

Abstract
Background
Endogenous adenosine can protect the overloaded heart against the development of hypertrophy and heart failure, but the contribution of A1 receptors (A1R) and A3 receptors(A3R) is not known.
Methods and Results
To test the hypothesis A1R and A3R can protect the heart against systolic overload, we exposed A3R gene deficient (A3R KO) mice and A1R KO mice to transverse aortic constriction (TAC). Contrary to our hypothesis, A3R KO attenuated 5 weeks TAC-induced left ventricular (LV) hypertrophy (ratio of ventricular mass/body weight increased to 7.6±0.3 mg/g in wild type (Wt) mice as compared with 6.3±0.4 mg/g in KO), fibrosis and dysfunction (LV ejection fraction decreased to 43±2.5% and 55±4.2% in Wt and KO mice, respectively). A3R KO also attenuated the TAC-induced increases of myocardial ANP and the oxidative stress markers 3′-nitrotyrosine (3′-NT) and 4-hydroxynonenal. In contrast, A1R-KO increased TAC-induced mortality, but did not alter ventricular hypertrophy or dysfunction compared to Wt mice. In mice in which extracellular adenosine production was impaired by CD73 KO, TAC caused greater hypertrophy and dysfunction, and increased myocardial 3′-NT. In neonatal rat cardiomyocytes induced to hypertrophy with phenylephrine, the adenosine analogue 2-chloroadenosine (CADO) reduced cell area, protein synthesis, ANP and 3′-NT. Antagonism of A3R significantly potentiated the anti-hypertrophic effects of CADO.
Conclusions
Adenosine exerts protective effects on the overloaded heart, but A3R act counter to the protective effect of adenosine. The data suggest that selective attenuation of A3R activity might be a novel approach to treat pressure overload-induced LV hypertrophy and dysfunction.
Keywords: hypertrophy, heart failure, oxidative stress, adenosine receptor
Introduction
Recently, we demonstrated that genetic deletion of CD73 (an ectonucleotidase that produces extracellular adenosine) exacerbated myocardial hypertrophy and heart failure resulting from LV pressure overload produced by transverse aortic constriction (TAC) 1, suggesting that endogenous extracellular adenosine can protect against maladaptive hypertrophy. Adenosine exerts multiple functions through activation of individual adenosine receptor subtypes 2-5. A1 receptors (A1R) and A3 receptors (A3R) are expressed in cardiomyocytes, and a substantial body of evidence indicates that adenosine can protect the heart during and after an ischemic insult 6, 7. Liao et. al. demonstrated that the adenosine analogue 2-chloroadenosine (CADO) also attenuated pressure overload induced LV hypertrophy through activation of the A1R 8. Similar to A1R, the A3R are Gi protein coupled receptors which have been shown to activate similar downstream signaling pathways 9, 10. A3R activation has also been reported to protect the heart against ischemic 11, 12 and doxorubicin induced damage 13. On the other hand, transgenic over-expression of the A1R 14 or A3R15, 16 promotes cardiac dilation and dysfunction, suggesting these receptors may also exert adverse effects on cardiac function. While we and others have demonstrated that adenosine protects against LV hypertrophy and maladaptive remodeling during pressure overload, the distinct contributions of the A1R and A3R to this protective effect are not known. Here we examined the effect of A1R KO and A3R KO on TAC-induced ventricular hypertrophy in vivo, and extended our examination to the roles of A1R and A3R in modulating hypertrophy in cultured neonatal rat cardiomyocytes, free from hemodynamic and neurohormonal factors that can influence the in vivo heart.
Methods
Mice
Male C57BL/6 (Taconic, Germantown, NY) body weight matched A3R KO mice 2 (crossed back to Taconic C57BL/6 mice at least 16 times), 8-12 weeks old, were used for TAC or control. A1R KO (129 background) and their control wild type mice (Wt) were generated as previously described 17. The CD73 KO strain and control Wt mice were generated as previously described 1, 18. This study was approved by the Institutional Animal Care and Use Committee of University of Minnesota.
Minimally invasive TAC Procedure
TAC of moderate (using a 26G needle to calibrate the degree of constriction) or severe (using a 27G needle) degree was created as previously described 19. To assure that similar pressure overload was produced in the KO and Wt mice, the TAC procedure was performed on KO and corresponding Wt mice on the same day by the same surgeon who was blinded as to the genotype of the mice.
Echocardiography
Mice were anesthetized with 1.5% isoflurane. Echocardiographic images were obtained with a Visualsonics Veve 770 system as previously described 19, 20.
Sample collection and Western blots
Myocardial samples for protein analysis were flash frozen in liquid nitrogen, weighted on an electronic balance, and stored in liquid nitrogen until transfer into a -80°C freezer where they were maintained until analysis. Samples for histological analysis were fixed in formaldehyde. Protein expression was analyzed using Western blots as previously described19 using antibodies against ANP (Penninsula Biolabs), 3-nitrotyrosine, 4-HNE (Millipore), cyclooxygenase-2 (COX-2), c-Jun N-terminal kinase (JNK), phosphorylated JNK (p-JNK Thr183/Tyr185) (Santa Cruz Biotechnology), eNOS (Transduction Laboratories) extracellular signal-regulated kinase (ERK), and phospho-ERKThr202/Tyr204, phospho-AktSer473 and phospho GSK-3βSer21/9 (Cell Signaling).
