

Abstract
Recessive mutations in parkin are the most common cause of familial early-onset Parkinson's disease (PD). Recent studies suggest that certain parkin mutants may exert dominant toxic effects to cultured cells and such dominant toxicity can lead to progressive dopaminergic (DA) neuron degeneration in Drosophila. To explore whether mutant parkin could exert similar pathogenic effects to mammalian DA neurons in vivo, we developed a BAC (bacterial artificial chromosome) transgenic mouse model expressing a C-terminal truncated human mutant parkin (Parkin-Q311X) in DA neurons driven by a dopamine transporter promoter. Parkin-Q311X mice exhibit multiple late-onset and progressive hypokinetic motor deficits. Stereological analyses reveal that the mutant mice develop age-dependent DA neuron degeneration in substantia nigra accompanied by a significant loss of DA neuron terminals in the striatum. Neurochemical analyses reveal a significant reduction of the striatal dopamine level in mutant mice, which is significantly correlated with their hypokinetic motor deficits. Finally, mutant Parkin-Q311X mice, but not wild-type controls, exhibit age-dependent accumulation of proteinase K-resistant endogenous α-synuclein in substantia nigra and colocalized with 3-nitrotyrosine, a marker for oxidative protein damage. Hence, our study provides the first mammalian genetic evidence that dominant toxicity of a parkin mutant is sufficient to elicit age-dependent hypokinetic motor deficits and DA neuron loss in vivo, and uncovers a causal relationship between dominant parkin toxicity and progressive α-synuclein accumulation in DA neurons. Our study underscores the need to further explore the putative link between parkin dominant toxicity and PD.
Keywords: Parkinson's disease, dopaminergic, transgenic, mice, neuron death, neuropathology
Introduction
Parkinson's disease (PD) is the second most common late-onset neurodegenerative disorder, with cardinal clinical features of resting tremor, rigidity, and bradykinesia (Fahn, 2003). Pathological hallmarks of PD include the loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc) and accumulation of cytoplasmic inclusions, called Lewy bodies (LBs) (McGeer et al., 1976; Trojanowski and Lee, 1998). To date, the mechanisms underlying the late-onset progressive DA neuron loss in PD remain unknown (Fahn and Sulzer, 2004). Although the vast majority of PD cases are sporadic, a small number of PD cases are familial, and among them, five disease-causing mutant genes have been identified: α-synuclein, parkin, PINK1, DJ-1, and LRRK2 (Dawson and Dawson, 2003; Moore et al., 2005; Hardy et al., 2006). Parkin (or PARK2) was identified as the most frequently mutated gene in familial early-onset recessive PD cases (Kitada et al., 1998; Hattori and Mizuno, 2004). Parkin encodes a ubiquitin E3 ligase, which exerts a variety of cellular functions, including proteasome-mediated degradation of critical neuronal substrates (Zhang et al., 2000; Chung et al., 2001; Imai et al., 2001; Huynh et al., 2003; Ko et al., 2005, 2006), EGF (epidermal growth factor) receptor trafficking (Fallon et al., 2006), and maintenance of mitochondrial integrity (Greene et al., 2003). Currently, it remains unclear which known function of parkin may be essential in protecting DA neurons from degeneration in vivo.
The loss of normal parkin function is an established genetic mechanism in the pathogenesis of early-onset PD, but the role of heterozygous parkin mutants as a risk factor for PD remains controversial and requires additional genetic studies (Klein et al., 2007). Multiple lines of evidence, including positron emission tomography and magnetic resonance imaging, transcranial sonography, and clinical exams consistently demonstrate that otherwise asymptomatic heterozygous parkin mutation carriers exhibit significant nigrostriatal dysfunction and mild extrapyramidal motor deficits (Hilker et al., 2001; Varrone et al., 2004; Khan et al., 2005). The genetic mechanism underlying the heterozygous parkin mutation effect on DA neurons remains unclear but could be attributable to either a partial loss of function (i.e., haploid insufficiency) or gain of function (i.e., dominant-negative or dominant gain of a toxic novel function) (West and Maidment, 2004; Klein et al., 2007). Consistent with the latter possibility, recent studies demonstrate that certain parkin mutants, but not wild-type (WT) parkin, exert dominant toxic effects in cultured cells (Hyun et al., 2002, 2005; Cookson et al., 2003; von Coelln et al., 2004b; Henn et al., 2005; Sriram et al., 2005; Wang et al., 2005a,b). Furthermore, truncated parkin mutations (with a stop codon at position 311 or parkin-Q311X) and two single-point mutations (T240R and R275W) provoke progressive DA neuron degeneration in transgenic (Tg) Drosophila (Sang et al., 2007; Wang et al., 2007). In both studies, the dominant toxic effects of the parkin mutants are selective to the DA neurons, because pan-neuronal expression of the mutants elicits DA neuron degeneration but spares several other neuronal types (Sang et al., 2007; Wang et al., 2007).
Despite the accumulating evidence in cell culture and fly models, no mammalian genetic studies have been conducted to address whether certain parkin mutants (e.g., a truncated parkin) can exert dominant toxicity in mammalian DA neurons. To address this question, we have developed a bacterial artificial chromosome (BAC)-mediated transgenic mouse model overexpressing parkin-Q311X in DA neurons. The parkin-Q311X mutation [replacing a glutamine residue at position 311 with a stop codon and resulting in a C-terminal truncation of 155 aa of parkin (see Fig. 1A)] has been found in a Turkish early-onset PD patient (Hattori et al., 1998). This patient had two mutant alleles, and because of the rarity of this mutation, no clinical or neuropathological information has been reported on whether parkin-Q311X carriers exhibit DA neuron dysfunction or neurodegeneration. However, because of the compelling cell culture studies (Hyun et al., 2002, 2005; Sriram et al., 2005) and Drosophila genetic studies (Sang et al., 2007; Wang et al., 2007) demonstrating the dominant toxic effects of this parkin mutant, we proceeded to test whether the same parkin mutant can elicit similar toxic effects in a mammalian model to cause progressive DA neuron degeneration. We show that these transgenic mice exhibit age-dependent hypokinetic motor deficits associated with significant DA neuron degeneration, thus demonstrating that this truncated mutant parkin can elicit cell-autonomous and dominant toxicity to mammalian DA neurons.
Figure 1.

Generation of Parkin-Q311X transgenic mice. A, Schematic representation of the mouse DAT gene structure. 5′-FLAG-tagged mutant parkin (Q311X) followed by a polyadenylation signal (PA) was inserted into exon 2 of the DAT gene preceding the endogenous translation initiation codon. B, Determination of transgene copy number using semiquantitative PCR. Equal amounts of genomic DNA from two Parkin-Q311X line A mice and two Parkin-Q311X line D mice were amplified using a human parkin-specific primer [A, Parkin-Q311X(A); D, Parkin-Q311X(D)]. The loading per lane was controlled using PCR amplicons of a ribosomal 18S DNA. Band densities were normalized to human genomic DNA (assumed to have 2 copies of endogenous parkin) amplification band. C, parkin-Q311X is a human mutation of parkin that lacks the second RING finger motif and IBR domain. This truncated parkin protein migrates in SDS-PAGE gel as a 30 kDa band as shown previously (Hyun et al., 2002; Henn et al., 2005). BAC transgenic mice (lines A, B, C, and D) and wild-type mice were killed at 3 months of age. Mouse striatal and midbrain sections were dissected and homogenized. Parkin was detected by Western blotting using a FLAG antibody. The asterisk (*) indicates nonspecific staining of FLAG antibody in wild-type mouse brain tissue. D, Immunoprecipitation of mutant protein using FlagM2 affinity gel and detected with FLAG-M2 antibody in brains of Parkin-Q311X(A) and wild-type littermates.
Materials and Methods
Generation of BAC transgenic mice.
A 200 kb mouse BAC (RP23-408F13) containing the 50 kb Slc6a3 [dopamine transporter (DAT)] coding region, 100 kb 5′-flanking region, and 40 kb 3′-flanking sequence was identified through a database search and obtained from the BACPAC Resource Center (Oakland Children's Hospital, Oakland, CA). FLAG-tagged mutant parkin carrying the Q311X mutation followed by a polyadenylation signal was inserted into exon 2 of the Slc6a3 gene preceding the endogenous translation initiation codon according to an established protocol (Gong and Yang, 2005). Maxiprep DNA was prepared from the modified Slc6a3-parkin(Q311X) BACs and purified through Sepharose CL-4B columns, and DNA from each fraction was separated on a pulsed field gel to select the fractions with intact BAC DNA. The intact BAC DNA fraction was selected and microinjected into fertilized FvB mouse zygotes to generate BAC transgenic mice. Two of the Parkin-Q311X lines, designated lines A and D, were subsequently expanded by crossing with wild-type FvB mice. The heterozygote transgenic mice and their wild-type littermates were used for the subsequent studies. Mouse care in the current study was in accordance with the Public Health Service's Guide for the Care and Use of Laboratory Animals. The procedures were approved by the Institutional Animal Care and Use Committee at University of California at Los Angeles (UCLA). Veterinarian care was provided by the UCLA Department of Laboratory Medicine. Animals were housed in a standard barrier facility at UCLA, with up to four mice per cage, with food and water available ad libitum. They were housed in a temperature-controlled environment with 12 h light/dark cycle.
Behavioral analyses.
Parkin-Q311X and wild-type littermate control mice were tested on a battery of behavioral tests sensitive to dopaminergic dysfunction (Fleming and Chesselet, 2005; Hwang et al., 2005). Tests included the challenging beam traversal, the cylinder test of spontaneous activity, adhesive removal, and locomotor activity in the open field. Two of the Parkin-Q311X lines, designated lines A and D, were used for behavioral studies. Line A male and female Parkin-Q311X (males, 7–8; females, 3–5) and wild-type (male, 1; females, 7–8) mice from a total of nine litters were repeatedly tested on the challenging beam, for spontaneous activity in the cylinder, and in the adhesive removal test. For the beam and cylinder tests, mice were tested at 3, 6–9, and 16–19 months of age, whereas for the adhesive removal test, Parkin-Q311X and wild-type mice were tested at 3, 12, and 16 months of age. Similarly for line D, male and female Parkin-Q311X (males, 6–9; females, 2–3) and wild-type (males, 2–3; females, 3) mice from a total of six litters were repeatedly tested on the challenging beam, for spontaneous activity in the cylinder, and adhesive removal at 3, 6, and 16 months of age. For locomotor activity in the open field, separate cohorts of naive line A Parkin-Q311X (males, 2–10; females, 2–6) and wild-type (males, 3–5; females, 0–4) mice from a total of eight litters were tested at 6, 12, 16, and 21 months of age. For line D, separate cohorts of naive Parkin-Q311X (males, 2–9; females, 1–7) and wild-type (males, 3–4; females, 1–4) mice from a total of seven litters were tested at 6, 12, 16, and 20 months of age.
Locomotor and spontaneous activity.
Locomotion was measured with the open-field test using an automated Truscan mouse activity monitor cage (Coulbourn Instruments). Naive Parkin-Q311X mice and wild-type littermates were put into the open field for 15 min during the dark phase. Parameters were automatically monitored and analyzed, which included but were not limited to, horizontal moving distance, time spent moving, moving velocity, rearing, and center versus periphery movement. In addition, the cylinder test was used to measure spontaneous motor activity (Fleming et al., 2004, 2006; Hwang et al., 2005). Animals were placed in a small transparent cylinder (height, 15.5 cm; diameter, 12.7 cm) and videotaped. The cylinder was placed on a piece of glass with a mirror that was positioned at an angle beneath the cylinder to allow a clear view of movements on the ground and along the walls of the cylinder. The number of rears, forelimb and hindlimb steps, and time spent grooming were measured. Videotapes were viewed and rated in slow motion by an experimenter blind to the mouse genotype.
Challenging beam traversal.
Motor performance and coordination were measured with the challenging beam traversal test (Fleming et al., 2004; Hwang et al., 2005). Briefly, the beam was constructed out of Plexiglas (Plastics Zone) and consisted of four sections (25 cm each, 1 m total length), each section having a different width. The beam started at a width of 3.5 cm and gradually narrowed to 0.5 × 1 cm increments. Animals were trained to traverse the length of the beam starting at the widest section and ending at the narrowest, most difficult, section. The narrow end of the beam led directly into the animal's home cage. Animals received 2 d of training before testing. Training consisted of having each mouse traverse the length of the beam for five trials. To increase difficulty further, on the day of the test a mesh grid (1 cm squares) of corresponding width was placed over the beam surface leaving a ∼1 cm space between the grid and the beam surface. Animals were then videotaped while traversing the grid-surfaced beam for a total of five trials. Videotapes were viewed by an investigator blind to the mouse genotype and rated in slow motion for errors, number of steps made by each animal, and time to traverse across five trials. Error per step scores, time to traverse, and number of steps were calculated for Parkin-Q311X and wild-type mice across all five trials and averaged.
Adhesive removal.
