9e, and analyzed the immunoprecipitated with antibodies directed against p63 or Raf1. Sample aliquots were used in a kinase assay by the addition of [γ-32P]ATP directly to the immunoprecipitates. As shown in Figure 9e, p63 was phosphorylated only in Raf1 immunocomplexes.
FIGURE 8. Dlx3 induces Ran phosphorylation.
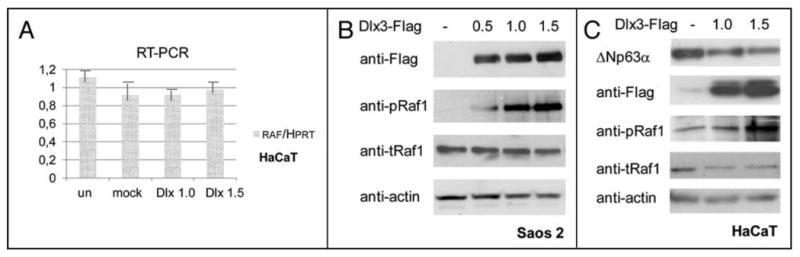
a) HaCaT cells were mock transfected or transfected with 1.0 or 1.5 μg of Dlx3 expression vector. Total RNA was prepared from untransfected, mock or Dlx3 transfected HaCaT cells at 48 hours post-transfection. Real Time PCR was performed with Raf1 and HPRT specific oligonucleotides as described in Material and Methods. The experiment was performed twice and samples were done in triplicate. HPRT was used for normalization. The results were expressed with the value relative to HPRT (set at 1) for each mRNA sample. Standard deviations are also shown. b) Saos2 cells were transiently transfected with the indicated amounts of Dlx3 plasmid. 24 hours after transfection cell lysates were blotted with antibodies against actin, total and phosphorylated Raf1. Dlx3 was detected with antibodies to Flag. c)HaCaT cells were mock transfected or transfected with 1.0 or 1.5 ug of Dlx3 expression vector. Equal amounts of total protein (40 μg) were blotted with antibodies against p63, Flag, total (α-tRaf1) or phosphorylated Raf1 (α-pSer338Raf1). Loading equivalence was assessed with antibodies against actin.
FIGURE 9. Raf-1 interacts with Dlx3 and ΔNp63α.
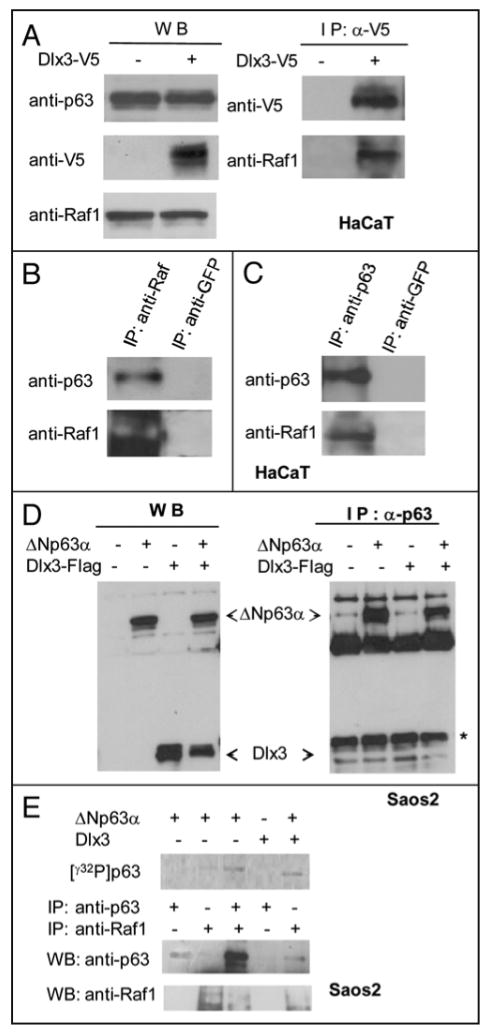
A) Western blot of HaCaT cells transiently transfected with V5-Dlx3 encoding plasmid and treated with 10 μM ALLNL. Equal amounts of total protein extracts from cells transfected or not were immunoblotted with anti-p63, anti-Raf and anti-V5 (left panel). Extracts from mock or V5-Dlx3 transfected cells were immunoprecipitated with anti-V5 antibody and the immunocomplexes were blotted and probed with anti-V5 or anti-Raf1 antibodies (right panel). b) Untransfected HaCaT cell lysates were immunoprecipitated with anti-Raf1 or anti-GFP antibodies. The immunocomplexes were blotted and probed with anti-p63 and anti-Raf1 antibodies. Immunoprecipitation with anti-GFP was performed as control. c) Untransfected HaCaT cell lysates were immunoprecipitated with anti-p63 or anti-GFP antibodies. The immunocomplexes were blotted and probed with anti-Raf1 and anti-p63 antibodies. d) Saos2 cells were transfected with 1 μg of ΔNp63α plasmid alone or together with 1 μg of Dlx3 plasmid. Cell extracts were immunoprecipitated with 2μg of anti-p63 antibodies. Dlx3 was revealed with antibodies to Flag. (*) Unspecific signal. e) Saos2 cells were transfected with p63 and Dlx3 vectors as indicated. Cells were treated with ALLNL. At 24 hrs extracts were immunoprecipitated with the indicated antibodies and equal aliquots incubated with 50 μM ATP and 5 μCi/sample of γ32P-ATP in kinase buffer (see materials and methods). Radiolabelled γ32P p63 (upper panel) was revealed using the PMI imaging system (BioRad). The lower panels show the immunoprecipitated p63 and Raf1 proteins as analyzed by Western blot followed by ECL detection.