Histological staining and measurement of fibrosis
Tissue sections (6μm) from the central portion of the LV were stained with Sirius Red (Sigma) for fibrosis 19, and FITC-conjugated wheat germ agglutinin (AF488, Invitrogen) to evaluate myocyte size. For mean myocyte size, the cross sectional area of at least 120 cells/sample and at least 4 samples/group were averaged.
Neonatal rat cardiomyocyte (NVM) isolation and culture
NVW were isolated from 2-day-old Sprague-Dawley rats as previously described1. To induce hypertrophy, cells were treated with 50μM phenylephrine for 48 hours. The stable adenosine analogue CADO(5μM) was used to activate adenosine receptors (the affinities of CADO at rat A1R and A3R are 9.3 nM and 1,890 nM, respectively) 22. The selective inhibitors DPCPX and MRS1191 were used at 5μM to block A1R and A3R, respectively. 5μM MRS1911 has been reported to selectively inhibit A3 receptor activation without affecting A1 receptor dependent responses 23. After treatment, cells were fixed with 4% paraformaldehyde and stained using Rhodamine conjugated Phalloidin (5 units/ml in PBS, Invitrogen), DAPI, ANP (Penninsula Biolabs) and 3′-NT (Millipore), followed by alexa fluor 488, or alexa fluor 633 labelled secondary antibodies (Invitrogen). Protein synthesis was measured over 48 hours of treatment in 96 well plates by H3-phenylalanine incorporation.
Data Analysis
All values are expressed as mean ± standard error. Kaplan-Meier survival analysis was performed with SigmaStat using the Gehan-Breslow test. Two-way analysis of variance (ANOVA) was used to test for differences among treatment groups, followed with pairwise multiple comparisons of Tuke’s Test. Statistical significance was defined as P< 0.05.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
A3R KO attenuated LV hypertrophy and dysfunction produced by moderate pressure overload
LV structure and function were not different between A3R KO and Wt mice under control conditions (Figure 1A-G), and histological staining of LV tissue showed no difference in cardiac myocyte size or relative fibrosis between A3R KO and Wt mice (Figure 1C-D). After 5 weeks of moderate TAC (using a 26G needle to calibrate the degree of TAC), ventricular weight and the ratio of ventricular weight to body weight were significantly lower in the A3R KO mice as compared with Wt mice (Figure 1A-B), indicating that loss of A3 receptors attenuated the TAC-induced myocardial hypertrophy. Histological staining showed that the A3R KO hearts had significantly less TAC induced increases of LV fibrosis and myocyte hypertrophy (Figure 1C-D, Figure S1). Thus, the lesser hypertrophy in the A3R KO hearts after TAC resulted from both reduced myocyte size and decreased fibrosis. The TAC induced mortality was not different between A3R KO and Wt mice (Figure S2).
Figure 1.

A3R KO significantly attenuates chronic moderate TAC-induced ventricular hypertrophy (A, B), cardiac myocyte hypertrophy (C), ventricular fibrosis (D), increased LV end-systolic diameter (E), ventricular dilation (F) and decreased ejection fraction (G). Heart rate was not different between Wt and A3R KO under corresponding conditions (H). *P<0.05 compared to the corresponding control; #p<0.05 compared to Wt-TAC.
Echocardiographic imaging of the heart 5 weeks after TAC demonstrated significant increases of LV end systolic diameter and LV end diastolic diameter in both A3R KO and Wt mice in comparison with mice of similar body weight without TAC (Figure 1E-F). However, TAC caused significantly less LV dysfunction in the A3R KO mice, as demonstrated by a higher ejection fraction and a smaller LV end systolic diameter (Figure 1E,1G). Myocardial ANP (biochemical marker for LV dysfunction) was increased in both Wt and A3R KO mice 5 weeks after TAC, but this increase was significantly less in the A3R KO mice (Figure 2). These data indicate that the presence of the A3 receptor exacerbated the LV hypertrophy and dysfunction in response to TAC.
Figure 2.

A3R KO significantly attenuates moderate TAC-induced increases of ventricular ANP, nitrotyrosine, 4-HNE and COX-2. TAC caused significant increases of ventricular TNFα in both A3R KO and wild type mice. A3R KO tended to decrease TNFα after TAC, but this difference was not significant (p=0.10). Data are normalized to Wt-TAC. *P<0.05 compared to the corresponding control; #p<0.05 compared to Wt-TAC.
Because recent studies using A3R KO mice demonstrated that attenuation of A3R signaling reduces the inflammatory response2, 24, 25 in several pathological conditions, we examined myocardial TNFα and COX-2. TAC resulted in significant increases of TNFα and COX-2 in the hearts of both Wt mice and in A3R KO mice (Figure 2). However, the increase of COX-2 was significantly less in the A3R KO mice as compared with Wt mice. The TAC-induced increase of TNFα tended to be less in the A3R KO mice (p=0.10). In addition, hearts from Wt mice had higher levels of 3′-NT and 4-HNE after TAC than did A3R KO hearts, implying that the A3R KO mice had lower levels of oxidative stress (Figure 2). eNOS uncoupling can be a source for increased oxidative stress19, 26, and we have found that the increase of myocardial eNOS protein after TAC was related to the degree of LV dysfunction19. Consistent with our previous report, myocardial eNOS protein was significantly increased in the Wt mice following TAC, and this response was attenuated in the A3R KO mice (Figure 2).