Motor response to sensory stimuli was measured with a stimulation test adapted from rats for mice (Fleming et al., 2004). Briefly, small adhesive stimuli (Avery adhesive-backed labels; 1/4 inch round) were placed on the snout of the mouse, and the time to make contact with the label and the time to remove the label were recorded. If the animal did not remove the label within 60 s, then the experimenter removed it, and the trial for the next mouse was initiated. The shortest time for each measure of three trials was used for analysis.
Histology and immunohistochemistry.
Parkin-Q311X mice and wild-type littermates were perfused transcardially with 4% paraformaldehyde in 0.1 m PBS. Mouse brains were removed and postfixed with 4% paraformaldehyde for 12 h at 4°C. The brains were then dehydrated in 30% sucrose and frozen at −80°C. Forty micrometer coronal sections were cut on a Leica 1800 cryostat and cryoprotected for additional usage. For tyrosine hydroxylase (TH) staining, sections were washed in 0.1 m PBS and treated with H2O2 (1%) for 20 min to block endogenous peroxidase activity. The sections were incubated with 3% bovine serum albumin and 2% normal goat serum (Vector Laboratories) in 0.1 m PBS at room temperature for 30 min. Sections were then incubated with anti-TH antibody (1:800) (Pel-Freez) in 0.1 m PBS with 3% bovine serum albumin and 2% normal goat serum, 0.08% sodium azide, and 0.2% Triton X-100 for 24 h at room temperature. After three 10 min washes in 0.1 m PBS, the sections were incubated with biotinylated goat anti-rabbit antibody (1:200) (Vector ABC Elite) for 2 h at room temperature. After another three 10 min washes in 0.1 m PBS, the sections were incubated for 2 h in avidin–biotin complex (Vector ABC Elite) in 0.1 m PBS containing 0.2% Triton X-100. The staining was visualized by incubation with 3,3′-diaminobenzidine tetrahydrochloride (DAB) (Sigma-Aldrich) and H2O2 in 0.05 m Tris buffer, pH 7.6. Sections were subsequently dehydrated and mounted with Eukitt (Calibrated Instruments). For quantification of TH immunofluorescence in striatum, 40-μm-thick free-floating sections were rinsed in 0.1 m PBS, blocked with 10% normal goat serum containing 0.5% Triton X-100, and incubated at 4°C overnight with primary anti-TH antibody (Millipore Bioscience Research Reagents; 1:400). Sections were rinsed three times in 0.1 m PBS and incubated with a Cy3-labeled secondary antibody for 1 h (Millipore Bioscience Research Reagents AP132C; 1:400). After rinsing in 0.1 m PBS, sections were mounted on slides. For proteinase K (PK)-resistant α-synuclein immunostaining and double immunofluorescence with nitrotyrosine, sections were incubated with 10 μg/ml proteinase K (Promega) in 0.1 m PBS before incubation with monoclonal α-synuclein antibody (1:250; BD Biosciences; catalog #610787) and 3-nitrotyrosine (3-NT) antibody (2.5 μg/ml; Millipore; catalog #06-284).
Stereology.
To estimate the total number of neurons and TH-immunoreactive cells in SNc, design-based stereological procedures (Gundersen et al., 1988; West et al., 1991) were performed by an investigator who was blind to mouse genotypes. Briefly, age- and gender-matched mice were transcardially perfused, and their brains were postfixed and cryoprotected as described above. For the first batch of stereology studies at 16 months, four Parkin-Q311X(A) (male, 1; females, 3) and wild-type (male, 1; females, 3) mice were used. For the second batch of studies, six Parkin-Q311X(A) (males, 4; females, 2) and six wild-type (males, 4; females, 2) mice were used. For additional stereology studies, Parkin-Q311X(A) and their wild-type littermates at 3 months of age, TH-GFP mice at 16 months of age, and Parkin-Q311X(D) and wild-type littermate mice at 16 months of age were analyzed. Only male mice were used for line D at 16 months. Sections were cut into 40 μm coronal sections with a Leica cryostat. The first section for stereology analysis was randomly chosen from the foremost four sections containing the SNc. This section and every fourth section thereafter were stained with TH antibody as described above. The sections were then counterstained using 0.5% cresyl violet (Sigma-Aldrich) for total neuron number analysis. The total number of neurons in SNc was defined by the combined counting of both TH-positive neurons and Nissl-positive (cresyl violet staining)/TH-negative neurons in the SNc. Sections were dehydrated and defatted in xylene, and mounted with Eukitt (Calibrated Instruments). StereoInvestigator 6.0 software (MicroBrightField) was used to estimate the number of neurons and TH-immunoreactive cells in the SNc. The region of the mouse SNc (excluding the pars reticulata and ventral tegmental areas) for stereology counting was delineated under a 5× objective. The optical fractionator method was used to estimate the cell number at higher magnification with a 100× (1.30 numerical aperture) oil objective. Coefficient of sampling error (Gundersen's CE, m = 1) was used to determine the precision of the estimates or sampling variation within each brain. Within the current study, the CE was controlled to be <10% (Gundersen and Jensen, 1987; Gundersen et al., 1999).
In situ hybridization. The detailed in situ hybridization (ISH) protocol has been described previously (Lobo et al., 2006). Briefly, human parkin-Q311X cDNA was inserted into pBluescript (Stratagene). Plasmids were linearized with AscI and antisense RNA probes were generated and labeled with [35S]UTP (PerkinElmer) by in vitro transcription using Stratagene RNAmaxx High Yield Transcription kit (Stratagene). Brains from Parkin-Q311X and wild-type mice at 3 months of age were fresh frozen on dry ice, and 20 μm of coronal sections were cut by cryostat and mounted directly onto slides, followed by fixation in 4% paraformaldehyde and a rinse in 0.1 m PB. Sections were blow dried and then rinsed in 7.5 mg/ml glycine in 0.1 m PB, followed with a 0.1 m PB rinse, and an acetic anhydride (2.5 μl/ml)/tetraethylammonium (13 μl/ml), pH 8.0, rinse. Sections were dehydrated through a series of graded alcohols for 1 min each [50% EtOH, 70% EtOH, 95% EtOH, 100% EtOH, chloroform (5 min), 100% EtOH, and 95% EtOH] and subsequently blow dried before hybridization. Sections were hybridized overnight at 60°C in hybridization solution containing 50% formamide, 35% Denhardt's solution, 0.14× SSC, 10% dextran sulfate, 0.3 mg/ml salmon sperm DNA, 0.15 mg/ml yeast tRNA, 40 mm dithiotreitol with 10 million counts/ml 35S probes. Next day, the sections were treated with 20 μg/ml RNase A in 0.1 m Tris, pH 8.0, 0.5 m NaCl, and 1 mm EDTA at 45°C for 35 min and underwent a graded series of stringent 20 min SSC rinses containing 2.5 mg/ml sodium thiosulfate at 60°C. The sections were blow dried at room temperature and exposed to film for 4–5 d, and then subsequently dipped in K.5D emulsion (Polysciences). After 4–5 weeks, sections were developed and analyzed.
Laser-capture microdissection and quantitative real-time PCR.
For all quantitative real-time PCR (qPCR) experiments, male animals [Parkin-Q311X(A,D); n = 5 in each group] were killed by cervical dislocation. The age of the animals varied between 6 and 8 months. Brains were quickly removed and frozen in isopentane on dry ice. Starting from the caudal end, the substantia nigra (SN) was cut in 10 μm sections, which were mounted on plain glass slides and stored at −80°C until additional use. Before immunohistochemical staining for TH, sections were fixed in 100% ethanol and rehydrated in decreasing ethanol concentrations (95, 70, and 50%). Sections were briefly equilibrated in 1× PBS, and primary and secondary antibodies (Millipore Bioscience Research Reagents) were incubated in 1× PBS containing 0.5% acetylated bovine serum albumin (Sigma-Aldrich) for 5 and 3 min, respectively. Detection of the biotinylated secondary antibody was achieved by using the Vector ABC and DAB staining kits according to the manufacturer's instructions (Vector Laboratories). To visualize surrounding non-TH-positive cells, the sections were counterstained with cresyl violet (Sigma-Aldrich) for 1 min. The counterstaining increases the purity of cell samples by minimizing the inclusion of non-TH-positive cells. A total of 600 TH-positive neurons was excised from the SNc of three adjacent sections of each animal by laser-capture microdissection using a PixCell II (Molecular Devices) and pooled to one sample. RNA extraction and cDNA synthesis were performed as previously described (Kamme et al., 2004) with one minor modification. QIAGEN RNeasy MinElute columns (QIAGEN) were used instead of the Microcon YM-100 columns. qPCR analyses were performed on a Roche LightCycler 480 using the Roche LightCycler FastStart DNA MasterPLUS SYBR Green I mix (Roche Diagnostics). All samples were run in triplicate. Standard curves for determining the amplification efficiency were generated by 10-fold serial dilutions of plasmids or gel-purified PCR fragments. Aldehyde reductase (Akr1a4) and subunit A of succinate dehydrogenase (Sdha) were used to normalize the overall cDNA content of all samples. Size and purity of all amplification products were verified by agarose gel electrophoresis. For the human and mouse parkin, each qPCR sample contained the equivalent volume of 70 neurons of the cDNA samples. Because of their higher expression levels, mRNA quantities of TH were assessed in 20 neurons per qPCR sample, whereas for Slc6a3, Sdha, and Akr1a4 15 neurons per qPCR sample were used.
Quantification of TH immunofluorescence in striatum.
TH-immunostained sections from 16-month-old mice were mounted on slides and scanned using an Agilent DNA microarray (Agilent Technologies) scanner at 10 μm resolution and analyzed with ImageJ software, version 1.38x (National Institutes of Health; http://rsb.info.nih.gov/ij). TH-immunoreactive fibers in the striatum were measured in three slices per animal. For the rostral (bregma, 1.1 mm) and medial (bregma, 0.14 mm) portions of the striatum, measurements were performed in the dorsolateral, dorsomedial, ventrolateral, and ventromedial quadrants of the striatum. Because of its different anatomical layout, the caudal striatum (bregma, −0.22 mm) was divided into a dorsal, medial, and ventral area. Measurements were performed on the left and right striatum and then averaged.
Measurement of striatal dopamine content.
Brains were removed from the skull and flash-frozen in liquid nitrogen. Striatal punches (2 mm diameter; 500 μm thick) were weighed, homogenized in 250 μl of 0.1 m PCA (perchloric acid)/0.1% EDTA, containing isoproterenol (1 μm) and DHBA (dihydroxybutyrate) (0.1 μm) as internal standards, and centrifuged at 10,000 rpm for 20 min at 4°C. The supernatant was filtered through a 0.22 μm filter by centrifugation at 10,000 rpm in a Microfuge for 5 min at 4°C and frozen at −80°C. Samples were analyzed for DA using HPLC with electrochemical detection (HPLC-ECD) (Antec Leyden) using a mobile phase consisting of sodium acetate (50 mm), sodium dodecane sulfonate (0.50 mm), EDTA (10 μm), acetonitrile (12%), methanol (9%), pH 5.5, pumped at a rate of 200 μl/min (model LC-10AD; Shimadzu) through a 100 × 2 mm column (2 μm; Super-ODS C18; Tosoh Bioscience). The system was calibrated at regular intervals and provided a limit of detection of 0.5 fmol for a 5 μl injection of sample. The data were collected and analyzed using ChromPerfect software (Justice Innovations).
Western blotting and nitrotyrosine ELISA.
Mouse brain tissues were homogenized in a modified radioimmunoprecipitation assay (RIPA) buffer followed by centrifugation at 4°C for 15 min at 16,100 × g. Protein samples were heated in NuPAGE LDS buffer (Invitrogen) for 10 min at 70°C before loading. Proteins were resolved in 10% Bis-Tris NuPAGE gels (Invitrogen) using MOPS [3-(N-morpholino)propanesulfonic acid] running buffer. The protein was transferred onto PVDF (polyvinylidene difluoride) membranes using NuPAGE transfer buffer (Invitrogen). Immunoblots were probed with anti-FLAG M2 (Sigma-Aldrich; 1:1000), anti-α-synuclein (Millipore) and anti-tyrosine hydroxylase at 1:8000 dilutions in 5% blocking solution, and anti-β-actin (Sigma-Aldrich) at a 1:4000 dilution in 5% blocking solution. ECL Plus (or ECL Advance for synuclein blots) Western Blotting Detection reagents (GE Healthcare) were used for chemiluminescent detection. FLAG immunoprecipatation was performed according to the instructions of the kit manufacturer (Sigma-Aldrich). For nitrotyrosine ELISA, mice nigra sections were dissected out and homogenized in 10 vol of ice-cold modified RIPA buffer containing protease and phosphatase inhibitors. Supernatant fractions of the tissue were prepared by centrifugation at 14,000 × g for 30 min at 4°C. The indirect ELISA protocol was used to detect 3-nitrotyrosine (Northwest Life Science). A standard curve was generated using SigmaPlot software.
Cyclic voltammetry.
