

Abstract
Background
Phosphorylation is a key regulatory event in controlling the function of the cardiac gap junction protein connexin43 (Cx43). Recently, three new phosphorylation sites (S296, S297 and S306) were identified on Cx43; two of which (S297 and S306) are dephosphorylated during ischemia. The functional significance of these new sites is currently unknown.
Objective
To examine the role of S296, S297 and S306 in the regulation of electrical intercellular communication.
Methods
To mimic constitutive dephosphorylation serine was mutated to alanine at the three sites and expressed in HeLa cells. Electrical coupling and single-channel measurements were performed by double patch clamp; protein expression levels were assayed by western blotting, localization of Cx43 and phosphorylation of S306 by immunolabeling. Free hemichannels were assessed by biotinylation.
Results
Macroscopic conductance in cells expressing S306A was reduced to 57 % compared to WT while coupling was not significantly changed in either S296A or S297A expressing cells. S306A expressing cells displayed similar protein and free hemichannel abundance compared to WT-Cx43, whereas the fractional area of plaques in cell to cell interfaces was increased. However, single-channel measurements showed a WT-Cx43 main state conductance of 119 pS, whereas the main state conductance of S306A channels was reduced to 95 pS. Furthermore, channel gating was affected in S306A channels.
Conclusions
Lack of phosphorylation at serine 306 results in reduced coupling, which can be explained by reduced single channel conductance. We suggest that dephosphorylation of S306 partly explains the electrical uncoupling seen in myocardial ischemia.
Keywords: gap junction, connexin43, phosphorylation, ischemia, single channel
Introduction
Cardiac arrhythmias may arise from inappropriate impulse formation or propagation. Impulse propagation is in part governed by the electrical coupling between cardiomyocytes, and alterations in this coupling have been linked to the development of arrhythmia (1–3). In acute ischemia there is a substantial increase in tissue resistance (4), which is likely caused by reduced intercellular coupling (5). The mechanisms reducing electrical coupling includes acidosis (6, 7), changes in membrane lipid composition (8–10) and changes in phosphorylation (5, 11, 12).
Connexin43 (Cx43) is the major gap junction protein in the heart and its regulation by phosphorylation is known in some detail. To date, 19 phosphorylation sites have been described in Cx43; of these, serine 368 (S368) has been most intensively investigated. S368 is phosphorylated by PKC and is dephosphorylated during acute ischemia (5, 11), although phosphorylation during ischemia has also been reported (13). Furthermore, Lampe et al. showed that the serines 325, 328 and 330 are dephosphorylated during ischemia and that this dephosphorylation is linked to a relocalization of Cx43 from intercalated discs to the lateral cell borders (12). Present evidence shows that regulation of Cx43 by phosphorylation is both important and complex. The complexity most likely arises from the large number of phosphorylation sites.
In a recent study, we demonstrated three new phosphorylation sites in the C-terminal part of rat Cx43, namely S296, S297 and S306, by mass spectrometry (11). The study showed that S297 and S306 were dephosphorylated in response to global ischemia. Dephosphorylation of S306 was observed after 7 minutes of ischemia whereas dephosphorylation of S297 coincided with electrical uncoupling and asystole between 15 and 30 minutes of no-flow ischemia. The third site, S296, remained phosphorylated throughout the entire period of the global ischemic insult. Exposure to the anti-arrhythmic peptide analog rotigaptide prevented dephosphorylation of S297 and S368 (11). Rotigaptide is known to increase electrical coupling in cardiomyocytes (14) and it prolonged the time to asystole during global ischemia (11), underlining the importance of phosphorylation for normal action potential propagation.
The relevance of the phosphorylation state of residues S296, S297 and S306 to channel function is unknown. The aim of the current study was to determine the effect that their loss of phosphorylation may have on electrical coupling. Therefore, we generated a series of serine-to-alanine substitutions in Cx43 (S296A, S297A and S306A) to mimic constitutively dephosphorylated forms of the protein. In order to test trafficking, and junctional and unitary conductance of the mutated Cx43 channels, we expressed the proteins in HeLa cells and performed confocal microscopy, western blotting and dual patch clamp. The results demonstrate a decreased unitary conductance of Cx43 channels lacking phosphorylation in position 306. We suggest that this change may be relevant to a loss of electrical coupling in the setting of myocardial infarction.