Activation of mitogen-activated protein kinases (MAPK) and the PI3K signaling pathway is often associated with increased oxidative stress27, 28 and the development of LV hypertrophy or heart failure 29-31. To examine signaling pathways related to the protective effect observed in the A3R KO mice after TAC, total-JNK and phosphorylated JNK, ERK, Akt and GSK-3β were determined (Figure 3). Under control conditions A3R KO had no effect on the myocardial content of total or phosphorylated ERK, JNK, Akt or GSK-3β. TAC caused significant increases of p-ERKThr202/Tyr204 and p-JNK Thr183/Tyr185, and the ratio to their total proteins in both KO and Wt mice. However, A3R KO significantly attenuated the TAC-induced increases of p-ERKThr202/Tyr204 and p-JNK Thr183/Tyr185 (Figure 3), indicating that A3R KO attenuated the TAC-induced activation of the MAPK signaling pathways. In addition, the TAC-induced increases of p-AktSer473 and p-GSK-3βSer21/9 were significantly attenuated in the A3R KO mice, suggesting decreased signaling through the PI3K-Akt pathway.
Figure 3.

Ventricular p-ERKThr202/Tyr204, total-ERK, p-JNK Thr183/Tyr185, total-JNK, p-AktSer473, total-Akt, p-GSK-3βSer21/9 and total-GSK-3β in A3R KO mice and Wt mice under control conditions and 5 weeks after moderate TAC. Data are normalized to Wt-TAC. *P<0.05 compared to the corresponding control; #p<0.05 compared to Wt-TAC.
A1R KO did not influence ventricular hypertrophy produced by TAC but exacerbated mortality following severe TAC
Although previous studies have demonstrated that either the adenosine analogue CADO 8, 32 or endogenous adenosine1 can protect the heart from pressure overload induced LV remodeling, the specific contribution of A1R activation has been controversial 8, 32. To determine whether A1R KO might exacerbate the degree of hypertrophy and myocardial dysfunction later during systolic overload, we studied mice 4 weeks after moderate TAC (using a 26G needle). This moderate systolic overload caused similar increases in the ratio of ventricular mass to body weight, LV end diastolic diameter, LV end-systolic diameter and LV wall thickness in A1R KO and Wt mice (Figure 4A-F). Moderate TAC of 4 weeks duration also caused similar decreases of LV ejection fraction in the two groups (Figure 4C). Although mortality tended to be higher in the A1R KO group during the 4 weeks following moderate TAC (5 out 17 mice died) as compared with Wt mice (2 out 17 wild type mice died), this difference was not significant (Figure 4H).
Figure 4.

A1R KO had no significant effect the increase of ventricular mass (A), the ratio of ventricular mass to body weight (B), decrease of LV ejection fraction (C), increase of LV diastolic diameter (D,E) or LV wall thickness (F,G) produced by moderate TAC of 4 weeks duration. A1R KO mice tended to have a higher mortality during four weeks following moderate TAC, but this difference was not significant (H)). *P<0.05 compared to the corresponding control.
As mice with severe LV dysfunction are more likely to die after TAC, the relatively higher TAC-induced mortality in the A1R KO mice than in the Wt mice might potentially have influenced ventricular weights of the surviving mice. That is, if the sicker A1R KO mice died early after TAC, the residual surviving animals might underestimate the overall response to systolic overload. Since the TAC-induced death occurred predominantly during the first 2 days after TAC, we determined the degree of hypertrophy 2 days after severe TAC when comparable numbers of A1R KO and wild type mice survived. As compared with sham surgery, at two days after severe TAC the ratio of ventricular weight to body weight was similarly increased in Wt (21± 2.5 %) and in A1R KO mice (22 ± 3.5%), indicating that A1R KO did not alter the acute hypertrophic response to severe pressure overload (Figure S3). Taken together, the data indicate that A1R KO had no significant influence on TAC-induced ventricular hypertrophy or dysfunction.
Because the A1R KO mice tended to have a higher mortality than their wild type controls in this initial study, we subsequently went on to examine whether this trend toward a higher mortality would be statistically significant when a more severe degree of systolic overload (using a 27G needle) was applied. When TAC of severe degree was applied, the excess mortality in the A1R KO animals did in fact become significant (Figure S4). To understand the nature of the TAC-induced increase in mortality in the A1R KO mice, ECG telemetry was performed in additional A1R KO and Wt mice. The results demonstrated that animals destined to die generally developed progressive sinus bradycardia with giant P-waves that progressed to high grade atrioventricular block with further bradycardia and death (Figure S5). Again, the degree of hypertrophy was not different between the surviving A1R KO and Wt mice after severe TAC.
Taken together, the data indicate that A1R KO had no significant influence on TAC-induced ventricular hypertrophy or dysfunction, but resulted in significantly greater mortality in mice subjected to severe TAC.
CD73 KO exacerbated oxidative stress and hypertrophy produced by moderate pressure overload
The reduction of extracellular adenosine production produced by CD73 KO significantly exacerbated the hypertrophy (Figure 5A-B), fibrosis (Figure 5D-E), myocyte hypertrophy (Figure 5C, 5E), LV dilation and decrease of LV ejection fraction produced by moderate TAC of 4 weeks duration (Figure S6). CD73 KO also exacerbated the TAC-induced increases of ventricular ANP and TNFα (Figure S7). In addition, CD73 KO exacerbated the TAC-induced increase of myocardial 3-nitrotyrosine (Figure S7), indicating increased oxidative stress. To validate these findings, we examined the ability of adenosine analogue CADO to rescue the increased ventricular hypertrophy produced by TAC in the CD73 KO mice. We found that CADO attenuated the myocardial hypertrophy produced by moderate TAC of 2 weeks duration in the CD73 KO mice (Figure S8).
Figure 5.