Electrochemical recording using cyclic voltammetry (CV) was adapted from that of Schmitz et al. (2001). Mice were killed and coronal brain slices (300 μm) were prepared on a vibratome. Slices were placed in a holding chamber in oxygenated artificial CSF (ACSF) at room temperature and were allowed to recover for at least 1 h before the start of the experiment. Slices were placed in a recording chamber and superfused with oxygenated ACSF at 36°C for the duration of the experiment. Electrochemical recordings were done using a carbon fiber electrode (CFE). The CFE was placed into the dorsal striatum ∼50 μm below the surface, and recordings were made using CV. During CV recordings, a triangular voltage wave (−400 to +900 mV at 280 V/s vs Ag/AgCl) was applied to the electrode every 100 ms. Striatal slices were stimulated with a bipolar stimulating electrode via an Iso-Flex stimulus isolating unit triggered by a Master-8 (A.M.P.I.). The current generated by the reduction and oxidation of dopamine on the electrode surface was converted to concentration based on a calibration in ACSF against a known concentration of dopamine. Slices were stimulated every 2 min by a single pulse stimulus or by paired stimulus with an interstimulus interval of 1, 2, 5, 10, 20, 30, or 60 s. To calculate the paired-pulse ratio (PPR), the response to the second pulse was normalized to the first, and the percentage recovery was plotted as shown in supplemental Figure S4 (available at www.jneurosci.org as supplemental material).
Statistical analysis.
Independent sample Student's t test and one-way ANOVA followed by Fisher's least significant difference (LSD) post hoc test were used for statistical analysis for open field, Western blot, RT-PCR, dopamine striatal tissue content, and stereology results. For the challenging beam traversal, a 2 × 3 mixed-design ANOVA was used to compare mean error per step scores, time to traverse, and number of steps across genotype (between factor) and age (repeated factor). Similarly, spontaneous activity in cylinder was analyzed using a 2 × 3 mixed-design ANOVA to compare mean number of rears, forelimb and hindlimb steps, and time spent grooming across genotypes and ages. Post hoc analysis for the challenging beam and spontaneous activity was done with Fisher's LSD. For adhesive removal, the best contact and removal times were compared for Parkin-Q311X and wild-type mice at each age using the Mann–Whitney U test. Statistical analyses were conducted with SPSS 13.0 (SPSS) for Windows or GB-STAT software (Dynamic Microsystems; 2000) for Macintosh. The level of significance was set at p < 0.05.
Results
Generation of BAC transgenic mice expressing parkin-Q311X in dopaminergic neurons
To test the potential dominant toxicity of a truncated mutant Parkin in a mammalian model, we used a transgenic approach in mice to express a human cDNA encoding a C-terminal truncated human mutant parkin-Q311X in DA neurons by using a murine Slc6a3 BAC (Fig. 1A). Because the human parkin locus is too large (∼1 Mb) to be accommodated in a single BAC, we chose a strategy to selectively express this mutant parkin in DA neurons, an experiment analogous to that conducted previously in Drosophila (Sang et al., 2007). In this design, we were able to address whether the cell-autonomous toxic effects of mutant parkin-Q311X is sufficient to elicit DA neuron dysfunction and/or degeneration in vivo. We obtained five independent Parkin-Q311X transgenic founders and selected two lines (lines A and D) with germ line transmission of the transgene and good breeding characteristics for subsequent colony expansion and phenotypic characterization. BAC transgene copy numbers in these lines were measured using a semiquantitative PCR method. As shown in Figure 1B, line A has two copies of the BAC transgene integrated in tandem, whereas line D has a single copy. To confirm the expression of truncated FLAG-tagged parkin-Q311X protein in the mice, we performed Western blot analyses using combined striatal and midbrain extracts from these mice using the FLAG-M2 antibody (Sigma-Aldrich). The truncated parkin-Q311X protein is expressed as a ∼30 kDa protein in the transgenic brains as shown by Western blot (Fig. 1C) and immunoprecipitation (detected using FLAG-M2 antibody) (Fig. 1D). Because the Slc6a3 promoter should drive restricted expression of the transgene in the DA neurons in the SNc and ventral tegmentum area (VTA), we next performed ISH using a 35S-labeled antisense RNA probe to detect human parkin-Q311X transcripts in the adult transgenic mice and wild-type littermates. Under a stringent hybridization condition, we detected parkin-Q311X expression in the SNc and VTA areas of transgenic, but not in the wild-type brains (Fig. 2A). This result is consistent with a recent study demonstrating that the same BAC (RP23-408F13) can drive the expression of Cre recombinase selectively in DA neurons in transgenic mice (Turiault et al., 2007). To confirm the expression of the FLAG-tagged parkin-Q311X protein in the DA neurons in SNc, we performed double immunofluorescent staining of 3-month-old brains with antibodies against murine Slc6a3 and FLAG, which tags the N-terminal of mutant parkin. As shown in Figure 2B, double-stained SNc DA neurons are only present in the transgenic mice but not in the wild-type controls.
Figure 2.
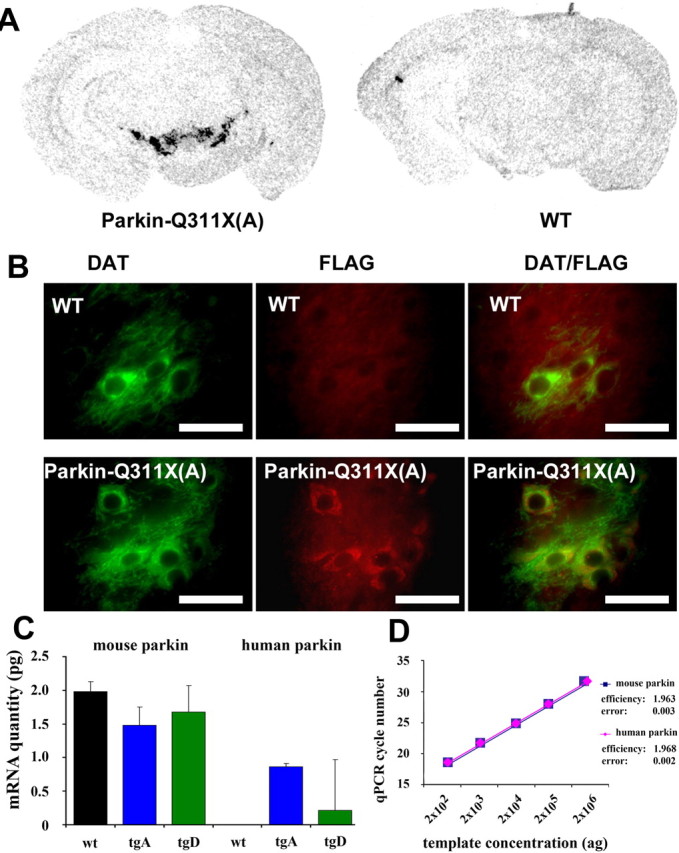
Selective expression of transgene in DA neurons. A, Representative photomicrograph of in situ hybridizations using 35S-labeled parkin-Q311X antisense RNA probe on midbrain sections from transgenic mice (line A) and wild-type mice at 3 months of age. B, DAT and FLAG double immunofluorescence. Midbrain sections from Parkin-Q311X(A) and wild-type mice were immunostained using DAT (green) and FLAG (red) antibodies. Scale bar, 12.5 μm. C, D, qPCR analyses of parkin expression in laser-capture microdissected dopaminergic neurons of the SNc. Error bars indicate SEM. C, Absolute quantification of human and mouse parkin mRNA levels in WT, transgenic line A (tgA), and line D (tgD) animals. mRNA quantities are given in picograms. Note that there is no human parkin PCR product in WT animals. D, The amplification efficiencies for the mouse and human parkin cDNAs, calculated as increase in PCR product for each PCR cycle, are virtually identical. Both values are close to the theoretical value of 2.0.
Finally, to rule out the possibility that any phenotypes in the Parkin-Q311X mice are simply attributable to an arbitrary high level of the transgene expression in the DA neurons, we determined the abundance of mutant parkin-Q311X mRNA in both lines A and D by quantitative real-time reverse transcriptase PCR (qRT-PCR) analyses using laser-captured TH(+) DA neurons from SNc (Fig. 2C,D). In line A, the expression level of the mutant human parkin-Q311X is at 42% of the murine endogenous murine parkin. Although the median level in line D is substantially lower than in line A, the difference does not reach statistical significance because of the relatively high variability in line D. The comparability of the human and mouse parkin expression data is ensured by the identical amplification efficiencies for both templates (Fig. 2D) (see Materials and Methods). In summary, these data demonstrate that the expression of the truncated human parkin in lines A and D in DA neurons is at a level that is equivalent to or below that expected from a heterozygous parkin allele, thus allowing us to test whether modest overexpression of mutant parkin is sufficient to elicit dominant toxic effects to the DA neurons in vivo.
Progressive motor phenotypes in Parkin-Q311X mice
We next used a well established battery of behavioral tests to assess nigrostriatal motor function in Parkin-Q311X and wild-type control mice (Fleming and Chesselet, 2005). These tests included monitoring spontaneous activity in a cylinder (the cylinder test), recording locomotor activity in an automated open-field box (the automated open-field test), examining motor performance and coordination (the challenging beam traversal test), and examining motor response to sensory stimuli (adhesive removal test) (Fleming et al., 2004, 2006; Fleming and Chesselet, 2005). These sensitive tests have been used to demonstrate motor dysfunction in variety of toxin and genetic rodent models of PD (Fleming et al., 2004, 2006; Fleming and Chesselet, 2005). For example, the Aphakia/Pitx3 knock-out mice (with developmental loss of DA in SNc) exhibit deficits in both the challenging beam test and cylinder test, and these deficits are reversible with l-DOPA (Hwang et al., 2005). As shown in supplemental Figure S1 (available at www.jneurosci.org as supplemental material), there was no significant difference between male and female mice in the open-field, cylinder, beam, and dot tests. Therefore, both male and female mice were included in the behavioral tests described in the following sections.
Our studies reveal that Parkin-Q311X mice exhibit multiple, late-onset hypokinetic motor deficits. A cohort of Parkin-Q311X (line A) and wild-type control littermate mice of mixed gender were tested independently at 3, 12, 16, and 21 months of age for spontaneous activity in the automated open-field test. Parkin-Q311X(A) mice demonstrate significantly progressive hypoactivity as measured in open field (independent groups measured at different ages) (Fig. 3A,B). For both floor plane entry and vertical plane entry measurement in open filed, two-way ANOVA analyzing time and genotype detected a main effect of genotype (F(1,55) = 12.093; p < 0.001) and time (F(3,55) = 16.073; p < 0.001). There was no significant genotype by time interaction (F(3,55) = 0.767; p = 0.517). In the cylinder and the challenging beam, mixed design 2 × 3 ANOVA revealed significant age-dependent deficits. A significant effect of age (F(2,38) = 8.52; p < 0.01) was observed in the challenging beam (Fig. 3C). Post hoc analysis with Fisher's LSD indicated that Parkin-Q311X mice made significantly more errors per step compared with the wild-type mice at 16–19 months of age (p < 0.01). A significant effect of age (F(2,38) = 4.13; p < 0.05) was also observed for rearing in the cylinder test (Fig. 3D). Post hoc analysis indicated that, at 16–19 months of age, rearing of Parkin-Q311X mice was significantly reduced compared with wild-type mice (p < 0.05). A main effect of genotype (F(1,19) = 6.49; p < 0.05) was observed for hindlimb stepping (Fig. 3E). Fisher's LSD indicated that at 3 and 16–19 months of age, Parkin-Q311X mice made significantly fewer hindlimb steps compared with wild-type mice (p < 0.05, p < 0.01, respectively). In addition, the adhesive removal test, which measures motor response to sensory stimuli, was used to evaluate sensorimotor function in the mutant and wild-type mice. Again, Parkin-Q311X mice were comparable with the controls at 3 and 6 months of age on repeated testing, but exhibited significant late-onset deficits in this test at 16–19 months of age (Fig. 3F,G) (Mann–Whitney U test, p < 0.05). Thus, four independent motor tests reveal that Parkin-Q311X(A) transgenic mice exhibit late-onset motor deficits that are consistent with nigrostriatal dysfunction.
Figure 3.