Discussion
The epidermis, the outermost component of the skin, is established during embryogenesis as a result of a complex and precisely coordinated stratification program. The role of p63 in skin development is essential and TA and ΔN isoforms cooperate to carry out the epidermal program of stratification and differentiation.22 Mutations in the p63 gene have been associated with ectodermal dysplasias and limb deformities that include ectrodactyly-ectodermal dysplasia-cleft/lip palate (EEC), limb mammary syndrome (LMS), split hand-foot malformation (SHFM) and ankyloblepharon-ectodermal dysplasia-clefting (AEC) syndrome.23,24 Instead, DIx3 gene mutations were linked to autosomal dominant tricho-dento-osseus (TDO) syndrome.25 We have previously shown that p63 is able to bind and transactivate Dlx3 gene promoter. In particular, we have found that both TAp63α and γ were able to transactivate Dlx3, at similar levels. Remarkably, mutant p63 proteins derived from AEC patients exhibit an impaired ability to transactivate Dlx3, indicating that the misregulation of the Dlx3 gene is involved in the pathogenesis of AEC syndromes.
In adult epidermis, ΔNp63α is abundantly expressed in the basal compartment where it sustains the proliferative potential of basal cells. In the suprabasal layers of epidermis TAp63 expression becomes detectable while 8Np63a protein is less abundant. 1 It is well known that p63 protein abundance is regulated both at transcriptional and post-translational level; however the mechanisms that control ΔNp63α protein abundance during keratinocyte differentiation are not fully understood. Recently, a micro-RNA (miR-203) was found to be induced in the skin concomitantly with stratification and differentiation.26 miR-203 was demonstrated to reduce ΔNp63α level under differentiation stimuli. However miR-203 was not sufficient per se to trigger full expression of differentiating epithelial markers.27 The observation that Dlx3, which is expressed exclusively in the suprabasal differentiated layers of epidermis.4,11 is induced by TA, but not ΔNp63α protein10 is perfectly in line with the hypothesis that ΔNp63α plays a major role in the basal layer of epidermis, at the interface between proliferative epithelial progenitor cells and the differentiation pathway.26.27 We have now observed that expression of Dlx3 causes a remarkable decrease of endogenous and transfected ΔNp63α protein. Furthermore, depletion of endogenous Dlx3 protein expression in HaCaT cells resulted in a reproducible increase of ΔNp63α protein level, confirming that basal Dlx3 levels are important in controlling basal ΔNp63α levels. These data support a previously unrecognized role for Dlx3 in the posttranslational regulation of ΔNp63α protein. We might hypothesize that TAp63 expression induces Dlx3 which activates a molecular mechanism that target ΔNp63α for proteasome-mediated degradation. Given the role of ΔNp63α in sustaining proliferation of basal keratinocytes, Dlx3-mediated ΔNp63α degradation may cooperate to accomplish the program of terminal skin differentiation. To this purpose, impaired expression of Dlx3 by mutated p63, in AEC/Hay Wells patients, might potentially explain the observation that AEC-mutated ΔNp63α proteins are not longer restricted to the proliferative cell layer and persists in the uppermost layer of epidermis where cells normally undergo terminal differentiation28 Interestingly, Dlx3 overexpression resulted in efficient ΔNp63α/β, but not γ, protein degradation. The evidence that ΔNp63γ is completely resistant to Dlx3-mediated degradation indicates that residues located in the p63 α and β tails are absolutely required for the mechanism of Dlx3-mediated p63 degradation. Furthermore, we also observed that TAp63α is more resistant than the ΔNα isoform, suggesting that the N-terminal region of p63 may in some way interfere with the phosphorylation or the degradation mechanism of p63, possibly due to the reduced accessibility of the three-dimensional structure of TAp63α. As mentioned before, TAp63α protein is undetectable in the basal layer of epidermis while it is present in the suprabasal layers3 