Materials and methods
Generation of rCx43 alanine substitutions
The mutations substituting serines 296, 297 and 306 for alanine were introduced using mutated oligonucleotide extension (PfuTurbo Polymerase, Stratagene, La Jolla, CA) from a plasmid template harboring the cDNA of interest (rCx43, GenBank Accession number NM_012567), digested with DpnI (Fermentas, St. Leon-Rot, Germany) and transformed into E.coli XL1 Blue cells. The construct was verified by complete DNA sequencing of the cDNA insert. Mutated versions of Cx43 were subsequently subcloned into pIRES-hrGFP-1a (Stratagene, La Jolla, CA) and re-sequenced. The following primers were used for exchange of the serines in question:
S296A; forward AGAAACAATGCCTCGTGCCGCAATTACAACAAGCAAGCTAGCGAGCAAAA,
reverse TTTTGCTCGCTAGCTTGCTTGTTGTAATTGCGGCACGAGGCATTGTTTCT,
S297A; forward AGAAACAATTCCGCGTGCCGCAATTACAACAAGCAAGCTAGCGAGCAAAA,
reverse TTTTGCTCGCTAGCTTGCTTGTTGTAATTGCGGCACGCGGAATTGTTTCT,
S306A; forward AGAAACAATTCCTCGTGCCGCAATTACAACAAGCAAGCTGGCGAGCAAAA,
reverse TTTTGCTCGCCAGCTTGCTTGTTGTAATTGCGGCACGAGGAATTGTTTCT
Cell culture and transfection
The day before transfection 2 × 105 HeLa cells were seeded in a Petri dish (Ø 3.5 cm) containing five circular glass cover slips. Cells were transfected with 0.25 µg of DNA using Effectene transfection reagent (Qiagen, Hilden, Germany) following manufacturer’s recommendations. HeLa cells were kept in D-MEM (32430, Invitrogen, Carlsbad, CA) supplemented with 10% FCS and 1% penicillin/streptomycin (cat. no. 10106 and 15070, Invitrogen, Carlsbad, CA).
Immunostaining and confocal microscopy
Transfected HeLa cells were fixed with 2% paraformaldehyde and permeabilized in PBS with 4% BSA and 0.2% Triton X-100. PBS with 4% BSA was used for all blocking steps. Cells were incubated overnight with a primary Cx43 antibody (C6219, 1:1500, Sigma-Aldrich, St. Louis, MO) in PBS with 4% BSA. Phosphorylation of S306 was detected by an affinity purified rabbit antibody directed against phosphorylated serine 306 in Cx43 (pS306 Ab, Pacific Immunology, Ramona, CA). The antibody was raised against the following sequence: CRNYNKQA-S(PO3)-EQNWAN, which spans the phosphorylated S306 site in Cx43. The pS306 Ab was used in a dilution of 1:50. For the immunofluorescence studies in rat heart the sections (thickness 10 µm) were boiled in citrate buffer (pH 6.0) and blocked with 5% skim milk. Total Cx43, was detected by use of a mouse anti-Cx43-antibody (35–5000, Invitrogen, Carlsbad, CA). After washing, cells were incubated with secondary ALEXA conjugated antibodies and counterstained with rhodamine-phalloidin/TO-PRO-3, or if biotinylated, incubated with an ALEXA-conjugated streptavidin, for 45 min at room temperature (all Invitrogen, Carlsbad, CA). Cells were washed and mounted in Prolong Gold mounting medium (Invitrogen, Carlsbad, CA) and imaged in a laser confocal microscope (Leica TCS SP2, Leica Microsystems, Wetzlar, Germany) using a 63X objective. For assessment of plaque numbers and sizes, proteins in the cell surface were labeled with biotin before permeabilization in order to visualize the plasma membrane using EZ-link Sulfo-NHS-SS Biotin (Thermo Fischer Scientific, Rockford, IL) according to the manufacturer’s recommendations. After staining for Cx43, transfected cell pairs were imaged. Only plasma membrane surfaces showing gap junctions were included in the analysis. Plaque sizes and numbers were analyzed by Leica LCS Lite software and data exported to Excel for further analysis.