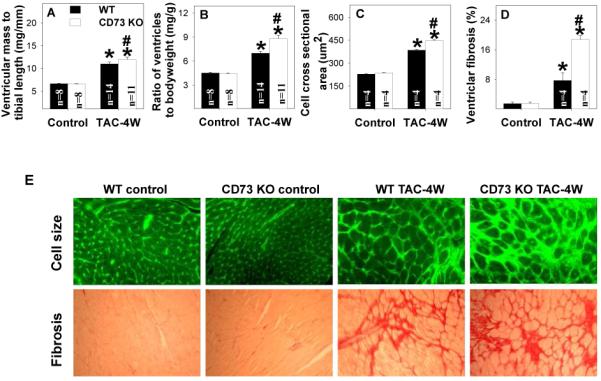
Disrupting extracellular adenosine production by CD73 KO exacerbated ventricular hypertrophy (A, B), cardiomyocyte hypertrophy (C,E) and ventricular fibrosis (D,E) produced by 4 weeks of moderate TAC. *P<0.05 compared to the corresponding control; #p<0.05 compared to Wt-TAC.
The A3R Antagonist MRS1911 Potentiates the Anti-hypertrophic Effect of CADO in Neonatal Cardiomyocytes
Understanding the role of A1R and A3R in the response of the cardiomyocytes to systolic overload in vivo may be complicated by effects of adenosine on blood flow, neurohormonal responses and inflammatory or paracrine responses. Therefore, we sought to determine the role of A1R and A3R in isolated cardiomyocytes in the setting of saturating levels of the non-selective adenosine analogue CADO. We previously demonstrated that CADO or adenosine reduced phenylephrine (PE) induced hypertrophy and ANP expression in neonatal cardiomyocytes 1. To examine the role of A1R and A3R in mediating anti-hypertrophic effects of CADO, we treated cells with 50μM PE and 5μM CADO in the presence or absence of selective A1R and A3R antagonists, and then measured cell area, protein synthesis and the oxidative stress marker 3′-NT. PE increased cardiomyocyte protein synthesis (Figure 6A, E), cell area (Figure 6B), ANP expression (Figure 6C) and 3′-NT production (Figure 6D-E) over 48 hours of treatment, while CADO significantly attenuated the PE-induced increases in these variables. Blocking A1R with DPCPX slightly reversed the CADO induced reductions of cell area (Figure 6B), protein synthesis (Figure 6A) and ANP levels (Figure 6C) in the PE treated cells. Inhibition of A1R caused a substantial increase in 3′-NT (Figure 6D), suggesting a role for A1R in modulating oxidative stress in the hypertrophying myocytes. Inhibition of the A3R with MRS1191 reduced protein synthesis, ANP expression and 3′-NT production beyond the reduction caused by CADO alone (Figure 6). The reduction in 3′-NT by MRS1191 was confirmed by western blot analysis (data not shown). The reduction in hypertrophy by the A3R antagonist was associated with reduced sustained activation of the MAP kinases ERK and JNK (Figure S9). These results suggest that A3R contributes to increased oxidative stress, higher sustained activation of ERK and JNK, and an increased hypertrophic response to PE.
Figure 6.

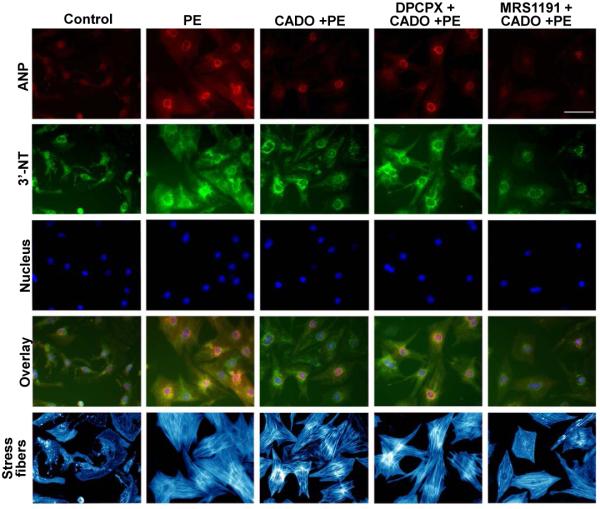
Addition of the A3R antagonist MRS1191 to CADO treated cells further attenuated the PE-induced increases of protein synthesis (A,E), and the expression of ANP (C,E) and 3′-NT (D,E) in cultured rat cardiomyocytes. MRS1191 did not further decrease the average cell area (B,E). *P<0.05 between the indicated groups.
Discussion
To the best of our knowledge, this is the first report assessing the effect of A1R KO and A3R KO on chronic pressure overload induced ventricular hypertrophy and contractile function. The major new finding is that A3R KO attenuated TAC-induced LV hypertrophy, fibrosis, oxidative stress and dysfunction. Since adenosine has been reported to be cardioprotective in the setting of chronic pressure overload 1, 8, the finding that disruption of A3R attenuated the TAC-induced ventricular hypertrophy and dysfunction is unexpected and intriguing. Our finding that the A3R antagonist MRS1191 augmented the anti-hypertrophic effect of CADO in PE treated isolated cardiomyocytes is in agreement with the concept that selective A3R blockade can enhance the beneficial effect of adenosine. These results support the novel concept that the adenosine A3R exerts adverse effects in the pressure overloaded heart, and suggest that A3R blockade may havepotential to protect the heart against pressure overload-induced oxidative stress, LV remodeling and contractile dysfunction.