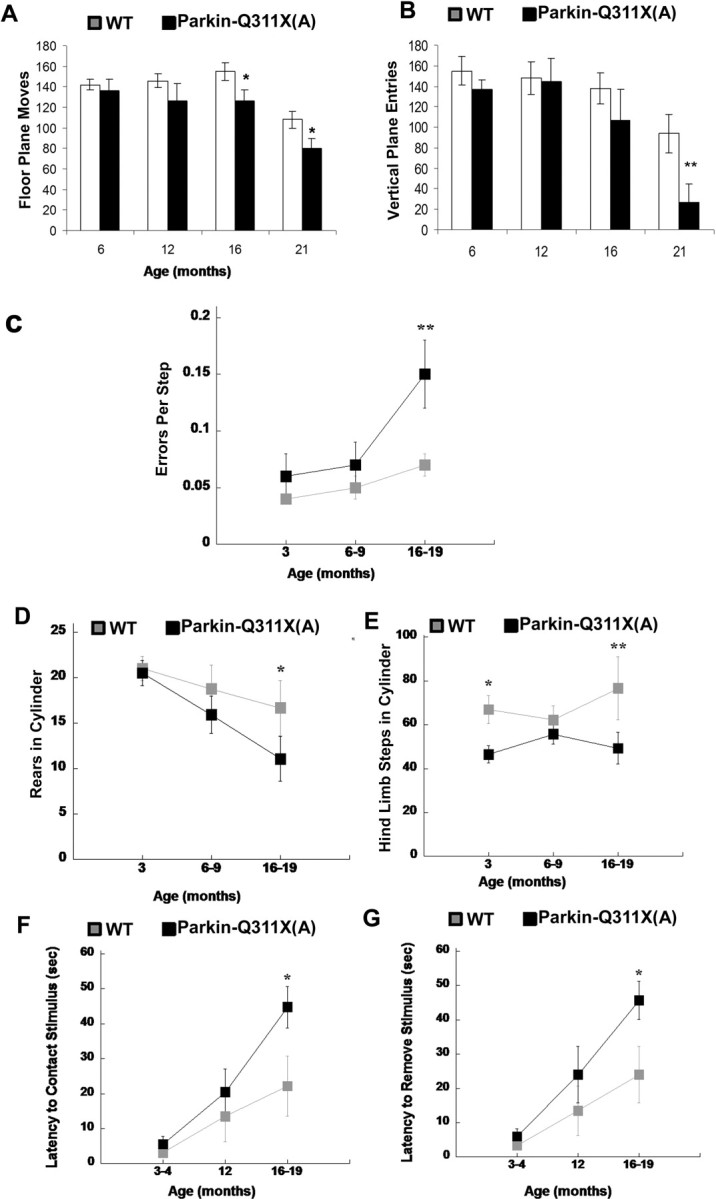
Progressive behavioral deficits in Parkin-Q311X transgenic mice. A, B, Locomotor activity in independent groups of naive Parkin-Q311X(A) (n = 12, 6, 7, 8 for different time points) and wild-type littermate (n = 6, 5, 7, 13 for different time points) mice was measured in the open field at 6, 12, 16, and 21 months of age. Parkin-Q311X mice demonstrate significant progressive hypoactivity as measured by floor plane moves and vertical plane entries in open field (significant genotype and time difference revealed by two-way ANOVA, p < 0.001; significant genotype difference at the same time point is indicated; *p < 0.05, independent sample Student's t test). C, Motor performance and coordination were repeatedly measured in Parkin-Q311X(A) (n = 12) and wild-type (n = 9) mice using the challenging beam. Errors per step were measured at 3, 6–9, and 16–19 months of age. Parkin-Q311X(A) mice made more errors per steps while traversing the beam compared with wild-type mice; **p < 0.01 compared with wild-type mice at the same age. D, E, Spontaneous activity of Parkin-Q311X(A) mice and wild-type littermates in the cylinder was measured at 3, 6–9, and 16–19 months. Rearing (D) and hindlimb (E) steps were measured. Transgenic mice were less active in each measure compared with wild-type mice at the older ages. *p < 0.05 compared with wild-type mice. F, G, Motor response to sensory stimuli was measured in the adhesive removal test at 3–4, 12, and 16–19 months of age [Parkin-Q311X(A) (n = 11) and wild-type (n = 8)]. Parkin-Q311X(A) mice had significantly slower contact (F) and removal time (G) compared with wild-type mice at 16–19 months of age (*p < 0.05, Mann–Whitney U test). Error bars indicate SEM.
To ensure that the progressive motor deficits observed in Parkin-Q311X(A) line mice were attributable to expression of the truncated mutant parkin and not attributable to artifacts of transgene integration in this particular mouse line, we administered a similar battery of behavioral tests to an independent Parkin-Q311X mouse line (line D). Because line D carries only a single copy of the BAC transgene, we expected the behavioral phenotype to be milder than in line A, which carries two copies of the BAC transgene (Fig. 1B). Consistent with this notion, Parkin-Q311X(D) mice also exhibited significant late-onset deficits in the automated open field at 20 months of age (but not before 16 months of age) (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material) (independent sample t test, p < 0.05 compared with wild-type littermates). They also exhibit deficits in the adhesive removal test at 13–16 months of age (supplemental Fig. S2E,F, available at www.jneurosci.org as supplemental material) (Mann–Whitney U test, p < 0.01 compared with wild-type littermates). Unlike line A mice, line D mice did not significantly differ from the wild-type controls in the challenging beam and the cylinder test up to 16 months of age (supplemental Fig. S2B,C, available at www.jneurosci.org as supplemental material). Thus, the late-onset behavioral deficits are present in two independent Parkin-Q311X lines; hence it is unlikely that these deficits are attributable to transgene integration site effects. Furthermore, line A with higher transgene copy number exhibits more robust and earlier onset hypokinetic motor deficits than line D, which is consistent with a dose-dependent, dominant mechanism elicited by mutant parkin.
To exclude the possibility that the late-onset behavioral deficits in the Parkin-Q311X mice are simply attributable to nonspecific long-term overexpression of any exogenous protein in DA neurons in the FvB inbred background, we used as a control another BAC transgenic mouse line in FvB background that overexpresses green fluorescent protein (GFP) in DA neurons using the TH promoter (Gong et al., 2003). We chose not to pursue, as a control model, BAC mice overexpressing wild-type parkin in DA neurons because wild-type human parkin has consistently been shown to be neuroprotective and not neurotoxic in vitro (Cookson, 2003; Henn et al., 2007) and in vivo (Yang et al., 2003; Lo Bianco et al., 2004). We tested a cohort of TH-GFP mice at 16 months using two of the behavioral tests, automated open field and adhesive removal test. TH-GFP mice did not differ from wild-type mice in locomotor activity as measured in open field (wild-type floor plane moves, 121.0 ± 4.76; TH-GFP floor plane moves, 129.2 ± 7.67; n = 5 for wild-type and n = 4 for TH-GFP mice; p > 0.05 compared with wild-type controls, independent sample Student's t test) and motor response to adhesive stimuli (wild-type reach time, 32.67 ± 13.02 s; TH-GFP mice reach time, 34.13 ± 6.50 s; n = 5 per genotype; p > 0.05 compared with wild-type controls, independent sample Student's t test) at 16 months of age. Thus, this control study confirms that overexpression of GFP, a neutral or mildly cytotoxic protein (Persons et al., 1998), in DA neurons of FvB mice fails to elicit the late-onset hypokinetic motor deficits seen in the Parkin-Q311X mice. Together, this study demonstrates that expression of a C-terminal truncated mutant parkin in DA neurons can elicit late-onset hypokinetic motor deficits consistent with nigrostriatal dysfunction in a mammalian model.
Age-dependent dopaminergic neuron phenotypes in Parkin-Q311X mice
To determine whether the progressive motor deficits in Parkin-Q311X mice could be associated with DA neuron degeneration, we performed a series of quantitative neuropathological analyses to study DA neurons in SNc and their terminals in the striatum. We first performed immunohistochemical staining with anti-TH antibodies to detect DA neurons and their processes in coronal midbrain sections from the Parkin-Q311X and wild-type mice. Age- and gender-matched transgenic and wild-type littermate mice at 3 and 16 months of age were used in this study. At 3 months, TH staining of SNc did not reveal any apparent differences between the Parkin-Q311X(A) mice and wild-type controls (Fig. 4A,B). At 16 months, the time point at which the Parkin-Q311X mice exhibit multiple motor deficits, we found that mutant but not wild-type mice exhibited loss of TH-immunoreactive neurons in the SNc (Fig. 4C,D). Consistent with these observations in the Parkin-Q311X(A) line, we also observed a similar reduction of TH-immunoreactive neurons in the SNc of Parkin-Q311X(D) line mice at 16 months of age. As an independent reporter to visualize such degeneration of DA neurons, we crossed TH-GFP mice with Parkin-Q311X(D) mice. As shown in supplemental Figure S3 (available at www.jneurosci.org as supplemental material), whereas the wild-type TH-GFP mice at 16 months of age (n = 2) have a full complement of GFP-expressing DA neurons in both SNc and VTA, the double transgenic mice (Parkin-Q311X(D) × TH-GFP; n = 2) exhibit marked loss of TH-GFP(+) DA neurons in the SNc, with relative sparing of those in the VTA (Fig. 4H; supplemental Fig. S2, available at www.jneurosci.org as supplemental material), a pattern resembling that observed in PD.
Figure 4.
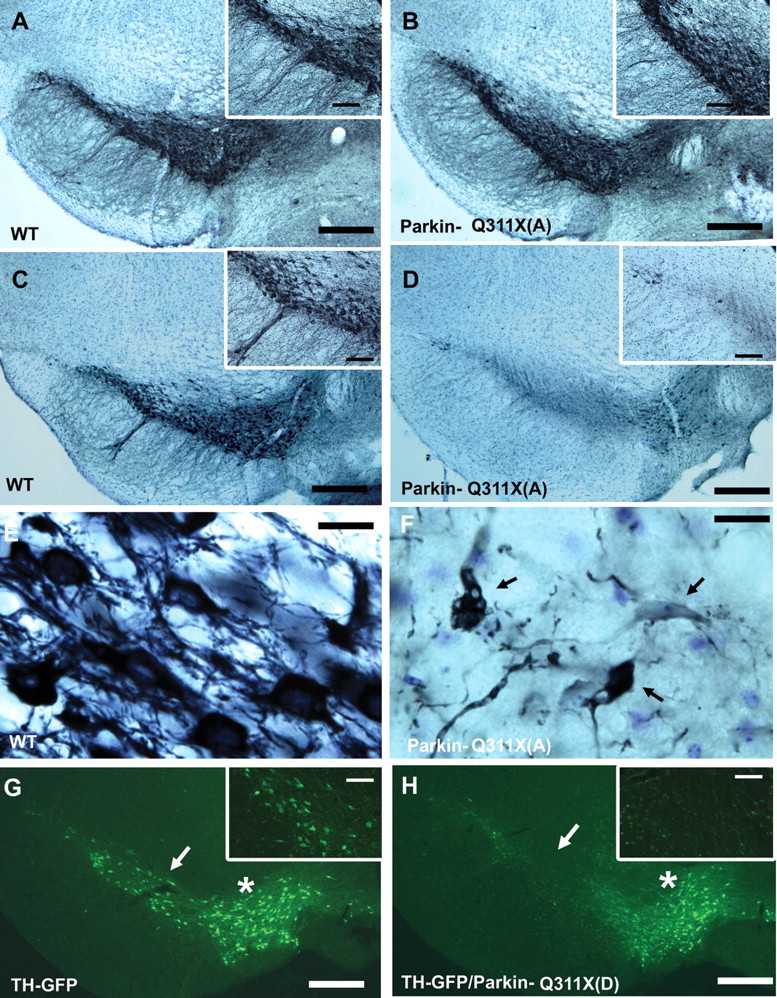
Progressive loss of SNc DA neurons in Parkin-Q311X transgenic mice. A, B, Photomicrographs of TH immunostaining of representative midbrain sections showing DA neurons in the SNc of Parkin-Q311X(A) and WT littermates at 3 months of age. Scale bar, 250 μm. The insets on the right top corner are higher-power views of the SNc area in A and B. Scale bar, 125 μm. C, D, Photomicrographs of TH immunostaining of midbrain sections for Parkin-Q311X(A) mice and wild-type littermates at 16 months of age (scale bar, 250 μm) and the higher-power view of the SNc area in C and D (scale bar, 50 μm). E, F, Higher-power view of the TH-immunoreactive neurons in Parkin-Q311X(A) mice and wild-type littermates at 16 months of age. Scale bar, 12.5 μm. G, H, Photomicrographs of direct visualization of GFP-expressing neurons in the representative midbrain sections from TH-GFP/Parkin-Q311X(D) mice and TH-GFP control mice at 16 months of age. Scale bar, 250 μm. The white arrows in the photomicrographs indicate SNc area. The white stars indicate the VTA.
To further determine whether the Parkin-Q311X mice exhibit progressive loss of TH(+) DA neurons versus loss of TH (or TH-GFP) expression but no cell loss, we performed unbiased stereological counting of both the TH(+) DA neurons and the total neurons in SNc at 3 and 16 months of age using age- and gender-matched mice. The total number of neurons in SNc is estimated via counting of both the TH(+) neurons and the TH(−)/Nissl (cresyl violet)-positive neurons in the SNc. At 3 months of age, the transgenic and wild-type mice have a comparable number of TH(+) and total neuronal cells, indicating that the Parkin-Q311X(A) mice do not exhibit developmental and early postnatal loss of DA neurons (Fig. 5A). At 16 months, however, the mutant mice show a significant reduction (∼40%) of TH-immunoreactive neurons in SNc, accompanied by a comparable reduction in the total number of neurons (∼30%) (Fig. 5B) (independent sample Student's t test, p < 0.05 compared with wild-type littermate mice). These data demonstrate that the Parkin-Q311X(A) mice exhibit late-onset DA neuron loss, with some reduction of TH gene expression. We then confirm the loss of DA neurons in the parkin-Q311X(A) mice using an independent cohort of age- and gender-matched mice at 16 months of age (n = 6 per genotype; independent sample Student's t test, p < 0.05 compared with wild-type littermates) (Fig. 5C).