Biotin immunoprecipitation
Transfected cells were washed twice in cold PBS and incubated for 30 min on ice at 4°C with 1 mg/mL EZ-link Sulfo-NHS-SS Biotin (Thermo Fisher Scientific, Rockford, IL) and then incubated for 20 min with PBS/glycine on ice at 4°C. Cells were scraped off the dish in PBS containing PMSF (1 mmol/L), sodium orthovanadate (1 mmol/L), Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktail 1 and 2 (Sigma-Aldrich, St. Louis, MO) in the concentrations recommended by the manufacturer. After centrifugation for 2 min at 2000 g cells were resuspended in RIPA buffer with protease and phosphatase inhibitor cocktails as above. 1 mmol/L MgCl2 and CaCl2 were added and the cells were incubated for 10 min at 37°C. NeutrAvidin Agarose Resin (150 µl) (Thermo Fisher Scientific, Rockford, IL) was added to the solution and the samples were incubated overnight at 4°C during constant mixing. After incubation, samples were centrifuged at 4°C at 2000 g for 2 min. The supernatants (unbound fraction) were saved for analysis. The pellets were washed once in 0.5 mol/L NaCl and once in 10 mmol/L Tris/0.2 % Triton X-100. All steps included protease and phosphatase inhibitors. NuPAGE LDS sample buffer (Cat. No. NP0007, Invitrogen, Carlsbad, CA) was added to the samples. These were heated to 95°C for 5 min, centrifuged for 5 min at 20,000 g and the supernatant saved for western blot analysis. The two fractions were adjusted to the same sample volume and applied to the SDS-PAGE gel in a ratio of 1:20, unbound:bound fractions. The western blot procedure was carried out and data quantified as described below.
Western blotting
Transfected HeLa cell cultures were harvested by addition of cold RIPA buffer containing protease and phosphatase inhibitors as described above. 20 µg protein per lane was applied to a 7% NuPAGE gel (Invitrogen, Carlsbad, CA). After gel electrophoresis the protein was transferred to a nitrocellulose membrane, blocked and incubated in 5% skim milk with the primary antibodies (Cx43: C6219, 1:1500, Sigma-Aldrich, St. Louis, MO, and β-tubulin: MAB3408, 1:20.000 Millipore, Billerica, MA). After blocking (5% skim milk) the membrane was incubated with a HRP-conjugated antibody (anti-mouse and - rabbit; Thermo Fisher Scientific, Rockford, IL) followed by SuperSignal West Femto HRP-substrate (Thermo Fisher Scientific, Rockford, IL) and imaged and quantified in a UVP Epi Chem II Darkroom (UVP Inc., Upland, CA).
Dual patch clamp experiments
Intercellular electrical coupling was measured in transiently transfected Hela-cells using the dual whole cell patch clamp method. 24 to 72 hours after transfection, coverslips were mounted in a chamber on the stage of an inverted microscope (DMIRB, Leica, Wetzlar, Germany). The chamber volume was 1 ml and the cells were superfused at 1 mL/min with a solution containing in (mmol/L 160 NaCl; 10 CsCl; 2 CaCl2; 0.6 MgCl2; 10 HEPES; pH 7.4. Transfected cell pairs were selected by hr-GFP fluorescence and each cell was patched in the fast whole cell configuration. Patch pipettes were pulled from borosilicate capillaries (GC150F, Harvard Apparatus, Edenbridge, UK) to a resistance of 4–7 MOhm using a PIP5 puller (Heka, Lambrecht/Pfalz, Germany). The pipettes were filled with a solution containing (in mmol/L): 130 CsCl; 0.5 CaCl2; 10 HEPES; 10 EGTA; 2 Na2ATP; 3 MgATP, pH adjusted to 7.2 using CsOH. Measurements were done using two EPC7 amplifiers (Heka, Lambrecht/Pfalz, Germany) and signals were filtered at 3 kHz using the intrinsic filters. Data were sampled at 10 kHz using Cellworks software, an analog-to-digital converter board (PCI1200, National Instruments, Austin, TX), and an INT-10 breakout box (NPI Electronic, Tamm, Germany).