The effect of the A1R on LV remodeling is controversial. CADO has been reported to attenuate TAC-induced LV hypertrophy in mice through A1R activation 8. Furthermore, an A1R antagonist was reported to attenuate the antihypertrophic effect of CADO in vitro 33. However, a subsequent study from the same group reported that A1R blockade had no effect on infarct-induced cardiomyocyte hypertrophy or LV remodeling in rats 32. We have observed that moderate A1R overexpression in mice failed to exert a beneficial effect on myocardial infarct induced ventricular remodeling (unpublished data). It is unclear why A1R blockade would attenuate the anti-hypertrophic effect of CADO, while A1R KO had no effect on TAC-induced hypertrophy. Nevertheless, the present finding that A1R KO exacerbated the death rate in mice exposed to severe TAC demonstrates that activation of the A1R can exert some degree of cardioprotection in the pressure overloaded heart.
Although no previous reports have directly examined the effect of A3R KO on systolic overload-induced ventricular remodeling, there is evidence that A3R signaling can affect cardiac structure and function. Thus, transgenic mice with cardiac specific over-expression of A3R developed a dilated cardiomyopathy characterized by increased ventricular mass, LV dilation, expression of biomarkers of hypertrophy, bradycardia and systolic dysfunction 15, 16, suggesting that chronically augmented A3R signaling in the heart is detrimental.
The MAPK and PI3K signaling pathways are often activated in response to extracellular stresses such as inflammation or oxidative stress 28, and have been shown to contribute to cardiac hypertrophy and heart failure. The increased myocardial oxidative stress after TAC in the present study, associated with activating phosphorylations of p-AktSer473, p-ERKThr202/Tyr204 and p-JNK Thr183/Tyr185 and inactivating phosphorylation of GSK3βSer21/9, is consistent with previous reports 19,34. The decreases in TAC induced oxidative stress, p-ERKThr202/Tyr204, p-JNK Thr183/Tyr185, p-AktSer473 and p-GSK3βSer21/9 as a result of A3R KO likely contributed to the lesser degrees of fibrosis and cardiac myocyte hypertrophy in the A3 KO mice. TAC-induced ventricular hypertrophy is associated with increased eNOS expression 19 and eNOS uncoupling 26, so that attenuation of the increase of eNOS in the A3R KO mice after TAC may have contributed to the decreased oxidative stress in this strain.
The effect of A3R on activation of myocardial PI3K/Akt signaling pathways in vivo has not been previously reported. However, the A3R agonist 2-chloro-N(6)-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide (Cl-IB-MECA) or adenosine dose- and time-dependently increased p-AktSer473 in cultured neonatal rat cardiomyocytes9 and A375 human melanoma cells 35, which is consistent with our finding that A3R KO attenuated the increase of myocardial p-AktSer473 and its downstream target p-GSKSer21/9 after TAC. Similarly, previous studies have reported that the A3R agonist Cl-IB-MECA or adenosine can activate p-ERKThr202/Tyr204 in cultured cardiomyocytes 36 and tumor cell lines 37, while the increase of p-ERKThr202/Tyr204 in response to A3R agonist is PI3K/Akt dependent 10. The finding that A3 receptor activation increased p-ERKThr202/Tyr204 in cultured cell lines 37 is conceptually consistent with our finding that A3R KO attenuated the TAC-induced increase of p-ERKThr202/Tyr204. The effect of A3R on p-JNK Thr183/Tyr185 has not been previously reported.
The mechanism by which A3R KO protected the heart against the LV hypertrophy and dysfunction produced by TAC is of considerable interest. A3R are expressed in both cardiac myocytes and inflammatory cells. Our data demonstrating that antagonism of the A3R further reduced oxidative stress and expression of ANP in CADO treated cardiomyocytes indicates that the A3R can contribute to oxidative stress and the hypertrophic response independent of the paracrine effects or inflammatory response that occur in vivo. The decrease in nitrotyrosine production in the isolated cardiomyocytes was accompanied by decreases of ERK and JNK activation, similar to the reduced activation of these enzymes in A3R KO mice. These results are consistent with numerous reports associating oxidative stress with activation of MAPK signaling 28, 38, 39.
In addition to a direct role of A3R on cardiomyocyte hypertrophy and oxidative stress, the A3R has also been demonstrated to modulate the inflammatory response. Specifically, the A3R appears important for mast cell degranulation 2, neutrophil chemotaxis 40, and infiltration of inflammatory cells 24, 25. It is possible that A3R mediated augmentation of the inflammatory response to the pressure overload produced by TAC could have exacerbated LV hypertrophy and dysfunction. Guo et al demonstrated that inflammatory cell accumulation and infarct area were decreased in A3R KO mice as compared to wild type mice 24 hours after ischemia-reperfusion injury 41, suggesting that the A3R can promote an increased inflammatory response in the heart. Our finding that A3R KO attenuated the TAC-induced increase of COX-2 and tended to decrease TNFα after TAC supports a role for the A3R in the TAC-induced myocardial inflammatory response.
The finding that A3R KO enhanced the antihypertrophic effect of the CADO in neonatal cardiomyocytes suggests the possibility of interactions between A3R and A1R. There is some previous support for such interactions. Thus, Norton et. al demonstrated that adenosine A2aR antagonists enhanced A1R-induced antiadrenergic responses in the heart, while A2aR agonists attenuated the antiadrenergic actions of A1R activation42. Although these investigators did not find interaction between A3R activity and A1R mediated antiadrenergic effects in the heart, interaction between A1R function and A3R has been demonstrated in the hippocampus, where A3R activation desensitized A1R dependent inhibition of excitatory neurotransmission by adenosine 43. Although examination of potential interactions between adenosine receptors was beyond the scope of the present report, this is clearly an area in need of further study.