Figure 5.
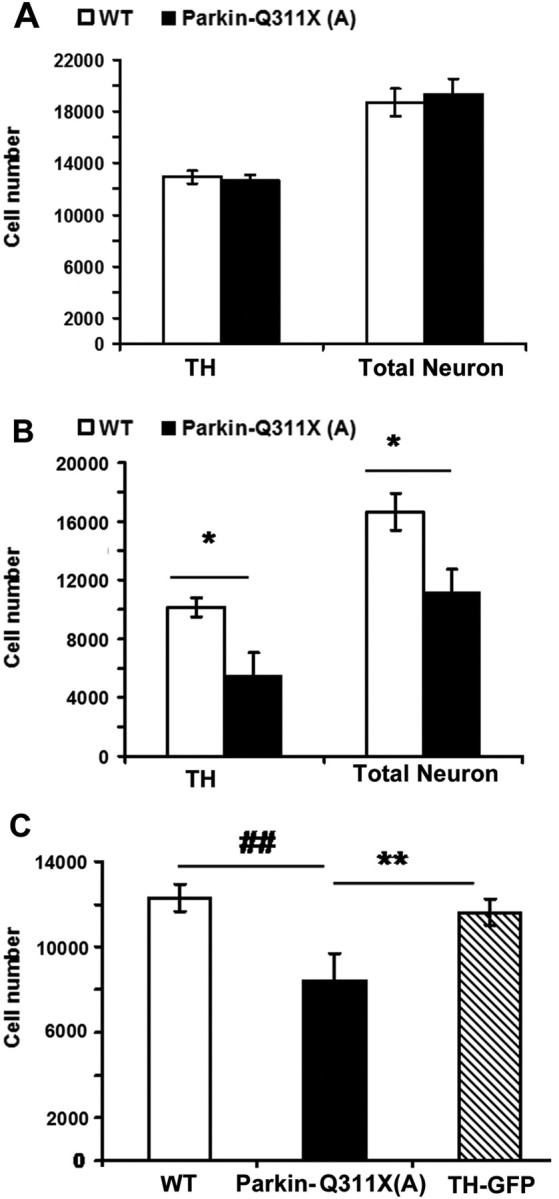
SNc DA neuron stereology analyses. A, TH-immunoreactive neurons were estimated using design-based stereology (Gundersen et al., 1988; West et al., 1991). Normal numbers of TH-expressing neurons in the SNc of Parkin-Q311X(A) mice at 3 months of age [n = 4 per genotype; TH, tyrosine hydroxylase-positive neuron numbers in the SNc; total number of neurons in SNc was estimated by counting of both TH(+) and Nissl(+)/TH(−) neurons in the SNc; p > 0.05 compared with wild-type littermates, independent sample Student's t test]. B, Significant loss of TH-immunoreactive neurons in the SNc of Parkin-Q311X(A) mice at 16 months of age (n = 4 per genotype; *p < 0.05 compared with wild-type littermates, independent sample Student's t test). C, Normal numbers of TH-immunoreactive neurons in the SNc of TH-GFP mice at 16 months of age [Parkin-Q311X(A), n = 6; WT, n = 6; TH-GFP, n = 3; **p < 0.01 compared with Parkin-Q311X(A) mice; ##p < 0.01 compared with Parkin-Q311X(A) mice, independent sample Student's t test]. Error bars indicate SEM.
To confirm that the loss of TH(+) DA neurons also occurs in the Parkin-Q311X(D) line mice, which have a single copy BAC transgene, we performed stereological counting of TH(+) DA neurons in the SNc [Parkin-Q311X(D): n = 11; wild-type: n = 10, male mice]. We find that the transgenic mice in line D also exhibit significant TH-immunoreactive neuron loss (independent sample Student's t test, p < 0.05 compared with wild-type littermates). However, the DA neuron loss at 16 months of age is milder (∼20% loss) in line D than in line A (∼40% reduction of TH-positive neurons), consistent with a transgene dosage-dependent neurodegeneration phenotype. Furthermore, we demonstrate that the TH-GFP mice in the same FvB inbred background do not exhibit any TH(+) DA neuron loss in the SNc at this age (independent sample Student's t test, p > 0.05 compared with wild-type littermates) (Fig. 5C), verifying that the progressive DA neuron pathology is not simply a phenomenon of transgenic overexpression of any exogenous protein in the DA neurons over a long period of time. Together, the analyses demonstrate that two independent lines of Parkin-Q311X mice exhibit the phenotype of late-onset DA neuron loss in SNc.
Striatal DA terminal phenotypes in Parkin-Q311X mice
Because asymptomatic heterozygous parkin mutation carriers exhibit evidence of striatal DA terminal deficits (Hilker et al., 2001; Binkofski et al., 2007), we hypothesized that Parkin-Q311X mice might have DA terminal deficits in the striatum, before or after the onset of motor deficits (i.e., beginning at 16 months of age).
To test the possibility that Parkin-Q311X mice may exhibit DA terminal dysfunction before the onset of motor deficits, we examined evoked dopamine release using CV, a functional test to examine striatal dopaminergic synaptic transmission (Schmitz et al., 2001; Mosharov and Sulzer, 2005), at 12–14 months of age (an upper age limit at which CV has been effectively performed before). Single pulse-evoked release, which is used as a model of normal release probability at DA terminals, did not differ between transgenics and their WT littermates (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material). This suggests that release kinetics is normal in these mice. We also measured the PPR to assess recovery at terminals. The PPR in transgenic mice did not differ from that in wild type (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material), suggesting that mutant Parkin-Q311X mice do not have deficits of recovery at this age.
We next examined the striatal expression of TH, a marker for DA neuron projections and terminals in the striatum. Using Western blot analyses with anti-TH antibodies, we found a modest but significant loss of striatal TH protein in the Parkin-Q311X(A) transgenic mice compared with the wild-type controls at 12–14 months of age (Fig. 6A) (Western blotting band were quantified using KODAK imaging software; independent sample Student's t test, p < 0.05), suggesting that detectable TH loss is present in the presymptomatic mutant mice. Consistent with the Western blot result, by quantitating TH immunoflurorescence in the striatum using a microarray scanner in 16-month-old mice (see Material and Methods), we again detected a significant loss of TH immunostaining in both the dorsolateral and ventrolateral regions in the rostral striatum of mutant mice compared with the wild-type littermates (Fig. 6B,C) (independent sample Student's t test, p < 0.05). Similar significant loss of TH immunofluorescence was observed in the middle but not caudal striatal sections (data not shown), suggesting that the caudal-striatum targeted subset of DA neuron terminals are relatively spared in Parkin-Q311X mice.
Figure 6.
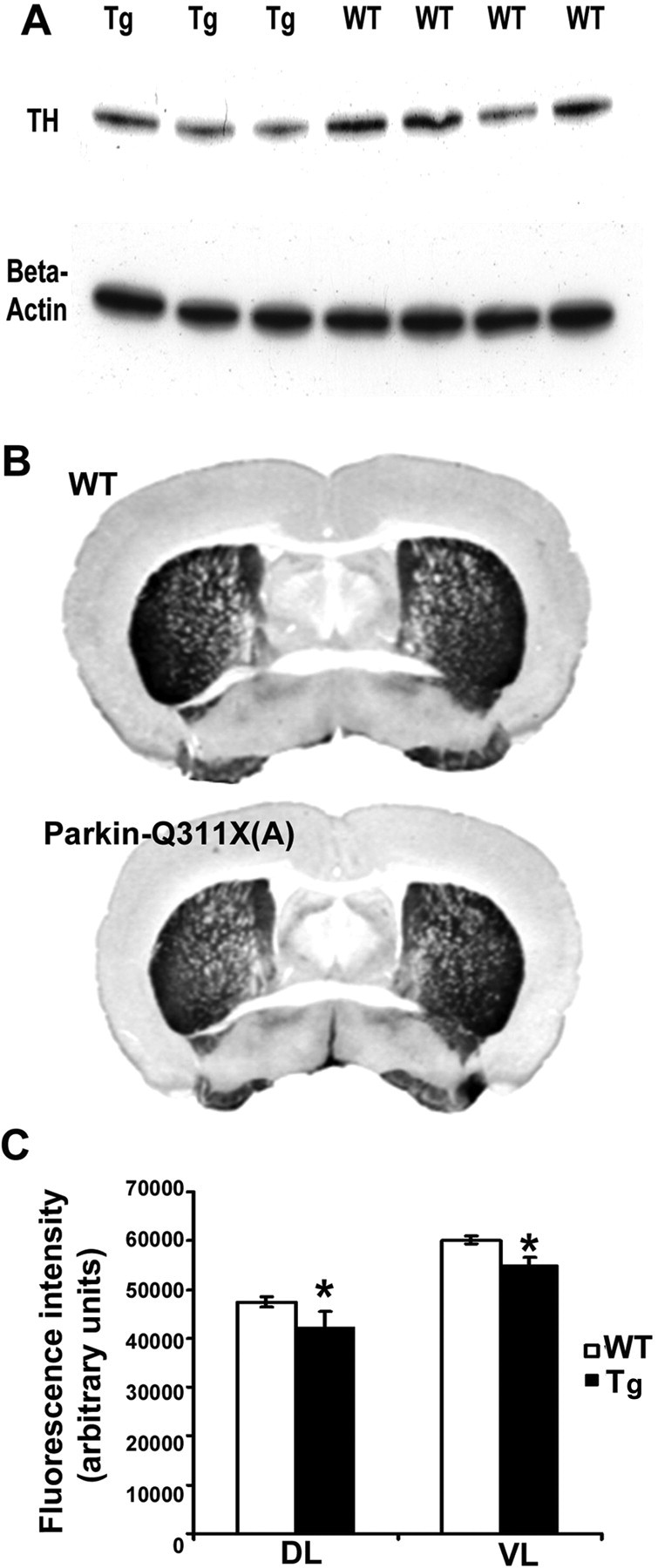
Striatum DA terminal loss in Parkin-Q311X mice. A, Representative Western blot demonstrating striatal TH levels in transgenic mice from line A and wild-type littermate controls at 12 months of age. Quantification of TH bands confirmed that striatal TH levels were significantly lower in Parkin-Q311X(A) mice than in wild-type littermate controls. n = 8 per genotype. *p < 0.05 compared with wild-type littermates. B, Representative photographs showing the TH immunohistochemistry staining in striatum of wild-type (top) and Parkin-Q311X(A) mice at 16–18 months of age. C, Quantification of TH immunofluorescence using a microarray scanner in 16-month-old mice. In rostral striatum, dorsolateral and ventrolateral striatal TH levels were significantly lower in Parkin-Q311X(A) mice than in wild-type littermate controls. n = 4 per genotype. *p < 0.05 compared with wild-type littermates. Error bars indicate SEM.
Finally, to obtain neurochemical evidence of DA terminal deficits in the striatum of symptomatic Parkin-Q311X mice, we measured striatal tissue DA and DA metabolite content in 19- to 21-month-old Parkin-Q311X and wild-type littermates using HPLC-ECD. We found significantly reduced DA levels in the Parkin-Q311X(A) mice compared with wild-type littermates (Fig. 7A) (WT, n = 10; Tg, n = 7; one-tailed independent sample Student's t test, p < 0.05). The concentration of the DA metabolite, 3,4-dihydroxyphenylacetic acid (DOPAC), was also significantly reduced in the striatum of Parkin-Q311X(A) mice (Fig. 7B) (WT, n = 10; Tg, n = 7; independent sample Student's t test, p < 0.05). Homovanillic acid (HVA) levels also trended lower but the difference failed to attain statistical significance (Fig. 7C). By comparing the striatal DA levels and motor performance, we found that the striatum DA levels in Parkin-Q311X(A) mice are significantly correlated with multiple hypokinetic motor deficits, including locomotor activity in the open-field test (Fig. 7D) (total number of floor plane moves and vertical plane entries in 15 min; Pearson's correlation coefficient, 0.590; p < 0.05), and vertical plane entries in the open-field test (Fig. 7E) (Pearson correlation coefficient, 0.651; p < 0.05).
Figure 7.

Reduced striatal DA and metabolite content in Parkin-Q311X mice and the correlation with behavioral deficits. A–C, Striatal DA, DOPAC, and HVA content measured using HPLC-ECD, in Parkin-Q311X(A) mice at 19–21 months of age. Results are expressed as picomoles per milligram of tissue [WT, n = 10; Parkin-Q311X(A), n = 7; *p < 0.05 compared with wild-type littermates]. Error bars indicate SEM. D, E, Total activity (D) and vertical plane entries (E) in open-field test were examined in wild-type littermates (blue squares) and Parkin-Q311X(A) mice (red squares) at 19–21 months of age and correlated with striatal DA levels.