For measurement of macroscopic conductance cells were clamped at −40 mV and a 1 s pulse to +60 mV was applied in one cell at 0.1 Hz. Data was analyzed using Cellworks Reader (NPI electronic, Tamm, Germany). Conductance was calculated as the resulting current deflection in the passive cell (non-pulsed) divided by 100 mV (14).
For single-channel measurements a constant voltage gradient of 60 mV was imposed between cells and octanol (1–2 mmol/L) was included in the bath solution. During wash in and wash out periods, single-channel activity was identified and analyzed using a custom written MatLab (The MathWorks, Natick, MA) routine. Data was low-pass filtered at 200 Hz by the MatLab FIR filter. All channel openings and closings were identified by eye and registered by mouse-clicking the closed and open states.
Chemicals
Chemical were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Statistical analysis
Data are presented as means ± SEM. Differences between multiple mean values were analyzed by one-way ANOVA followed by Bonferroni’s post-hoc test. Comparisons between pairs of mean values were done using Student’s t-test for paired or unpaired data as appropriate. P<0.05 was considered to be statistically significant. All statistical analyses were performed using STATISTICA 7.0 (StatSoft Inc., Tulsa, OK).
Results
Macroscopic conductance in serine to alanine mutants of Cx43
To analyze the junctional conductance in WT-Cx43 and Cx43 alanine mutants, we measured conductance between transiently transfected cell pairs by the dual whole cell patch clamp technique (figure 1). The average junctional coupling measured between HeLa cell pairs transfected with WT-Cx43 was 27.9±3.3 nS (n=21). In cells transfected with S296A or S297A electrical coupling was 29.7±4.5 nS (n=11) and 26.1±3.1 nS (n=12), respectively. These values were not significantly different from the coupling found in WT-Cx43 transfected cells. In contrast, coupling was significantly reduced to 16.0±2.7 (n=16, P<0.001) in cells transfected with S306A. The data are summarized in figure 1.
Figure 1.
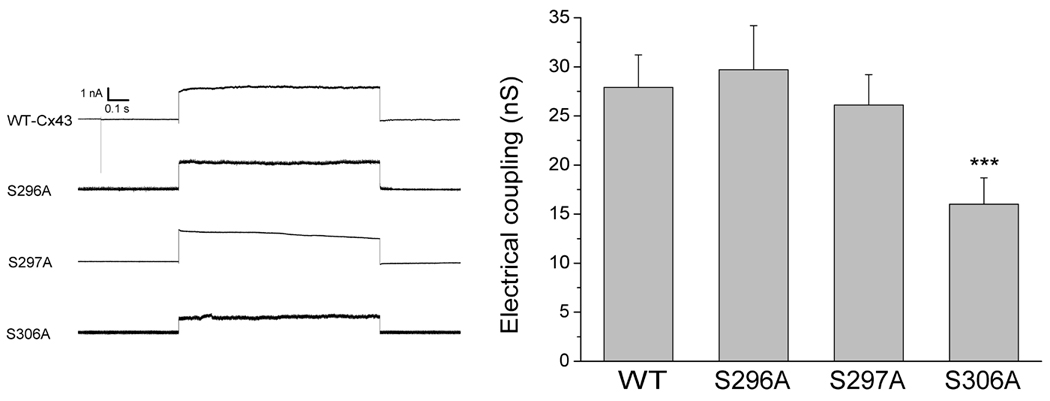
Effect of alanine substitutions on intercellular coupling. Left panel: Representative traces from cells expressing either WT-Cx43, S296A, S297A or S306A channels. Traces show the transjunctional current resulting from a 100 mV pulse. Right panel: Macroscopic electrical coupling in WT-Cx43 (n=21), S296A (n=11), S297A (n=12) and S306A (n=16) expressing cells. Alanine substitutions at S296 or S297 did not change electrical coupling as compared to WT-Cx43 cells whereas substitution of S306 caused a 43% reduction in junctional conductance (***P<0.001 vs. WT-Cx43).