Unfortunately, there are no highly potent and selective A3R antagonists available for mice. Therefore, a limitation of the present study is that the protective effect of A3R KO on the pressure overloaded heart could not be further confirmed by selective A3R inhibition with pharmacological compounds in an in vivo model.
In summary, A3R KO had no effect on LV structure or function in the unstressed heart, but significantly attenuated TAC-induced LV hypertrophy, fibrosis and dysfunction. Deletion of A3R also attenuated the TAC-induced increases of ventricular oxidative stress, COX-2, and the phosphorylation of p-ERKThr202/Tyr204, p-JNK Thr183/Tyr185, p-AktSer473 and p-GSK-3βSer21/9, suggesting that A3R mediated increases of oxidative stress and/or inflammation exacerbate detrimental ventricular remodeling by activation of the MAPK and PI3K-Akt pathways. A3R agonists are currently under development to treat tumors 44 inflammation 45 or cardiac injury. The present findings suggest that careful evaluation of the effect of selective A3R agonists on ventricular hypertrophy and dysfunction in the overloaded or diseased heart will be of importance.
Supplementary Material
Acknowledgments
Sources of Funding: This study was supported by NHLBI Grants HL71790 (YC) and HL21872 (RJB) from the National Institutes of Health. Dr. Xu is a recipient of an AHA Postdoctoral fellowship.
Footnotes
Disclosures: No conflicts declared.
Clinical Perspective
Adenosine A3 receptors (A3R) participate in cardioprotection against ischemia-reperfusion injury, and are involved in regulation of cell growth, neutrophil chemotaxis and activation of inflammatory cells. We examined whether the A3R can facilitate the adaptation of the left ventricle (LV) to pressure overload produced by transverse aortic constriction (TAC). Contrary to our expectation, mice with genetic ablation of the A3R developed less severe myocardial hypertrophy and LV dysfunction in response to TAC than did wild type mice, implying that A3R activation during pressure overload has a deleterious effect on the heart. In support of these functional data, A3R deletion decreased myocardial oxidative stress and the expression of pro-inflammatory cytokines. The findings suggest that selective A3R inhibition might have potential for treatment of pressure overload induced LV hypertrophy and dysfunction.
References
- (1).Xu X, Fassett J, Hu X, Zhu G, Lu Z, Li Y, Schnermann J, Bache RJ, Chen Y. Ecto-5′-Nucleotidase Deficiency Exacerbates Pressure-Overload-Induced Left Ventricular Hypertrophy and Dysfunction. Hypertension. 2008;51:1557–1564. doi: 10.1161/HYPERTENSIONAHA.108.110833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Salvatore CA, Tilley SL, Latour AM, Fletcher DS, Koller BH, Jacobson MA. Disruption of the A(3) adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Biol Chem. 2000;275:4429–4434. doi: 10.1074/jbc.275.6.4429. [DOI] [PubMed] [Google Scholar]
- (3).Donato M, Gelpi RJ. Adenosine and cardioprotection during reperfusion--an overview. Mol Cell Biochem. 2003;251:153–159. [PubMed] [Google Scholar]
- (4).Ashton KJ, Peart JN, Morrison RR, Matherne GP, Blackburn MR, Headrick JP. Genetic modulation of adenosine receptor function and adenosine handling in murine hearts: insights and issues. J Mol Cell Cardiol. 2007;42:693–705. doi: 10.1016/j.yjmcc.2006.12.012. [DOI] [PubMed] [Google Scholar]
- (5).Peart JN, Headrick JP. Adenosinergic cardioprotection: multiple receptors, multiple pathways. Pharmacol Ther. 2007;114:208–221. doi: 10.1016/j.pharmthera.2007.02.004. [DOI] [PubMed] [Google Scholar]
- (6).Tracey WR, Magee WP, Oleynek JJ, Hill RJ, Smith AH, Flynn DM, Knight DR. Novel N6-substituted adenosine 5′-N-methyluronamides with high selectivity for human adenosine A3 receptors reduce ischemic myocardial injury. Am J Physiol Heart Circ Physiol. 2003;285:H2780–H2787. doi: 10.1152/ajpheart.00411.2003. [DOI] [PubMed] [Google Scholar]
- (7).Jacobson KA, Costanzi S, Kim SK, Roh E, Joshi BV, Tchilibon S, Duong HT, Gao ZG. Action of nucleosides and nucleotides at 7 transmembrane-spanning receptors. Nucleosides Nucleotides Nucleic Acids. 2006;25:1425–1436. doi: 10.1080/15257770600919027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liao Y, Takashima S, Asano Y, Asakura M, Ogai A, Shintani Y, Minamino T, Asanuma H, Sanada S, Kim J, Ogita H, Tomoike H, Hori M, Kitakaze M. Activation of adenosine A1 receptor attenuates cardiac hypertrophy and prevents heart failure in murine left ventricular pressure-overload model. Circ Res. 2003;93:759–766. doi: 10.1161/01.RES.0000094744.88220.62. [DOI] [PubMed] [Google Scholar]
- (9).Germack R, Griffin M, Dickenson JM. Activation of protein kinase B by adenosine A1 and A3 receptors in newborn rat cardiomyocytes. J Mol Cell Cardiol. 2004;37:989–999. doi: 10.1016/j.yjmcc.2004.08.001. [DOI] [PubMed] [Google Scholar]