Accumulation of proteinase K-resistant α-synuclein and colocalization with 3-nitrotyrosine in the midbrain of Parkin-Q311X(A) mice
Our analyses thus far provide strong genetic evidence that Parkin-Q311X can elicit dominant toxicity to the mammalian DA neurons that is sufficient to cause progressive motor deficits and late-onset DA neuron loss. To explore a potential mechanistic link between the dominant parkin toxicity and other known causes of DA neuron degeneration, we considered whether Parkin-Q311X can elicit progressive accumulation of α-synuclein, which is a known etiological factor in PD. α-Synuclein is known to accumulate in substantia nigra as well as other brain regions in PD patients not only in the form of Lewy bodies and Lewy neurites (Spillantini et al., 1997; Braak et al., 2003) but also in the form of misfolded, relatively insoluble α-synuclein that can be detected in situ after PK treatment (Neumann et al., 2002, 2004). Although autosomal recessive juvenile PD caused by loss of parkin function may (Farrer et al., 2001; Sasaki et al., 2004; Pramstaller et al., 2005) or may not (Hayashi et al., 2000) present with Lewy body pathology, the relationship between the dominant parkin toxicity and α-synuclein remains unclear. Because parkin was shown to play a role in the ubiquitination and degradation of α-synuclein, and possibly in the formation of α-synuclein-positive inclusion bodies (von Coelln et al., 2006), we hypothesized that parkin dominant toxicity could elicit progressive accumulation of PK-resistant α-synuclein in substantia nigra.
To test this hypothesis, we examined the accumulation of PK-resistant endogenous murine α-synuclein in the midbrain of Parkin-Q311X(A) mice at 16–18 months of age. PK treatment in the wild-type mice almost completely destroyed α-synuclein immunohistochemistry staining using our established protocol (Fernagut et al., 2007; McCormack et al., 2008). However, α-synuclein-positive neurons were abundantly detected in cells in the SN of Parkin-Q311X(A) mice (Fig. 8E,F) at 16 months of age, but not at 3 months (Fig. 8A,B). Interestingly, most of the residual, PK-resistant α-synuclein staining is diffuse within the cytoplasm, which is distinct from the inclusion body-like α-synuclein aggregates described in α-synuclein transgenic mice (Fernagut et al., 2007). To quantify the α-synuclein-positive cells, the entire SN areas were delineated using StereoInvestigator software and the number of positive cells were counted in 3- and 16-month-old Parkin-Q311X(A) mice and their wild-type littermate controls (n = 4 for each genotype). There was a significant increase of α-synuclein-positive cell number in the middle and caudal areas of the SN of Parkin-Q311X(A) mice at 16 months of age (independent sample Student's t test, p < 0.05), but not at 3 months of age (independent sample Student's t test, p > 0.05). Although aggregated α-synuclein could also be detected by Western blot analyses, our initial analyses using midbrain protein extracts from aged Parkin-Q311X(A) mice (16–18 months) did not reveal significant increase in the insoluble α-synuclein levels in the mutant mice compared with their wild-type littermate controls (data not shown). The latter result is likely attributable to the fact that Western blot with extracts prepared from all the midbrain cells may not offer sufficient sensitivity to detect aggregated α-synuclein present only in a subset of midbrain neurons. In summary, chronic expression of Parkin-Q311X resulted in progressive accumulation of PK-resistant α-synuclein in the midbrain neurons, a pathological phenotype mimicking those see in the PD patients (Neumann et al., 2002, 2004).
Figure 8.
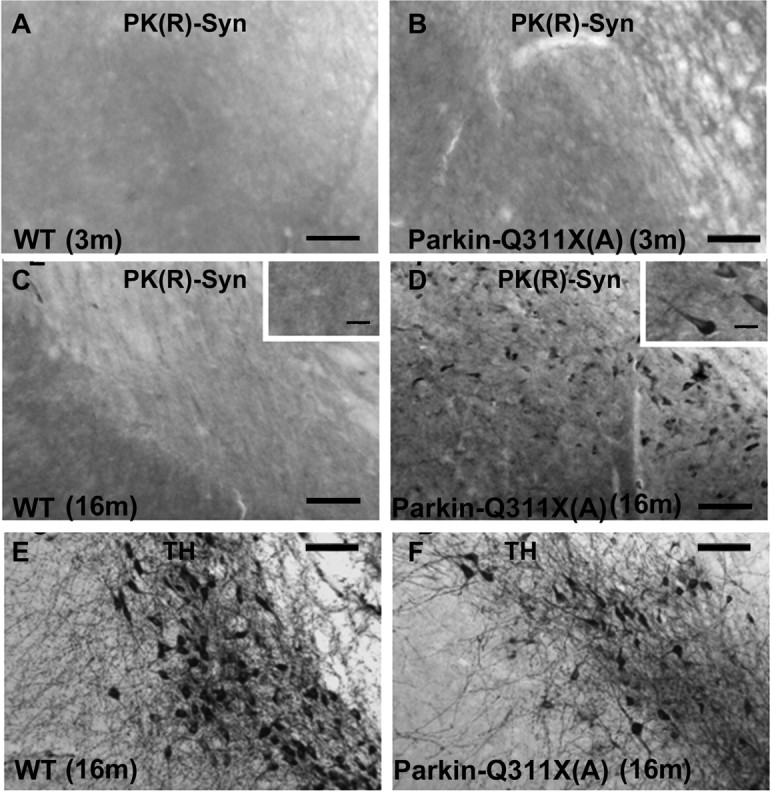
Accumulation of proteinase K-resistant α-synuclein in nigra of Parkin-Q311X(A) mice. A, B, Photomicrographs of the midbrain sections of Parkin-Q311X(A) mice and wild-type littermates at 3 months of age immunostained for proteinase K-resistant α-synuclein. Scale bar, 50 μm. C–F, Adjacent midbrain sections of Parkin-Q311X(A) mice and wild-type littermates at 16–18 months of age were immunostained for TH (C, D) (scale bar, 50 μm), proteinase K-resistant α-synuclein (E, F) (scale bar, 50 μm), and higher-magnification microphotographs of α-synuclein-immunopositive cells in nigra (insets in C and D) (scale bar, 12.5 μm).
Because α-synuclein oligomerization and aggregation can be greatly enhanced by various toxic insults, including oxidative stress (Giasson et al., 2000; Takahashi et al., 2007), and overexpression of several parkin mutants including Parkin-Q311X, but not wild-type Parkin, in cultured mammalian cells induces oxidative damage and increases vulnerability to oxidative stress (Hyun et al., 2002, 2005), we next sought evidence of oxidative damage via quantitative measurement of nigral nitrotyrosine (an indicator of oxidative damage as well as of the production of NO) using an ELISA method. Substantia nigra tissues were dissected out from wild-type and Parkin-Q311X(A) mice at the age of 19–21 months and used for quantitative measurement of nitrotyrosine [wild type, 4.58 ± 0.53 nmol/mg; Parkin-Q311X(A), 8.80 ± 3.14 nmol/mg; WT, n = 9; Parkin-Q311X(A), n = 7; p < 0.05 compared with wild-type littermates; single-tailed independent sample t test]. Because α-synuclein lesions in many synucleinopathies are also immunoreactive for 3-NT, a marker for oxidative (nitrative) protein damage (Good et al., 1998; Duda et al., 2000), we also stained PK-treated midbrain sections from 19- to 21-month-old Parkin-Q311X(A) and 16-month-old wild-type mice using the anti-3-NT antibody. As shown in the confocal images in Figure 9B, PK-resistant α-synuclein is colocalized with 3-NT staining in the SN of mutant, but not wild-type mice.
Figure 9.

Colocalization of nitrotyrosine and proteinase K-resistant α-synuclein in nigra of the Parkin-Q311X(A) mice. Confocal microphotographs of double immunofluorescence labeling proteinase K-resistant α-synuclein (green) and 3-NT (red) in nigra of Parkin-Q311X(A) (D–F) mice and wild-type littermates (A–C) at 16–18 months of age. Scale bar, 18 μm.
Together, our studies demonstrate that the dominant toxicity of mutant Parkin-Q311X can elicit two molecular pathogenic events with etiological connections to PD, the progressive accumulation of insoluble α-synuclein and oxidative protein damage within the DA neurons in SN (Maries et al., 2003; Lee and Trojanowski, 2006; Thomas and Beal, 2007).
Discussion
Our genetic study demonstrates that expression of a C-terminal truncated mutant parkin in DA neurons can elicit progressive DA neuron dysfunction and degeneration in mice. The Parkin-Q311X BAC transgenic model represents the first parkin-based genetic mouse model that exhibits such a neurodegenerative phenotype in vivo. The BAC transgenic approach is useful for modeling the dominant toxicity of a disease protein. BACs have large genomic inserts (average, ∼200 kb) and are likely to confer accurate, endogenous-like transgene expression in vivo (Yang et al., 1997; Heintz, 2001; Yang and Gong, 2005). In this model, truncated mutant parkin was driven by a Slc6a3 BAC and was expressed at ∼50% of the endogenous parkin level in the DA neurons. This approach excludes the possibility of nonspecific artifacts caused by gross overexpression and/or positional effects (e.g., ectopic or mosaic expression), which may occur with smaller promoter transgenic constructs. Because Parkin-Q311X mice have restricted expression of the mutant parkin in the DA neurons only, our current study was meant to investigate the cell-autonomous dominant toxic effects of this mutant parkin in DA neurons. Our study was not designed to address whether mutant parkin dominant toxic effects are selectively toward the DA neurons or they could affect other cell types in the brain. Future mouse genetic studies with ubiquitous expression of mutant parkin, preferably driven by its endogenous promoter and regulatory elements, are necessary to address such question.
Our Parkin-Q311X mice offer a novel mammalian genetic model with which to study the mechanisms underlying an age-dependent and slowly progressive form of DA neuron degeneration. With one exception (Fleming and Chesselet, 2006; Chesselet, 2008), existing genetic mouse models based on known PD mutations (i.e., parkin, Pink1, DJ-1 loss of function, and α-synuclein overexpression) recapitulate aspects of DA neuron dysfunction without exhibiting progressive degeneration of DA cell bodies. Other genetic mouse models, based on genes not linked to familial PD, do exhibit a DA neuron degeneration phenotype (e.g., loss-of-function mutants for En1/En2, Pitx3, and Ret, and knock-down mutant for VMAT2) and/or l-DOPA-reversible parkinsonism (e.g., DA-neuron-specific deletion of Tfam) (Hwang et al., 2005; Sgadò et al., 2006; Caudle et al., 2007; Ekstrand et al., 2007; Kramer et al., 2007), but, in most cases, the loss of DA neurons in these models occurs early in development, and/or their direct relevance to PD remains hypothetical.
Another important finding in this study was the progressive accumulation of PK-resistant α-synuclein in the substantia nigra of Parkin-Q311X mice. The absence of LBs in the brains of certain PD patients with parkin mutation prompted the suggestion that parkin may regulate α-synuclein aggregation in LB formation (Matsumine, 1998; Mori et al., 1998). However, recent neuropathological studies of the brains of certain PD patients with parkin mutation (Farrer et al., 2001; Sasaki et al., 2004; Pramstaller et al., 2005) and mouse genetic studies of α-synuclein transgenic mice in a parkin-null background (von Coelln et al., 2006) argue that α-synuclein aggregation and LB formation are independent of parkin function. Finally, several studies suggest that wild-type parkin function can protect primary midbrain neurons (Petrucelli et al., 2002), Drosophila (Yang et al., 2003) and rodent (Lo Bianco et al., 2004) DA neurons from neurodegeneration induced by α-synuclein overexpression. Hence previous studies suggest that at least part of the normal function of parkin is to protect against the toxic effects of α-synuclein, and loss of both copies of parkin in ARJP can result in loss of parkin-mediated protection to α-synuclein toxicity as well as other toxic insults to DA neurons (Cookson, 2003). Because parkin loss-of-function mutant mice do not exhibit the α-synuclein pathology and DA neuron degeneration phenotypes observed in the Parkin-Q311X mice (Goldberg et al., 2003; Itier et al., 2003; Lockhart et al., 2004; Palacino et al., 2004; von Coelln et al., 2004a; Fleming and Chesselet, 2005; Perez and Palmiter, 2005), we conclude that the progressive α-synuclein accumulation represents a novel dominant toxic mechanism elicited by the truncated parkin mutant.