Gap junction formation is unaltered in alanine mutants of Cx43
To investigate if reduced coupling in S306A transfected cells was caused by a lower protein expression level of Cx43 we compared the abundance of Cx43 by immunoblotting. Figure 2 shows a representative Western blot using total lysates of transfected HeLa cells. Expression levels of the alanine mutants were not significantly different (296A: 92.4±21.4; 297A: 77.5±16.3; 306A: 82.7±15.4) from WT-Cx43 (set to 100%, n=4). We next applied confocal immunofluorescence to assess if reduced intercellular coupling for the mutant S306A was due to an altered intracellular distribution of connexins. We did not find obvious changes in intracellular distribution, number or size of gap junctions in any of the mutants when compared to WT-Cx43 (Fig. 3). As S306A was the only mutation resulting in decreased junctional conductance, we examined this in more detail by quantifying size and number of gap junctions between S306A transfected cells compared to WT-Cx43. The relative coverage of connexin plaques in the cell membranes between neighboring cells was somewhat higher in S306A expressing cells (43.5±3.3 %, average of 191 plaques in 53 interfaces) compared to WT-Cx43 (33.6±2.8 %, average of 190 plaques in 55 interfaces, P<0.05). Average plaque sizes in S306A and WT-Cx43 transfected cells were 1.32±0.10 µm and 1.47±0.16 µm (n.s.), respectively. These results suggest that reduced coupling in S306A transfected cells was not caused by a decreased amount of gap junctions in cell surfaces between neighboring cells.
Figure 2.
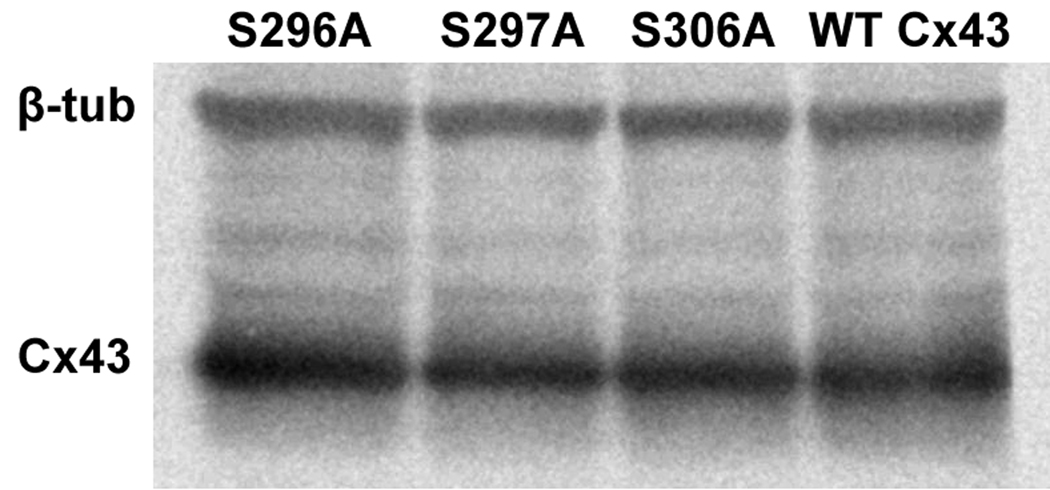
Western blotting was used for quantification of total amounts of Cx43. Representative western blot showing expression levels of different alanine substitutions and WT-Cx43.
Figure 3.
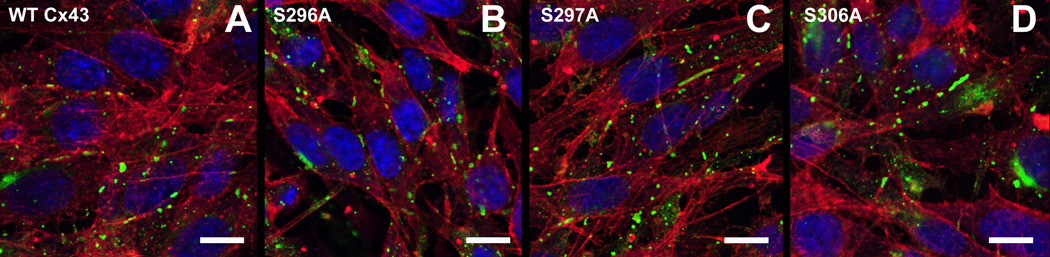
Confocal immunoflourescence microscopy of gap junctions between cells expressing WT and mutated Cx43. Cx43 is shown in green, F-actin in red and nuclei in blue. Scale bars are 10 µm.