where it could potentially contribute to the activation of Dlx3 gene expression.11 Interestingly, the evidence that TAp63α has a significantly lower sensitivity to Dlx3-mediated degradation compared to ΔNp63α, provides a reasonable explanation to its persistence in the granular layer of epidermis where Dlx3 is actually expressed. Moreover, terminally differentiated keratinocytes lack completely p63α expression while a smaller ΔN-form, reputed to be ΔNp63γ persists, and has been described to be compatible with the expression of terminal differentiation markers.3 Accordingly, we have shown that ΔNp63γ is completely resistant to Dlx3-induced degradation. Multisite phosphorylation is a common feature of many protein kinase substrates, which may enable docking interactions, integration of different kinase pathway signals or changes in the subcellular localization.29 An important finding in this report is that Dlx3 is unable to promote p63 degradation in Raf1 depleted MEF cells or upon pharmacological knockdown of Raf1. Remarkably, introduction of RafBxB, an activated form of Raf1, in Raf1 null MEFs, stabilizes Dlx3 and restores its ability to induce p63 degradation. These data strongly suggest that Raf1 is a crucial player in Dlx3-mediated p63 protein degradation. Interestingly, we observed a functional interplay between Dlx3 and Raf1. Indeed, Raf1 activation increases Dlx3 protein abundance while Dlx3 expression resulted in a significant enhancement of Raf1 phosphorylation. Altogether our observations suggest that Dlx3 and Raf1 can establish a positive regulatory loop through which Dlx3 induces Raf1 activation and activated Raf1 stabilizes Dlx3. Raf1 kinase activity appears to play a crucial role in epidermal differentiation being at the cross road between signals driving cell proliferation and differentiation. The mammalian Raf family of serine/threonine kinases consists of three highly conserved members, i.e., A-Raf, B-Raf and Raf1. Whereas Raft is ubiquitously expressed, A-Raf and B-Rafdisplay a more tissue-specific expression. The role of Raf1 in the epidermal differentiation is demonstrated by the phenotype of Raf1 genetically modified mice. Homozygote mice for a hypomorphic Raf1 allele (Raf1tmlZim) die during organogenesis. Only 5% of homozygotes are viable and they display underdeveloped hair follicles, thin epidermis and abnormal epidermal layer morphology.30 Interestingly, they also show abnormal placental labyrinth morphology strongly reminiscent of the placental defects of Dlx3 null mice.8 Our data show that substitution of Ser383 and Thr397 with alanine impairs Dlx3-mediated ΔNp63α degradation. Moreover, in presence of Dlx3 and proteasome inhibitors, wild type ΔNp63α appeared as a slowly migrating band and this effect was reversed by λ-PPase. Remarkably, we have observed that Raf1 can interact with Dlx3 and Dlx3 induces Raf1 phosphorylation at Serine 338, thus suggesting that Dlx3 can induce Raf1 kinase activation through their direct physical association. By in vitro kinase assay, we have also shown that Raf1 can bind and phosphorylate p63. However, further experiments need to be performed in order to uncover the mechanistic aspects of this functional interaction. Actually, Raf1 may directly phosphorylate p63 or alternatively, in addition to interacting with one another, Raf1 and p63 might recruit other kinases responsible for p63 phosphorylation. A similar mechanism has already been described for the recruitment of Akt by Raf1 to phosphorylate ASK1, the apoptosis signalregulating kinase 1.31 Finally, although we have found association between Raf1 and Dlx3, we failed to detect p63 in Dlx3-Raf1 immunocomplexes pointing against the existence of a ternary complex. These observations support a model in which Dlx3 activates Raf1 kinase that either directly or indirectly induces p63 phosphorylation thereby targeting p63 to proteasome-mediated degradation. All together, our data highlight the existence of a novel regulatory mechanism, relying on Dlx3 gene expression, which can reduce ΔNp63α at protein level. Misfunction of such mechanism might significantly contribute to the AEC phenotype.