The mechanisms linking mutant parkin action, late-onset accumulation of PK-resistant α-synuclein, and eventual DA neuron degeneration remain to be elucidated. However, several lines of evidence suggest that altered oxidative stress or cellular response to oxidative stress may play a role in this pathogenic process. First, oxidative stress is known to increase with aging (Lin and Beal, 2006); hence it could account for the late-onset (>16 months) motor and pathological phenotypes in Parkin-Q311X mice. Second, overexpression of several parkin mutants, including parkin-Q311X, but not wild-type parkin, can increase oxidative stress in transfected mammalian cells (Hyun et al., 2002) and decrease cellular protection against a variety of toxic oxidative insults [i.e., H2O2, MPP+ (1-methyl-4-phenylpyridinium), and HNE (4-hydroxynonenal)] (Hyun et al., 2005). Second, in Drosophila parkin-Q311X and parkin-T240R transgenic overexpression models, the DA neuron degeneration phenotype can be partially ameliorated by overexpressing vesicular monoamine transporter (VMAT) and can be enhanced by the loss of VMAT (Sang et al., 2007). This result suggests that the dominant toxicity of parkin mutants is sensitive to the relative levels of cytosolic versus vesicular DA, consistent with a previous hypothesis that cytosolic DA contributes to oxidative damage to DA neurons and vulnerability in PD (Sulzer et al., 2000). Our study provides additional support for this idea to the extent that PK-resistant α-synuclein in substantia nigra is colocalized with 3-NT, a marker of oxidative protein damage (Gow et al., 1996; Ischiropoulos, 1998). Furthermore, 3-NT-immunoreactive α-synuclein lesions have been observed in PD postmortem brains (Good et al., 1998; Duda et al., 2000), and oxidative stress can enhance α-synuclein oligomerization and aggregation (Giasson et al., 2000). Together, these data suggest that one possible dominant toxic mechanism of DA neuron insult from a parkin mutant could be increased oxidative stress leading to oligomerization and aggregation of α-synuclein.
The hypothesis that parkin mutant may exert a gain-of-function toxicity is supported by a series of human imaging (Hilker et al., 2001; Binkofski et al., 2007; Hagenah et al., 2007; Schneider et al., 2008) and genetic studies (Khan et al., 2005; I. E. Clark et al., 2006; Lesage et al., 2007). These studies suggest that heterozygous parkin mutations may manifest as clinically asymptomatic DA neuron dysfunction and degeneration or as sporadic cases of early-onset PD (West et al., 2002; Foroud et al., 2003; Beffert and Rosenberg, 2006; L. N. Clark et al., 2006; Sun et al., 2006). However, data derived from the human genetic studies are suggestive but not conclusive on this issue (Lincoln et al., 2003; Khan et al., 2005; I. E. Clark et al., 2006; Lesage et al., 2007). Available experimental evidence suggests that at least a subset of parkin mutations including truncation and point mutations could exert dominant toxicity. First, overexpression of multiple parkin mutants but not wild-type parkin induces dominant effects in transfected cells, including altered substrate binding, aggregation/oligomerization and subcellular mislocalization, and induction of oxidative stress (Hyun et al., 2002, 2005; Cookson et al., 2003; Henn et al., 2005; Sriram et al., 2005; Wang et al., 2005a,b). Second, in Drosophila, overexpression of three different parkin mutants, one truncation mutation (parkin-Q311X) and two point mutations (T240R and T275W), all result in progressive motor deficits and DA neuron loss (Sang et al., 2007; Wang et al., 2007).
Although the present study was not designed to resolve the controversial human genetic link between heterozygous parkin carriers and PD, it provides a proof-of-principle in a mammalian model, for toxic gain of function of parkin mutants. Furthermore, our data provide an additional mechanistic link between parkin dominant toxicity and two known etiological factors in PD, α-synuclein and oxidative stress. Together, these results underscore the important need to further study the potential dominant toxicity of certain parkin mutants in mammals and to perform more comprehensive human genetic studies and longitudinal clinical studies to conclusively assess the putative link between heterozygous parkin and PD.
Footnotes
This work was supported by Public Health Service Grants P50 NS38367 and U54 ES12078, the University of California at Los Angeles (UCLA) American Parkinson Disease Association Advanced Center, and a 2003 Stein–Oppenheimer Endowment Award (UCLA) (X.W.Y.). Dr. Sheila M. Fleming was also supported by a Chen Family Scholar Award and Dr. Fazard Mortazavi by The Michael J. Fox Foundation. We thank Dr. Nathaniel Heintz and Gene Expression Nervous System Atlas at Rockefeller University for TH-GFP mice, Dr. Pierre-Olivier Fernagut for assistance with the stereology, and Dr. David Krantz for critically reading this manuscript.
References
- Beffert U, Rosenberg RN. Increased risk for heterozygotes in recessive Parkinson disease. Arch Neurol. 2006;63:807–808. doi: 10.1001/archneur.63.6.807. [DOI] [PubMed] [Google Scholar]
- Binkofski F, Reetz K, Gaser C, Hilker R, Hagenah J, Hedrich K, van Eimeren T, Thiel A, Büchel C, Pramstaller PP, Siebner HR, Klein C. Morphometric fingerprint of asymptomatic Parkin and PINK1 mutation carriers in the basal ganglia. Neurology. 2007;69:842–850. doi: 10.1212/01.wnl.0000267844.72421.6c. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS, McCormack AL, Colebrooke RE, Di Monte DA, Emson PC, Miller GW. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. 2007;27:8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesselet MF. In vivo alpha-synuclein overexpression in rodents: a useful model of Parkinson's disease? Exp Neurol. 2008;209:22–27. doi: 10.1016/j.expneurol.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med. 2001;7:1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Clark LN, Afridi S, Karlins E, Wang Y, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K. Case-control study of the parkin gene in early-onset Parkinson disease. Arch Neurol. 2006;63:548–552. doi: 10.1001/archneur.63.4.548. [DOI] [PubMed] [Google Scholar]
- Cookson MR. Neurodegeneration: how does parkin prevent Parkinson's disease? Curr Biol. 2003;13:R522–R524. doi: 10.1016/s0960-9822(03)00446-9. [DOI] [PubMed] [Google Scholar]
- Cookson MR, Lockhart PJ, McLendon C, O'Farrell C, Schlossmacher M, Farrer MJ. RING finger 1 mutations in Parkin produce altered localization of the protein. Hum Mol Genet. 2003;12:2957–2965. doi: 10.1093/hmg/ddg328. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Chen Q, Gur TL, Hurtig HI, Stern MB, Gollomp SM, Ischiropoulos H, Lee VM, Trojanowski JQ. Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. Am J Pathol. 2000;157:1439–1445. doi: 10.1016/S0002-9440(10)64781-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S. Description of Parkinson's disease as a clinical syndrome. Ann N Y Acad Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- Fahn S, Sulzer D. Neurodegeneration and neuroprotection in Parkinson disease. NeuroRx. 2004;1:139–154. doi: 10.1602/neurorx.1.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon L, Bélanger CM, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, Voortman J, Haber M, Rouleau G, Thorarinsdottir T, Brice A, van Bergen En Henegouwen PM, Fon EA. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI3K-Akt signalling. Nat Cell Biol. 2006;8:834–842. doi: 10.1038/ncb1441. [DOI] [PubMed] [Google Scholar]
- Farrer M, Destée A, Levecque C, Singleton A, Engelender S, Becquet E, Mouroux V, Richard F, Defebvre L, Crook R, Hernandez D, Ross CA, Hardy J, Amouyel P, Chartier-Harlin MC. Genetic analysis of synphilin-1 in familial Parkinson's disease. Neurobiol Dis. 2001;8:317–323. doi: 10.1006/nbdi.2000.0326. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Hutson CB, Fleming SM, Tetreaut NA, Salcedo J, Masliah E, Chesselet MF. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression. Synapse. 2007;61:991–1001. doi: 10.1002/syn.20456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Chesselet MF. Phenotypical characterization of genetic mouse models of Parkinson's disease. In: LeDoux M, editor. Animal models of movement disorders. New York: Elsevier; 2005. pp. 183–192. [Google Scholar]
- Fleming SM, Chesselet MF. Behavioral phenotypes and pharmacology in genetic mouse models of Parkinsonism. Behav Pharmacol. 2006;17:383–391. doi: 10.1097/00008877-200609000-00004. [DOI] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human α-synuclein. J Neurosci. 2004;24:9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Hutson CB, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Behavioral effects of dopaminergic agonists in transgenic mice overexpressing human wildtype alpha-synuclein. Neuroscience. 2006;142:1245–1253. doi: 10.1016/j.neuroscience.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foroud T, Uniacke SK, Liu L, Pankratz N, Rudolph A, Halter C, Shults C, Marder K, Conneally PM, Nichols WC. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology. 2003;60:796–801. doi: 10.1212/01.wnl.0000049470.00180.07. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Gong S, Yang XW. Modification of bacterial artificial chromosomes (BACs) and preparation of intact BAC DNA for generation of transgenic mice. Curr Protoc Neurosci. 2005;5:5–21. doi: 10.1002/0471142301.ns0521s31. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Good PF, Hsu A, Werner P, Perl DP, Olanow CW. Protein nitration in Parkinson's disease. J Neuropathol Exp Neurol. 1998;57:338–342. doi: 10.1097/00005072-199804000-00006. [DOI] [PubMed] [Google Scholar]
- Gow AJ, Duran D, Malcolm S, Ischiropoulos H. Effects of peroxynitrite-induced protein modifications on tyrosine phosphorylation and degradation. FEBS Lett. 1996;385:63–66. doi: 10.1016/0014-5793(96)00347-x. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB. The efficiency of systematic sampling in stereology and its prediction. J Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Bagger P, Bendtsen TF, Evans SM, Korbo L, Marcussen N, Møller A, Nielsen K, Nyengaard JR, Pakkenberg B. The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS. 1988;96:857–881. doi: 10.1111/j.1699-0463.1988.tb00954.x. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB, Kiêu K, Nielsen J. The efficiency of systematic sampling in stereology—reconsidered. J Microsc. 1999;193:199–211. doi: 10.1046/j.1365-2818.1999.00457.x. [DOI] [PubMed] [Google Scholar]
- Hagenah JM, König IR, Becker B, Hilker R, Kasten M, Hedrich K, Pramstaller PP, Klein C, Seidel G. Substantia nigra hyperechogenicity correlates with clinical status and number of Parkin mutated alleles. J Neurol. 2007;254:1407–1413. doi: 10.1007/s00415-007-0567-y. [DOI] [PubMed] [Google Scholar]
- Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson's disease and parkinsonism. Ann Neurol. 2006;60:389–398. doi: 10.1002/ana.21022. [DOI] [PubMed] [Google Scholar]
- Hattori N, Mizuno Y. Pathogenetic mechanisms of parkin in Parkinson's disease. Lancet. 2004;364:722–724. doi: 10.1016/S0140-6736(04)16901-8. [DOI] [PubMed] [Google Scholar]
- Hattori N, Matsumine H, Asakawa S, Kitada T, Yoshino H, Elibol B, Brookes AJ, Yamamura Y, Kobayashi T, Wang M, Yoritaka A, Minoshima S, Shimizu N, Mizuno Y. Point mutations (Thr240Arg and Gln311Stop) [correction of Thr240Arg and Ala311Stop] in the Parkin gene. Biochem Biophys Res Commun. 1998;249:754–758. doi: 10.1006/bbrc.1998.9134. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Wakabayashi K, Ishikawa A, Nagai H, Saito M, Maruyama M, Takahashi T, Ozawa T, Tsuji S, Takahashi H. An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord. 2000;15:884–888. doi: 10.1002/1531-8257(200009)15:5<884::aid-mds1019>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Heintz N. BAC to the future: the use of bac transgenic mice for neuroscience research. Nat Rev Neurosci. 2001;2:861–870. doi: 10.1038/35104049. [DOI] [PubMed] [Google Scholar]
- Henn IH, Gostner JM, Lackner P, Tatzelt J, Winklhofer KF. Pathogenic mutations inactivate parkin by distinct mechanisms. J Neurochem. 2005;92:114–122. doi: 10.1111/j.1471-4159.2004.02854.x. [DOI] [PubMed] [Google Scholar]
- Henn IH, Bouman L, Schlehe JS, Schlierf A, Schramm JE, Wegener E, Nakaso K, Culmsee C, Berninger B, Krappmann D, Tatzelt J, Winklhofer KF. Parkin mediates neuroprotection through activation of IκB kinase/nuclear factor-κB signaling. J Neurosci. 2007;27:1868–1878. doi: 10.1523/JNEUROSCI.5537-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilker R, Klein C, Ghaemi M, Kis B, Strotmann T, Ozelius LJ, Lenz O, Vieregge P, Herholz K, Heiss WD, Pramstaller PP. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann Neurol. 2001;49:367–376. [PubMed] [Google Scholar]