To ensure that connexin in the plasma membrane was junctional connexin and that the similar values of gap junction formation and sizes obtained were not masked by an increase in the number of free hemichannels in the S306A mutant, we performed immunoprecipitation experiments of biotinylated Cx43. Cell surface biotinylation was performed using a membrane-impermeant reagent that biotinylates extracellular domains of transmembrane proteins. It has been shown that connexins in gap junction plaques are inaccessible to the biotinylation reagent in the tight space between docked connexons and therefore only free hemichannels are labeled using this procedure (16). After biotinylation the cells were lysed and Cx43 was immunoprecipitated with total anti-Cx43 antibody (C6219)(see supplementary figure 1). In the WT-Cx43 cells the unbound fraction of Cx43 was 19.7±3.8 fold (n=5) larger than the biotinylated fraction. In the S306A mutant the corresponding value was 26.6±8.3 (n=4) (n.s). Hence, there was no significant change in the amount of free hemichannels upon introduction of the S306A mutation. This result indicated, in line with the analysis of gap junction formation, an overall unaltered trafficking for the S306A mutant.
S306 is phosphorylated in both HeLa cells and rat heart tissue
To test whether S306 is phosphorylated in HeLa cells and rat heart tissue, we used an antibody (pS306 Ab) that only recognizes this residue in its phosphorylated form. When staining WT-Cx43 transfected HeLa cells with pS306 Ab and an antibody that recognizes total Cx43 (Fig. 4A), it is seen that Cx43 in plaques is recognized by both antibodies, indicating that Cx43 in plaques is phosphorylated in HeLa cells. When this combination of antibodies is used with S306A-transfected HeLa cells, no staining with pS306 Ab is observed (Fig. 4B). In normal rat heart tissue, clear staining with both antibodies is observed in the intercalated discs (Fig. 4C), confirming our previous mass spectrometry results (11).
Figure 4.
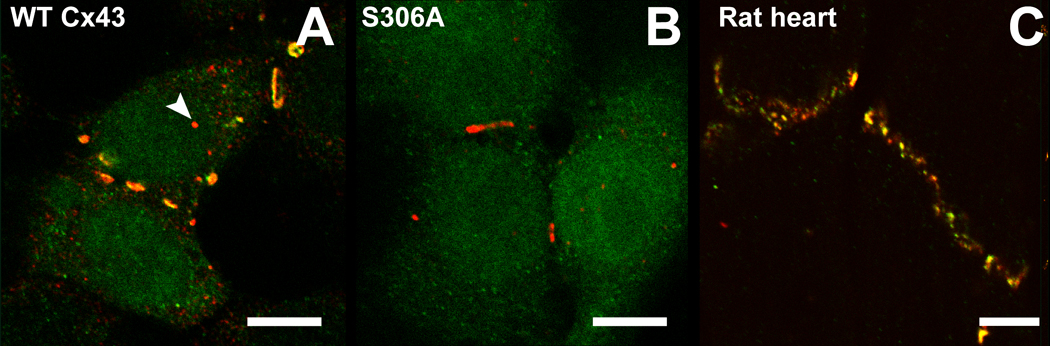
Immunofluorescence images showing total Cx43 (in red) and Cx43 phosphorylated at serine 306 (in green) in HeLa cells (A &B) and rat heart ventricular tissue (C). (A) HeLa cells transfected with WT-Cx43 display co-localization (in yellow) of the two antibodies mainly in gap junction plaques. Discrete spots only stain for total Cx43 (e.g. arrow) indicating that not all Cx43 is phosphorylated at S306. (B) S306A is not recognized by pS306 Ab in HeLa cells and therefore only total Cx43 is stained. (C) Intercalated discs in rat ventricular heart tissue are recognized by both antibodies showing that most of this Cx43 is phosphorylated at S306. Scalebars are 10 µm.