- (10).Hammarberg C, Fredholm BB, Schulte G. Adenosine A3 receptor-mediated regulation of p38 and extracellular-regulated kinase ERK1/2 via phosphatidylinositol-3′-kinase. Biochem Pharmacol. 2004;67:129–134. doi: 10.1016/j.bcp.2003.08.031. [DOI] [PubMed] [Google Scholar]
- (11).Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, Auchampach JA. Cl-IB-MECA [2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J Pharmacol Exp Ther. 2006;319:1200–1210. doi: 10.1124/jpet.106.111351. [DOI] [PubMed] [Google Scholar]
- (12).Liu GS, Richards SC, Olsson RA, Mullane K, Walsh RS, Downey JM. Evidence that the adenosine A3 receptor may mediate the protection afforded by preconditioning in the isolated rabbit heart. Cardiovasc Res. 1994;28:1057–1061. doi: 10.1093/cvr/28.7.1057. [DOI] [PubMed] [Google Scholar]
- (13).Shneyvays V, Mamedova L, Zinman T, Jacobson K, Shainberg A. Activation of A3 adenosine receptor protects against doxorubicin-induced cardiotoxicity. J Mol Cell Cardiol. 2001;33:1249–1261. doi: 10.1006/jmcc.2001.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Funakoshi H, Chan TO, Good JC, Libonati JR, Piuhola J, Chen X, MacDonnell SM, Lee LL, Herrmann DE, Zhang J, Martini J, Palmer TM, Sanbe A, Robbins J, Houser SR, Koch WJ, Feldman AM. Regulated overexpression of the A1-adenosine receptor in mice results in adverse but reversible changes in cardiac morphology and function. Circulation. 2006;114:2240–2250. doi: 10.1161/CIRCULATIONAHA.106.620211. [DOI] [PubMed] [Google Scholar]
- (15).Black RG, Jr., Guo Y, Ge ZD, Murphree SS, Prabhu SD, Jones WK, Bolli R, Auchampach JA. Gene dosage-dependent effects of cardiac-specific overexpression of the A3 adenosine receptor. Circ Res. 2002;91:165–172. doi: 10.1161/01.res.0000028007.91385.ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fabritz L, Kirchhof P, Fortmuller L, Auchampach JA, Baba HA, Breithardt G, Neumann J, Boknik P, Schmitz W. Gene dose-dependent atrial arrhythmias, heart block, and brady-cardiomyopathy in mice overexpressing A3 adenosine receptors. Cardiovasc Res. 2004;62:500–508. doi: 10.1016/j.cardiores.2004.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sun D, Samuelson LC, Yang T, Huang Y, Paliege A, Saunders T, Briggs J, Schnermann J. Mediation of tubuloglomerular feedback by adenosine: evidence from mice lacking adenosine 1 receptors. Proc Natl Acad Sci U S A. 2001;98:9983–9988. doi: 10.1073/pnas.171317998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Castrop H, Huang Y, Hashimoto S, Mizel D, Hansen P, Theilig F, Bachmann S, Deng C, Briggs J, Schnermann J. Impairment of tubuloglomerular feedback regulation of GFR in ecto-5′-nucleotidase/CD73-deficient mice. J Clin Invest. 2004;114:634–642. doi: 10.1172/JCI21851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang P, Xu X, Hu X, van Deel ED, Zhu G, Chen Y. Inducible nitric oxide synthase deficiency protects the heart from systolic overload-induced ventricular hypertrophy and congestive heart failure. Circ Res. 2007;100:1089–1098. doi: 10.1161/01.RES.0000264081.78659.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lu Z, Xu X, Hu X, Zhu G, Zhang P, van Deel ED, French JP, Fassett JT, Oury TD, Bache RJ, Chen Y. Extracellular superoxide dismutase deficiency exacerbates pressure overload-induced left ventricular hypertrophy and dysfunction. Hypertension. 2008;51:19–25. doi: 10.1161/HYPERTENSIONAHA.107.098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhang W, Anger T, Su J, Hao J, Xu X, Zhu M, Gach A, Cui L, Liao R, Mende U. Selective loss of fine tuning of Gq/11 signaling by RGS2 protein exacerbates cardiomyocyte hypertrophy. J Biol Chem. 2006;281:5811–5820. doi: 10.1074/jbc.M507871200. [DOI] [PubMed] [Google Scholar]
- (22).van Galen PJ, van Bergen AH, Gallo-Rodriguez C, Melman N, Olah ME, IJzerman AP, Stiles GL, Jacobson KA. A binding site model and structure-activity relationships for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:1101–1111. [PMC free article] [PubMed] [Google Scholar]
- (23).Dunwiddie TV, Diao L, Kim HO, Jiang JL, Jacobson KA. Activation of hippocampal adenosine A3 receptors produces a desensitization of A1 receptor-mediated responses in rat hippocampus. J Neurosci. 1997;17:607–614. doi: 10.1523/JNEUROSCI.17-02-00607.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Spruntulis LM, Broadley KJ. A3 receptors mediate rapid inflammatory cell influx into the lungs of sensitized guinea-pigs. Clin Exp Allergy. 2001;31:943–951. doi: 10.1046/j.1365-2222.2001.01087.x. [DOI] [PubMed] [Google Scholar]
- (25).Young HW, Molina JG, Dimina D, Zhong H, Jacobson M, Chan LN, Chan TS, Lee JJ, Blackburn MR. A3 adenosine receptor signaling contributes to airway inflammation and mucus production in adenosine deaminase-deficient mice. J Immunol. 2004;173:1380–1389. doi: 10.4049/jimmunol.173.2.1380. [DOI] [PubMed] [Google Scholar]