Materials and Methods
Plasmids
Raf1BxB32 encoding plasmid was provided by A. Costanzo. The p63 wild type cDNAs in the pcDNA3-myc vector were provided by H. van Bokhoven. The plasmid encoding the truncated Δ408 p63 protein has been previously described33. The Δ373 carboxyterminal truncated p63 protein, the ΔNp63αS383A and Thr397A point mutants and the ΔNp63αS383T397A double mutant were produced using the GcneEditor in vitro Site-Directed Mutagenesis System (Promega, Milan, Italy) following the manufacturer's instructions. The sequences of the mutagenic oligonucleotides were:
S383A FW S′CAG CAT GAA CAA GCT GCC TGC CGT GAG CCA GCT TAT CAA CCC AC3′
S383A REV S′GTG GGT TGA TAA GCT GGC TCA CGG CAG GCA GCT TGT TCA TGC TG 3′
T397A FW S′CGC AAT GCC CTC GCA CCC ACC ACC ATG 3′
T397A REV S′CAT GGT GGT GGG TGC GAG GGA ATT GCG 3′
Cell culture and transfections
We utilized an inducible PAM2l2-tetOn cell line developed in the laboratory. PAM2l2 is a naturally immortalized murine keratinocyte cell line.33 These cells were stably transfected with prtTA2-M2/IRES-Neo, clones were isolated, amplified and screened for low basal activity and a high sensitivity to Doxycycline (Dox), using a luciferase reporter construct. The best PAM2l2-TetOn clone isolated from the stable transfection described above was transfected with a bi-directional plasmid co-expressing Dlx3 (tagged with a V5 epitope) and GFP under the control of a unique tetracycline-responsive element (pBi-V5Dlx3/GFP) using Amaxa nucleofactor. Stable clones were also established and a clone showing no basal expression of V5Dlx3 in the absence of Dox and a strong induction of V5Dlx3 in the presence of Dox (2 μg/ml) was selected for subsequent experiments (PAM-TetOn-V5Dlx3/GFP). GFP expression in PAM-TetOn-V5Dlx3/GFP cells was observed using the Axio Observer inverted microscope (Zeiss) and images were acquired with the AxioCam MRm (Zeiss). Human osteosarcoma-derived Saos2 cells were maintained in RPMI 1640 medium and 10% fetal calf serum. Spontaneously immortalized human keratinocytes (HaCaT) were grown in RPMI (0.45 mM Ca2+) supplemented with 10% Chelex -treated Fetal Bovine Serum. For induction of Dlx3 expression in HaCaT, 1.8 mM Ca2+ was added to RPMI-IO% Chelex-treated FBS. HeLa, and MEF cells were grown in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (Euroclone, Life Science) at 37°C in a humidified atmosphere of 5% (v/v) C02 in air. HaCaT cells were transiently transfected with 0.5, 1.0 or 1.5 μg of pcDNAFlag-Dlx3 expression vector using LipofectAMINE 2000 reagent (In Vitrogen, Life Technologies. lnc.), HeLa, H1299, U20S and MEF cells were seeded at a density of about 70% confluence and co-transfected with 0.2μg of each pcDNAmyc-p63 expression plasmids and 0.5, 1.0 or 1.5 ug of pcDNAFlag-Dlx3 expression vector with LipofectAMINE 2000 reagent (In Vitrogen, Life Technologies. Inc.). The total amount of transfected DNA was kept constant by using an empty expression vector when necessary. At 24 hrs post-transfection, cells were lysed and total protein quantified with the BioRad protein assay. Forty micrograms of extracts were resolved by SDS-polyacrylamide gel electrophoresis for Western blot analysis. Half-life of p63 protein was determined by addition of 40 μg/ml cycloheximide (Sigma-Aldrich, Germany), 16 hours upon transfection. MG132, ALLNL, NH4Cl and Chloroquine treatments were performed the day after transfection with 10 and 20 μM MG132 (Sigma-Aldrich, Germany), 10 and 20 μM ALLN (Sigma-Aldrich, Germany), 20 mM NH4Cl, 100 μM Chloroquine (Sigma-Aldrich, Germany) or solvent alone for 6 hours. Pharmacological inhibition of Raf kinase activity was performed by treating cells with 5 and 10 μM GW5074 (Calbiochem, La Jolla, CA).
Luciferase assay
The -1291p57K1P2 luciferase construct was previously described21 Saos2 cell were transfected with -1291p57K1P2 luciferase construct (0.2 μg) and increasing amount (0.1, 0.3 and 0.5 μg) of ΔNp63α or ΔNp63αSer383Thr397A expression plasmid. Cells were lysed after 36 hours and luciferase activity determined.