Unitary conductance in S306A
Our data suggest that the reduced coupling in S306A was not caused by changes in the abundance or distribution of Connexin43. We therefore investigated the unitary conductance of the S306A and WT-Cx43 channels. To allow observation of single channel events in cells transfected with Cx43, we applied the gap junction uncoupler octanol (1–2 mmol/L) to the perfusate. Untransfected HeLa or vector-transfected HeLa cells were devoid of electrical coupling when tested by dual patch clamp, showing absence of endogenous connexin expression (data not shown).
Figure 5A top panel shows an original recording of single channel activity in cells expressing WT-Cx43 channels. The distance between the horizontal lines represents the size of a 100 pS event. Channel openings to the main open state are around 120 pS (black arrows), and frequently a partial closure to a residual state of around 75 pS (grey arrows) is observed. Occasionally, channels closed directly from the residual state (red arrow). The average of 655 events in 6 experiments is summarized in the frequency histogram (Fig. 5B). The distribution is based on absolute conductance changes, where openings (negative steps) and closures (positive steps) of the same size are placed in the same bin. The data is described by two Gaussian distributions with means of 119 pS (95% confidence interval 118–120 pS) and 73 pS (95% confidence interval 70–76pS). The 119 pS peak corresponds to transitions between the closed and the main open state, whereas the events around 73 pS represent transitions between the main open and the residual state. Closures directly from the residual state occurred at too low frequency to appear in the histogram.
Figure 5.
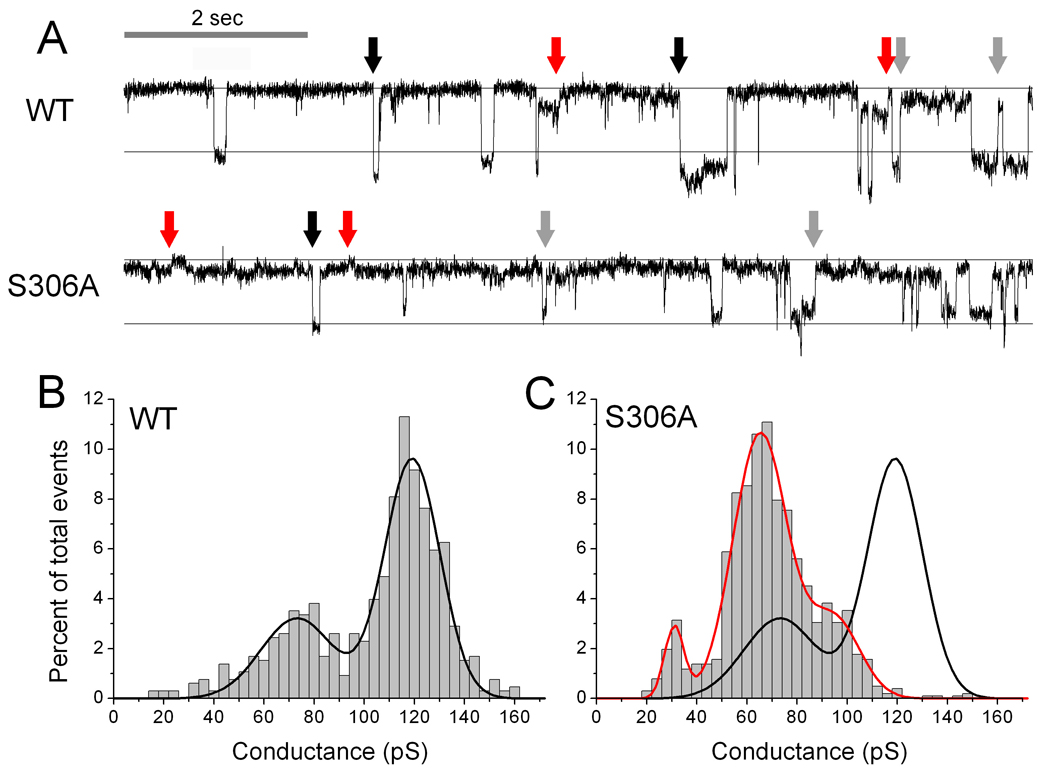
(A) Representative traces showing a 10 second recording of single channel events for WT-Cx43 and S306A, respectively. The guidelines show a difference of 100 pS and the upper line indicate the closed state of the channel. Black arrows indicate openings to the main state, gray arrows closures to the residual state and red arrows closures from the residual state. Panels B (WT-Cx43, n=655) and C (S306A, n=1015) show histograms of single channel openings and closures in bins of 5 pS. The black line in panel B shows a Gaussian fit of the distribution of WT-Cx43 events. In panel C, a Gaussian fit of the S306A distribution of events is shown in red with the Gaussian fit of the WT-Cx43 distribution superimposed in black.