- Huynh DP, Scoles DR, Nguyen D, Pulst SM. The autosomal recessive juvenile Parkinson disease gene product, parkin, interacts with and ubiquitinates synaptotagmin XI. Hum Mol Genet. 2003;12:2587–2597. doi: 10.1093/hmg/ddg269. [DOI] [PubMed] [Google Scholar]
- Hwang DY, Fleming SM, Ardayfio P, Moran-Gates T, Kim H, Tarazi FI, Chesselet MF, Kim KS. 3,4-Dihydroxyphenylalanine reverses the motor deficits in Pitx3-deficient aphakia mice: behavioral characterization of a novel genetic model of Parkinson's disease. J Neurosci. 2005;25:2132–2137. doi: 10.1523/JNEUROSCI.3718-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun DH, Lee M, Hattori N, Kubo S, Mizuno Y, Halliwell B, Jenner P. Effect of wild-type or mutant Parkin on oxidative damage, nitric oxide, antioxidant defenses, and the proteasome. J Biol Chem. 2002;277:28572–28577. doi: 10.1074/jbc.M200666200. [DOI] [PubMed] [Google Scholar]
- Hyun DH, Lee M, Halliwell B, Jenner P. Effect of overexpression of wild-type or mutant parkin on the cellular response induced by toxic insults. J Neurosci Res. 2005;82:232–244. doi: 10.1002/jnr.20638. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H. Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch Biochem Biophys. 1998;356:1–11. doi: 10.1006/abbi.1998.0755. [DOI] [PubMed] [Google Scholar]
- Itier JM, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier L, Ret G, Joubert C, Periquet M, Araujo F, Negroni J, Casarejos MJ, Canals S, Solano R, Serrano A, Gallego E, et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet. 2003;12:2277–2291. doi: 10.1093/hmg/ddg239. [DOI] [PubMed] [Google Scholar]
- Kamme F, Zhu J, Luo L, Yu J, Tran DT, Meurers B, Bittner A, Westlund K, Carlton S, Wan J. Single-cell laser-capture microdissection and RNA amplification. Methods Mol Med. 2004;99:215–223. doi: 10.1385/1-59259-770-X:215. [DOI] [PubMed] [Google Scholar]
- Khan NL, Scherfler C, Graham E, Bhatia KP, Quinn N, Lees AJ, Brooks DJ, Wood NW, Piccini P. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology. 2005;64:134–136. doi: 10.1212/01.WNL.0000148725.48740.6D. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007;6:652–662. doi: 10.1016/S1474-4422(07)70174-6. [DOI] [PubMed] [Google Scholar]
- Ko HS, von Coelln R, Sriram SR, Kim SW, Chung KK, Pletnikova O, Troncoso J, Johnson B, Saffary R, Goh EL, Song H, Park BJ, Kim MJ, Kim S, Dawson VL, Dawson TM. Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J Neurosci. 2005;25:7968–7978. doi: 10.1523/JNEUROSCI.2172-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HS, Kim SW, Sriram SR, Dawson VL, Dawson TM. Identification of far upstream element-binding protein-1 as an authentic Parkin substrate. J Biol Chem. 2006;281:16193–16196. doi: 10.1074/jbc.C600041200. [DOI] [PubMed] [Google Scholar]
- Kramer ER, Aron L, Ramakers GM, Seitz S, Zhuang X, Beyer K, Smidt MP, Klein R. Absence of Ret signaling in mice causes progressive and late degeneration of the nigrostriatal system. PLoS Biol. 2007;5:e39. doi: 10.1371/journal.pbio.0050039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Lesage S, Magali P, Lohmann E, Lacomblez L, Teive H, Janin S, Cousin PY, Dürr A, Brice A. Deletion of the parkin and PACRG gene promoter in early-onset parkinsonism. Hum Mutat. 2007;28:27–32. doi: 10.1002/humu.20436. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lincoln SJ, Maraganore DM, Lesnick TG, Bounds R, de Andrade M, Bower JH, Hardy JA, Farrer MJ. Parkin variants in North American Parkinson's disease: cases and controls. Mov Disord. 2003;18:1306–1311. doi: 10.1002/mds.10601. [DOI] [PubMed] [Google Scholar]
- Lo Bianco C, Schneider BL, Bauer M, Sajadi A, Brice A, Iwatsubo T, Aebischer P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson's disease. Proc Natl Acad Sci U S A. 2004;101:17510–17515. doi: 10.1073/pnas.0405313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat Neurosci. 2006;9:443–452. doi: 10.1038/nn1654. [DOI] [PubMed] [Google Scholar]
- Lockhart PJ, O'Farrell CA, Farrer MJ. It's a double knock-out! The quaking mouse is a spontaneous deletion of parkin and parkin co-regulated gene (PACRG) Mov Disord. 2004;19:101–104. doi: 10.1002/mds.20000. [DOI] [PubMed] [Google Scholar]
- Maries E, Dass B, Collier TJ, Kordower JH, Steece-Collier K. The role of alpha-synuclein in Parkinson's disease: insights from animal models. Nat Rev Neurosci. 2003;4:727–738. doi: 10.1038/nrn1199. [DOI] [PubMed] [Google Scholar]
- Matsumine H. A loss-of-function mechanism of nigral neuron death without Lewy body formation: autosomal recessive juvenile parkinsonism (AR-JP) J Neurol. 1998;245:P10–P14. doi: 10.1007/pl00007741. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Mak SK, Shenasa M, Langston WJ, Forno LS, Di Monte DA. Pathologic modifications of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated squirrel monkeys. J Neuropathol Exp Neurol. 2008;67:793–802. doi: 10.1097/NEN.0b013e318180f0bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer EG, Innanen VT, McGeer PL. Evidence on the cellular localization of adenyl cyclase in the neostriatum. Brain Res. 1976;118:356–358. doi: 10.1016/0006-8993(76)90726-5. [DOI] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Mori H, Kondo T, Yokochi M, Matsumine H, Nakagawa-Hattori Y, Miyake T, Suda K, Mizuno Y. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology. 1998;51:890–892. doi: 10.1212/wnl.51.3.890. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, Sulzer D. Analysis of exocytotic events recorded by amperometry. Nat Methods. 2005;2:651–658. doi: 10.1038/nmeth782. [DOI] [PubMed] [Google Scholar]
- Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Müller V, Odoy S, Fujiwara H, Hasegawa M, Iwatsubo T, Trojanowski JQ, Kretzschmar HA, Haass C. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest. 2002;110:1429–1439. doi: 10.1172/JCI15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Müller V, Kretzschmar HA, Haass C, Kahle PJ. Regional distribution of proteinase K-resistant alpha-synuclein correlates with Lewy body disease stage. J Neuropathol Exp Neurol. 2004;63:1225–1235. doi: 10.1093/jnen/63.12.1225. [DOI] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U S A. 2005;102:2174–2179. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persons DA, Allay JA, Riberdy JM, Wersto RP, Donahue RE, Sorrentino BP, Nienhuis AW. Use of the green fluorescent protein as a marker to identify and track genetically modified hematopoietic cells. Nat Med. 1998;4:1201–1205. doi: 10.1038/2704. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, O'Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- Pramstaller PP, Schlossmacher MG, Jacques TS, Scaravilli F, Eskelson C, Pepivani I, Hedrich K, Adel S, Gonzales-McNeal M, Hilker R, Kramer PL, Klein C. Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol. 2005;58:411–422. doi: 10.1002/ana.20587. [DOI] [PubMed] [Google Scholar]
- Sang TK, Chang HY, Lawless GM, Ratnaparkhi A, Mee L, Ackerson LC, Maidment NT, Krantz DE, Jackson GR. A Drosophila model of mutant human parkin-induced toxicity demonstrates selective loss of dopaminergic neurons and dependence on cellular dopamine. J Neurosci. 2007;27:981–992. doi: 10.1523/JNEUROSCI.4810-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Shirata A, Yamane K, Iwata M. Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology. 2004;63:678–682. doi: 10.1212/01.wnl.0000134657.25904.0b. [DOI] [PubMed] [Google Scholar]
- Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider SA, Talelli P, Cheeran BJ, Khan NL, Wood NW, Rothwell JC, Bhatia KP. Motor cortical physiology in patients and asymptomatic carriers of parkin gene mutations. Mov Disord. 2008;23:1812–1819. doi: 10.1002/mds.22025. [DOI] [PubMed] [Google Scholar]
- Sgadò P, Albéri L, Gherbassi D, Galasso SL, Ramakers GM, Alavian KN, Smidt MP, Dyck RH, Simon HH. Slow progressive degeneration of nigral dopaminergic neurons in postnatal Engrailed mutant mice. Proc Natl Acad Sci U S A. 2006;103:15242–15247. doi: 10.1073/pnas.0602116103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Sriram SR, Li X, Ko HS, Chung KK, Wong E, Lim KL, Dawson VL, Dawson TM. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum Mol Genet. 2005;14:2571–2586. doi: 10.1093/hmg/ddi292. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Zecca L. Intraneuronal dopamine-quinone synthesis: a review. Neurotox Res. 2000;1:181–195. doi: 10.1007/BF03033289. [DOI] [PubMed] [Google Scholar]
- Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA, Golbe LI, Mark MH, Racette BA, Perlmutter JS, Parsian A, Guttman M, Nicholson G, Xu G, Wilk JB, Saint-Hilaire MH, DeStefano AL, Prakash R, Williamson S, Suchowersky O, et al. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol. 2006;63:826–832. doi: 10.1001/archneur.63.6.826. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Ko LW, Kulathingal J, Jiang P, Sevlever D, Yen SH. Oxidative stress-induced phosphorylation, degradation and aggregation of alpha-synuclein are linked to upregulated CK2 and cathepsin D. Eur J Neurosci. 2007;26:863–874. doi: 10.1111/j.1460-9568.2007.05736.x. [DOI] [PubMed] [Google Scholar]
- Thomas B, Beal MF. Parkinson's disease. Hum Mol Genet. 2007;16:R183–R194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Aggregation of neurofilament and alpha-synuclein proteins in Lewy bodies: implications for the pathogenesis of Parkinson disease and Lewy body dementia. Arch Neurol. 1998;55:151–152. doi: 10.1001/archneur.55.2.151. [DOI] [PubMed] [Google Scholar]
- Turiault M, Parnaudeau S, Milet A, Parlato R, Rouzeau JD, Lazar M, Tronche F. Analysis of dopamine transporter gene expression pattern—generation of DAT-iCre transgenic mice. FEBS J. 2007;274:3568–3577. doi: 10.1111/j.1742-4658.2007.05886.x. [DOI] [PubMed] [Google Scholar]
- Varrone A, Pellecchia MT, Amboni M, Sansone V, Salvatore E, Ghezzi D, Garavaglia B, Brice A, Brunetti A, Bonavita V, De Michele G, Salvatore M, Pappatà S, Barone P. Imaging of dopaminergic dysfunction with [123I]FP-CIT SPECT in early-onset parkin disease. Neurology. 2004;63:2097–2103. doi: 10.1212/01.wnl.0000145765.19094.94. [DOI] [PubMed] [Google Scholar]
- von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM. Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci U S A. 2004a;101:10744–10749. doi: 10.1073/pnas.0401297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Coelln R, Dawson VL, Dawson TM. Parkin-associated Parkinson's disease. Cell Tissue Res. 2004b;318:175–184. doi: 10.1007/s00441-004-0924-4. [DOI] [PubMed] [Google Scholar]
- von Coelln R, Thomas B, Andrabi SA, Lim KL, Savitt JM, Saffary R, Stirling W, Bruno K, Hess EJ, Lee MK, Dawson VL, Dawson TM. Inclusion body formation and neurodegeneration are parkin independent in a mouse model of α-synucleinopathy. J Neurosci. 2006;26:3685–3696. doi: 10.1523/JNEUROSCI.0414-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Tan JM, Ho MW, Zaiden N, Wong SH, Chew CL, Eng PW, Lim TM, Dawson TM, Lim KL. Alterations in the solubility and intracellular localization of parkin by several familial Parkinson's disease-linked point mutations. J Neurochem. 2005a;93:422–431. doi: 10.1111/j.1471-4159.2005.03023.x. [DOI] [PubMed] [Google Scholar]
- Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP, Ho MW, Lim TM, Soong TW, Pletnikova O, Troncoso J, Dawson VL, Dawson TM, Lim KL. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin's protective function. Hum Mol Genet. 2005b;14:3885–3897. doi: 10.1093/hmg/ddi413. [DOI] [PubMed] [Google Scholar]
- Wang C, Lu R, Ouyang X, Ho MW, Chia W, Yu F, Lim KL. Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J Neurosci. 2007;27:8563–8570. doi: 10.1523/JNEUROSCI.0218-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A, Periquet M, Lincoln S, Lücking CB, Nicholl D, Bonifati V, Rawal N, Gasser T, Lohmann E, Deleuze JF, Maraganore D, Levey A, Wood N, Dürr A, Hardy J, Brice A, Farrer M. Complex relationship between Parkin mutations and Parkinson disease. Am J Med Genet. 2002;114:584–591. doi: 10.1002/ajmg.10525. [DOI] [PubMed] [Google Scholar]
- West AB, Maidment NT. Genetics of parkin-linked disease. Hum Genet. 2004;114:327–336. doi: 10.1007/s00439-003-1074-6. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Yang XW, Gong S. An overview on the generation of BAC transgenic mice for neuroscience research. Curr Protoc Neurosci. 2005;5:5–20. doi: 10.1002/0471142301.ns0520s31. [DOI] [PubMed] [Google Scholar]
- Yang XW, Model P, Heintz N. Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat Biotechnol. 1997;15:859–865. doi: 10.1038/nbt0997-859. [DOI] [PubMed] [Google Scholar]
- Yang Y, Nishimura I, Imai Y, Takahashi R, Lu B. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron. 2003;37:911–924. doi: 10.1016/s0896-6273(03)00143-0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci U S A. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]