Western blot analysis and coimmunoprecipitation
At 24 hrs after transfection cells were lysed in 10 mM Tris-HCI (pH 7.5), 1 mM EOTA, 150 mM NaCI, 0.5% NP-40, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 0.5% sodium deoxycholate and protease inhibitors. Cell lysates were incubated on ice for 30 min, and the extracts were centrifuged at 13,000 rpm for 10 min to remove cell debris. Protein concentrations were determined by the BioRad protein assay. After the addition of 2× loading buffer (2% sodium dodecyl sulfate [SOS], 30% glycerol, 300 mM ∼-mercaptoethanol, 100 mM Tris-HCI [pH 6.8]), the samples were incubated at 95°C for 5 min and resolved by SDS-polyacrylamide gel electrophoresis. Proteins were transferred to a PVDF (Millipore, Milan, Italy) and probed with the following antibodies: anti-p63 (4A4; Santa Cruz, Biotechnology, Inc.), anti-FLAG M2 (Sigma-Aldrich, Germany), anti-V5 (AbD Serotec, Oxford, UK), anti-GFP (sc-8334; Santa Cruz, Biotechnology, Inc.), anti-actin (1-19; Santa Cruz, Biotechnology, Inc.), anti-tRaf1 (C-12; sc-133; Santa Cruz, Biotechnology, Inc.), anti-pRaf1(Ser 338; sc-12385; Santa Cruz, Biotechnology, Inc.). Proteins were visualized by an enhanced chemiluminescence method (Amersham Pharmacia Biotech, Uppsala, Sweden). To detect p63 and Dlx3 interaction in Saos2 cells, 5.0 ×105 cells were plated in 60 mm dishes and transfected with 1μg of pcDNA ΔNp63α alone or together with lμg of Dlx3-Flag expression vector. To detect interaction between Raf1 and p63, I×106 Saos2 cells were cultured in 100mm dishes and transfected with 3μg of pcDNA ΔNp63α or 3μg of pCI-V5-DIx3 encoding plasmids. For immunoprecipitation in HaCaT keratinocytes, 1.5×106 cells were seeded in 100 mm dishes and transfected with 5 μg of pCI-VSDlx3 plasmid. After transfection, 10 μM ALLN was added to the cells to prevent Dlx3-mediated p63 degradation. Cells were harvested 24 hrs post-transfection and cell lysates were prepared as described above. Whole cell extracts were precleared with 30 μl of protein A-agarose (50% slurry; Roche, Manheim, Germany) and then incubated overnight at 4°C with the following antibodies: 2 μg of anti-p63 (4A4; Santa Cruz, Biotechnology, Inc.), 3 μg of anti-V5 (AbD Serotec, Oxford, UK), 3 μg of anti-GFP (sc-8334; Santa Cruz, Biotechnology, Inc.), 6 μg of anti-Raf1 (C-12; sc-133; Santa Cruz). Immunocomplexes were collected by incubation with protein A-agarose (Roche, Manheim, Germany) at 4°C for 4 hrs. The beads were washed with Co-IP buffer (50 mM Tris-HCI pH 7.5; 150 mM NaCI; 5 mM EDTA; 0.5% NP40; 10% glycerol), resuspended in 2× loading buffer (Sigma) and loaded in a SDS-10% polyacrylamide gel. Proteins were then transferred onto a PVDF membrane (Millipore, Milan, Italy) and probed with the iftdieated primary antibodies. Proteins were visualized with an enhanced chemiluminescence detection system (Amersham).
siRNA design and transfection
RNA duplexes of 21 nucleotides targeting the human Dlx3 mRNA were designed, chemically synthesized and supplied by Sigma-Aldrich (Sigma's MISSION siRNA predesigned). HaCaT cells seeded at 50% confluency were grown in RPMI-1.8 mM Ca2 supplied with 10% Chelex/FBS. Hyperfect (Quiagen, GmbH, Hilden, Germany)-mediated transient transfection of siRNA was done in 60 mm plates according to the manufacturer instructions. Scrambled oligonucleotides were used as a control. Monoclonal antibodies (4F8) from Sigma-Aldrich were used to detect endogenous Dlx3.
λ-phosphatase (λ-PPase) treatment and immunoblotting
Total cell lysates prepared as duplicate samples were incubated with or without 4 units of λ-phosphatase (New England Biolabs, GmbH, Germany) for 1 h at 30°C as suggested by the manufacturer. Extracts were analysed by SDS-PAGE using a high resolving 8% gel (37.5:1 Acryl/Bis Acrylamide).