- (26).Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–1231. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Das S, Otani H, Maulik N, Das DK. Redox regulation of angiotensin II preconditioning of the myocardium requires MAP kinase signaling. J Mol Cell Cardiol. 2006;41:248–255. doi: 10.1016/j.yjmcc.2006.03.009. [DOI] [PubMed] [Google Scholar]
- (28).Sugden PH, Clerk A. Oxidative stress and growth-regulating intracellular signaling pathways in cardiac myocytes. Antioxid Redox Signal. 2006;8:2111–2124. doi: 10.1089/ars.2006.8.2111. [DOI] [PubMed] [Google Scholar]
- (29).Petrich BG, Wang Y. Stress-activated MAP kinases in cardiac remodeling and heart failure; new insights from transgenic studies. Trends Cardiovasc Med. 2004;14:50–55. doi: 10.1016/j.tcm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- (30).Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- (31).Clerk A, Cullingford TE, Fuller SJ, Giraldo A, Markou T, Pikkarainen S, Sugden PH. Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. J Cell Physiol. 2007;212:311–322. doi: 10.1002/jcp.21094. [DOI] [PubMed] [Google Scholar]
- (32).Wakeno M, Minamino T, Seguchi O, Okazaki H, Tsukamoto O, Okada K, Hirata A, Fujita M, Asanuma H, Kim J, Komamura K, Takashima S, Mochizuki N, Kitakaze M. Long-term stimulation of adenosine A2b receptors begun after myocardial infarction prevents cardiac remodeling in rats. Circulation. 2006;114:1923–1932. doi: 10.1161/CIRCULATIONAHA.106.630087. [DOI] [PubMed] [Google Scholar]
- (33).Headrick JP, Willems L, Ashton KJ, Holmgren K, Peart J, Matherne GP. Ischaemic tolerance in aged mouse myocardium: the role of adenosine and effects of A1 adenosine receptor overexpression. J Physiol. 2003;549:823–833. doi: 10.1113/jphysiol.2003.041541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- (35).Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, Maclennan S, Borea PA. A3 adenosine receptor activation inhibits cell proliferation via phosphatidylinositol 3-kinase/Akt-dependent inhibition of the extracellular signal-regulated kinase 1/2 phosphorylation in A375 human melanoma cells. J Biol Chem. 2005;280:19516–19526. doi: 10.1074/jbc.M413772200. [DOI] [PubMed] [Google Scholar]
- (36).Germack R, Dickenson JM. Adenosine triggers preconditioning through MEK/ERK1/2 signalling pathway during hypoxia/reoxygenation in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 2005;39:429–442. doi: 10.1016/j.yjmcc.2005.06.001. [DOI] [PubMed] [Google Scholar]
- (37).Gessi S, Merighi S, Varani K, Cattabriga E, Benini A, Mirandola P, Leung E, Mac LS, Feo C, Baraldi S, Borea PA. Adenosine receptors in colon carcinoma tissues and colon tumoral cell lines: focus on the A3 adenosine subtype. J Cell Physiol. 2007;211:826–836. doi: 10.1002/jcp.20994. [DOI] [PubMed] [Google Scholar]
- (38).McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal. 2006;8:1775–1789. doi: 10.1089/ars.2006.8.1775. [DOI] [PubMed] [Google Scholar]
- (39).Takano H, Zou Y, Hasegawa H, Akazawa H, Nagai T, Komuro I. Oxidative stress-induced signal transduction pathways in cardiac myocytes: involvement of ROS in heart diseases. Antioxid Redox Signal. 2003;5:789–794. doi: 10.1089/152308603770380098. [DOI] [PubMed] [Google Scholar]
- (40).Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–1795. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- (41).Guo Y, Bolli R, Bao W, Wu WJ, Black RG, Jr., Murphree SS, Salvatore CA, Jacobson MA, Auchampach JA. Targeted deletion of the A3 adenosine receptor confers resistance to myocardial ischemic injury and does not prevent early preconditioning. J Mol Cell Cardiol. 2001;33:825–830. doi: 10.1006/jmcc.2001.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Norton GR, Woodiwiss AJ, McGinn RJ, Lorbar M, Chung ES, Honeyman TW, Fenton RA, Dobson JG, Jr., Meyer TE. Adenosine A1 receptor-mediated antiadrenergic effects are modulated by A2a receptor activation in rat heart. Am J Physiol. 1999;276:H341–H349. doi: 10.1152/ajpheart.1999.276.2.H341. [DOI] [PubMed] [Google Scholar]
- (43).Dunwiddie TV, Diao L, Kim HO, Jiang JL, Jacobson KA. Activation of hippocampal adenosine A3 receptors produces a desensitization of A1 receptor-mediated responses in rat hippocampus. J Neurosci. 1997;17:607–614. doi: 10.1523/JNEUROSCI.17-02-00607.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ohana G, Bar-Yehuda S, Arich A, Madi L, Dreznick Z, Rath-Wolfson L, Silberman D, Slosman G, Fishman P. Inhibition of primary colon carcinoma growth and liver metastasis by the A3 adenosine receptor agonist CF101. Br J Cancer. 2003;89:1552–1558. doi: 10.1038/sj.bjc.6601315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Baharav E, Bar-Yehuda S, Madi L, Silberman D, Rath-Wolfson L, Halpren M, Ochaion A, Weinberger A, Fishman P. Antiinflammatory effect of A3 adenosine receptor agonists in murine autoimmune arthritis models. J Rheumatol. 2005;32:469–476. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.