Subcellular distribution assay
Saos 2 cells (5.0 ×105) were plated in 35 mm dish and grown on micro cover glasses (BDH) and transfected with 0.2 μg of ΔNp63α or ΔNp63αSer383Thr397A plasmid with or without 1 μg of Dlx3 plasmid. ALLNL was added to inhibit p63 degradation. At 24 hrs after transfection with the indicated vectors, cells were washed with cold phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde (Sigma-Aldrich, Germany) for 15 min at 4°C. Cells were permeabilized with ice-cold 0.1% Triton X-IOO for 10 min and then washed with PBS. P63 or Dlx3 subcellular localization were determined by using a 1:200 dilution of the monoclonal antibody 4A4 against p63 or an anti-FLAG monoclonal antibody against Dlx3-Flag (Sigma-Aldrich, Germany) diluted 1:2000. After extensive washing in PBS, the samples were incubated with a Cy3-conjugated anti-mouse immunoglobulin G (ImmunoResearch Laboratory) at room temperature for 30 min. After PBS washing, the cells were incubated with DAPI (4′,6′-diamidino-2-phenylindole; 10 mg/ml [Sigma-Aldrich, Germany]) for 3 min. After PBS washing, the glasses were mounted with Moviol (Sigma-Aldrich, Germany) and examined under a fluorescence microscope (Nikon). Images were digitally processed by Adobe Photoshop software.
PCR analyses
For the analysis of Dlx3 expression in HaCaT cells, total RNA from HaCaT cultured in low (0.45 mM) or high (1.8 mM) Ca2+ was isolated using the RNA Mini Extraction Kit (Qiagen, GmbH, Hilden, Germany) according to the manufacturer's instructions. 1μg of total RNA was used to generate reverse transcribed using SuperScript III (InVitrogen Life technologies, Inc.). The following oligonucleotides were used:
Dlx3 (F) 5′ACCTACGGAGCCTCCTACCG
Dlx3 (R) 5′ACTCAGGTTCTGTGCGTGAT
Saos2 cells were transfected with a fixed amount of ΔNp63α expression vector alone (0.2 μg) or with increasing amount of pcDNA Dlx3-Flag (1 and 1.5 μg), Total RNA was isolated as described above. Reverse-transcripts were amplified with the following primers:
For the analysis of p63 gene expression:
(F) S′CCACAGTACACGAACCTGGGG
(R) S′CCGGGTAATCTGTGITGGAG
Human Hypoxanthine Phosphoribosyl Transferase (HPRT) gene was amplified using the following primers:
(F) 5′CCTGCTGGAITACAITAAAGC
(R) 5′ CTTCGTGGGGTCCTTTTC
The amplification sequence consisted of 35 cycles of 98°C/1′, 54°C/1′, 72°C/1′. PCR products were resolved by 2% agarose electrophoresis. RT-PCR amplification results were analyzed by Quantity One software (Biorad).
For analysis of the expression of Raf1 in HaCaT cells, RNA was isolated as described above. The following oligonucleotides were used: Raf1 (F) 5′-TTTCCTGGATCATGTTCCCCT and Raf1 (R) 5′- ACTITGGTGCTACAGTGCTCA. Real Time PCR was performed with a 7500 RT-PCR Thermo Cycler (Applied Biosystem) using SYBR GREEN Master Mix (Applied Biosystem). All samples were done in triplicate. HPRT was used for normalization. The results were expressed with the value relative to HPRT (set at I) for each mRNA sample.
Kinase assay
Saos2 cells were transfected and incubated with ALLNL. At 24 hours after transfection cells were lysed in 20 mM Tris-HCl, 150 mM NaCI, 25 mM β-glycerophosphate, 2 mM EDTA, 2 mM pyrophosphate, I mM orthovanadate, 1% Triton, 1mM OTT and 1mM NaF. Protease Inhibitors were added. Cell lysates were cleared by centrifugation. Aliquots were kept a -20°C for Western Blot analyses. Antibodies and Protein A Sepharose beads were added to the lysates. After extensive washes, beads were resuspended in 20 mM Tris-HCl, 10 mM MgCl2, 1mM OTT, 50 μM ATP and 5 μCi/sample of γ32P-ATP. Samples were incubated at 30°C for 30 minutes. Reactions were stopped with SDS-sample buffer and analyzed by SDS-polyacrylamide gel electrophoresis. Proteins were transferred to a PVDF (Millipore, Milan, Italy) membrane. Radiolabelled p63 was revealed using a PMI imaging system (BioRad, Milan, Italy).
Acknowledgments
We thank A. Pollice for helpful discussion and critical reading. G. Napolitano for technical support. A. Costanzo for providing the RaffixB plasmid. We are grateful to Catrin Pritchard who provided us with the Raft (-/-) mouse fibroblast cell line. This work was supported by Telethon GGP030326, AIRC and MIUR to G.L.M. L.G. was supported by Telethon GGP05056.
Abbreviations
- Dlx3
Distal-less homeobox 3
- ALLNL
N-acctyl-Ieucyl norlucinal
- PPase
protein phosphatase
References
- 1.Carroll OK, Carroll JS, Leong CO, Cheng F, Brown M, Mills AA, et al. p63 regulates an adesion programme and cell survival in epithelial cells. Nature Cell Biology. 2006;8:551–561. doi: 10.1038/ncb1420. [DOI] [PubMed] [Google Scholar]
- 2.Truong AB, Kretz M, Ridky TW, Kimmel R, Khavari PA. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Development. 2006;20:3185–3197. doi: 10.1101/gad.1463206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death inducing, and dominant-negative activities. Molecular Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 4.King KE, Ponnamperuma RM, Yamashita T, Tokino T, Lee LA, Young MF, et al. Unique domain functions of p63 isotypes that differentially regulate distinct aspects of epidermal homeostasis. Oncogene. 2003;22:3635–3644. doi: 10.1093/carcin/bgi200. [DOI] [PubMed] [Google Scholar]
- 5.King KE, Ponnamperuma RM, Gerdes MJ, Tokino T, Yamashita T, Baker CC, et al. DeltaNp63alpha functions as both a positive and a negative transcriptional regulator and blocks in vitro differentiation of murine keratinocytes. Carcinogenesis. 2006;27:53–63. doi: 10.1038/sj.onc.1206536. [DOI] [PubMed] [Google Scholar]
- 6.Hassan MQ, Javed A, Morasso MI, Karlin J, Montecino M, van Wijnen AJ, et al. Dlx3 transcriptional regulation of osteoblast differentiation: temporal recruitment of Msx2, Dlx3 and Dlx5 homcodomain proteins to chromatin of the ostcocalcin gene. Molecular Cell Biology. 2004;24:9248–61. doi: 10.1128/MCB.24.20.9248-9261.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morasso MI, Grinberg A, Robinson G, Sargent TO, Mahon KA. Placental failure in mice lacking the homeobox gene Dlx3. Proceedings of the National Academy of Sciences of the United States of America Natl Acad Sci USA. 1999;96:162–167. doi: 10.1073/pnas.96.1.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park GT, Morasso MI. Regulation of the Dlx3 homeobox gene upon differentiation of mouse keratinocytes. Journal of Biological Chemistry. 1999;274:26599–608. doi: 10.1074/jbc.274.37.26599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morasso MI, Markova NG, Sargent TO. Regulation of epidermal differentiation by a Distal-less homeodomain gene. Journal of Cell Biology. 1996;135:1879–1887. doi: 10.1083/jcb.135.6.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Radoja N, Guerrini L, Lo Iacono M, Merlo GR, Costanzo A, Weinberg W, et al. Homeobox gene Dlx3 is regulated by p63 during ectoderm development: relevance in the pathogenesis of ectodermal dysplasia. Development. 2007;134:13–18. doi: 10.1242/dev.02703. [DOI] [PubMed] [Google Scholar]
- 11.Koster MI, Roop O. Mechanisms regulating epithelial stratification. Annual Review of Cell Developmental Biology. 2007;23:93–113. doi: 10.1146/annurev.cellbio.23.090506.123357. [DOI] [PubMed] [Google Scholar]
- 12.Park GT, Denning MF, Morasso MI. Phosphorylation of murine homeodomain protein Dlx3 by protein kinase C. FEBS Letters. 2001;496:60–65. doi: 10.1016/s0014-5793(01)02398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westfall MO, Mays OJ, Sniezek JC, Pietenpol JA. The i.\Np63a phosphoprotein binds the p21 and 14.3.3cr promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome derived mutations. Molecular and Cellular Biology. 2003;23(7):2264–2276. doi: 10.1128/MCB.23.7.2264-2276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9(1):45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 15.Ghioni P, D'Alessandra Y, Mansueto G, Jaffray G, Hay RT, La Mantia G, et al. The protein stability and transcriptional activity of p63alpha are regulated by SUMO-I conjugation. Cell Cycle. 2005;4:183–90. doi: 10.4161/cc.4.1.1359. [DOI] [PubMed] [Google Scholar]
- 16.Okada Y, Osada M, Kurata S, Sato S, Aisaki K, Kageyama Y. p53 gene family p51(63)-encoded secondary transactivator pSIS (TAp63alpha) occurs without forming un immunoprecipitable complex with MDM2, but responds to genotoxic stress by accumulation. Experimental Cell Research. 2002;276:194–200. doi: 10.1006/excr.2002.5535. [DOI] [PubMed] [Google Scholar]
- 17.Rossi M, De Simone M, Pollice A, Santoro R, La Mantia G, Guerrini L. Itch/AlP4 associates with and promotes p63 protein degradation. Cell Cycle. 2006;5:18161822. doi: 10.4161/cc.5.16.2861. [DOI] [PubMed] [Google Scholar]
- 18.Liefer KM, Koster MI, Wang XJ, Yang A, McKeon F, Roop DR. Down-regulation of p63 is required for epidermal UV-B-induced apoptosis. Cancer Research. 2000;60:4016–4020. [PubMed] [Google Scholar]
- 19.Finlan LE, Hupp TR. p63: the phantom of the tumor suppressor. Cell Cycle. 2007;6:1062–1071. doi: 10.4161/cc.6.9.4162. [DOI] [PubMed] [Google Scholar]
- 20.Balckers J, Camacho-Carvajal M, Novak C, Kramer C, Danger B, Hammerschmidt M. Destabilization of DeItaNp63alpha by Nedd4-mediated ubiquitination and Ubc9-mediated sumoylation, and its implications on dorsoventral patterning of the zebrafish embryo. Cell Cycle. 2005;4:790–800. doi: 10.4161/cc.4.6.1694. [DOI] [PubMed] [Google Scholar]
- 21.Beretta C, Chiarelli A, Testoni B, Mantovani R, Guerrini L. Regulation of the ciclyn-dependent kinase inhibitor p57Kip2 expression by p63. Cell Cycle. 2005;4(11):1625–1631. doi: 10.4161/cc.4.11.2135. [DOI] [PubMed] [Google Scholar]
- 22.Yang A, Schweitzer R, Sun O, Kaghad M, Walker N, Bronson RT, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 23.Van Bokhoven H, McKeon F. Mutations in the p53 homolog p63: allele-specific developmental syndromes in humans. Trends Mol Med. 2002;8(3):133–139. doi: 10.1016/s1471-4914(01)02260-2. [DOI] [PubMed] [Google Scholar]
- 24.McGrath JA, Duijf PH, Doetsch V, Irvine AD, de Wall R, Vanmolkot KR, et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet. 2001;10:221–229. doi: 10.1093/hmg/10.3.221. [DOI] [PubMed] [Google Scholar]
- 25.Price JA, Wright JT, Kula K, Bowden OW, Hart TC. A common DLX3 gene mutation is responsible for tricho-dento-osseous syndrome in Virginia and North Carolina families. J Med Genet. 1998;35(10):825–828. doi: 10.1136/jmg.35.10.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rui Yi, Poy MN, Stoffel M, Fuchs E. A skin microRNA promotes differentiation by repressing “sternness”. Nature. 2008;452:225–229. doi: 10.1038/nature06642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lena AM, Shalom-Feuerstein R, Rivetti di Val Cervo P, Aberdam O, Knight RA, et al. miR-203 represses “sternness” by repressing ∼Np63. Cell Death and Differentiation. 2008;15:1187–1195. doi: 10.1038/cdd.2008.69. [DOI] [PubMed] [Google Scholar]
- 28.Fomenkov A, Huang YP, Topaloglu O, Brechman A, Motonobo O, Fomenkova T, Yuriditsky E, Trink B, Sidransky D, Ratovinski E. J Biol Chem. 2003;278(26):23906–23914. doi: 10.1074/jbc.M300746200. [DOI] [PubMed] [Google Scholar]
- 29.Cohen P. The regulation of protein function by multisite phosphorylation- a 25 year update. Trends in Biochemical Sciences. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 30.Wojnowski L, Stancato LF, Zimmer AM, Hahn H, Beck TW, Lamer AC, et al. Craf-l protein kinase is essential for mouse development. Mechanisms of Development. 1998;76:141–149. doi: 10.1016/s0925-4773(98)00111-7. [DOI] [PubMed] [Google Scholar]
- 31.Chen J, Fujii K, Zhang L, Roberts T, Haian F. Raf-l promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proceedings of the National Academy of Sciences of the United States of America Nat1 Acad Sci USA. 2001;98:7783–7788. doi: 10.1073/pnas.141224398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pearson G, Bumesteir R, Henry DO, Cobb MH, White MA. Uncoupling Raft from MEK1I2 impairs only a subset of cellular responses to Raf activation. Journal of Biological Chemistry. 2000;275:37303–37306. doi: 10.1074/jbc.C000570200. [DOI] [PubMed] [Google Scholar]
- 33.Yuspa SH, Hawley-Nelson P, Koehler B, Stanley JR. A survey of transformation markers in differentiating epidermal cell lines in culture. Cancer Research. 1980;40(12):469–703. [PubMed] [Google Scholar]