Channel activity in cells expressing the S306A mutant was quite different, and a representative trace is shown in Fig. 5A, lower panel. The 120 pS events, which dominated in WT-Cx43 channels, were completely absent, and the largest events were around 95 pS (black arrows). Smaller events around 30 and 65 pS were observed in the trace and some of these are marked with red and grey arrows, respectively. The sum of the smaller events equals that of the larger openings, indicating that these events are transitions between a residual state and the closed (30 pS) or between a residual state and the main state (95 pS) of the channels.
The frequency distribution of 1015 events from 7 experiments is shown in Fig. 5C. The S306A channel data was fitted by three Gaussian distributions (red curve) with peaks at 95 pS (91–99 pS), 65 pS (64–67 pS) and 31 pS (30–33 pS). The number of positive and negative events in each group was identical and therefore represents equal numbers of openings and closures. For comparison the Gaussian fit for WT-Cx43 is shown in black. The peaks in the S306A distribution correspond to the events pointed out in the trace in Fig. 5A lower panel, showing that both single channel conductance and gating of S306A channels are altered significantly in comparison to WT-Cx43. Both the main and residual states of S306A channels are smaller than WT-Cx43 channels. Furthermore, whereas WT-Cx43 channels mainly moved to and from the residual state via the main open state, S306A channels moved more freely between all states.
Discussion
We characterized the functional significance of three newly described Cx43 phosphorylation sites in the C-terminal part of Cx43, S296, S297 and S306. These sites were originally identified by mass spectrometry of Cx43 from rat hearts subjected to global ischemia (11). Mimicking the unphosphorylated state, we show that exchange of serine with alanine in position 296 and 297 does not affect macroscopic conductance. In contrast, alanine substitution of serine 306 reduced electrical coupling to 57% without reducing the formation of gap junction plaques. The main and residual state conductances of S306A channels were 80% and 67% of WT-Cx43 channels, respectively. The reduction of channel conductance in combination with altered channel kinetics may account for the observed decrease in macroscopic electrical coupling.
Reduction of electric coupling between cells to 57% for the S306A mutant could theoretically be explained by: 1) a change in the number of channels residing in gap junctional plaques; 2) a change in their open probability; or 3) a change in single channel conductance. It is well known that phosphorylation regulates trafficking of Cx43 to the cell membrane (15), and a reduction in plaque size and/or number could be the main mechanism behind the reduced coupling. Our results, however, show that total Cx43 content was similar regardless of the Cx43 construct employed. Also, immunostaining of HeLa cell cultures showed that the relative area occupied by gap junctions was slightly larger in S306A expressing cells. Although the number and size of plaques were not statistically different in S306A and WT-Cx43, the combined effect of these parameters resulted in a slightly larger relative area occupied by gap junctions in S306A cells. However, if the concentration of Cx43 was the same in both WT and S306A plaques, the average current per channel would be even lower than the 57 % of WT-Cx43 observed at the macroscopic level.
Channels in the cell membrane are either docked to channels of the neighboring cell or present as free hemichannels. Therefore, electrical coupling can be reduced by an increase in hemichannel number. We applied biotinylation followed by immunoprecipitation and quantified the biotin bound and unbound Cx43 fractions by immunoblotting. WT-Cx43 and S306A expressing cells did not display any difference. Since the number of free hemichannels did not reveal a statistically significant difference, we conclude that the number of available channels is, within the resolution of our detection methods, either similar or larger in S306A compared to WT-Cx43 expressing cells.
We then investigated if the reduced coupling in S306A was due to reduced open channel probability (Po) and/or a reduced single channel conductance. Under octanol treatment the main state single channel conductance of S306A channels was reduced to 95 pS compared to 119 pS in WT-Cx43. Phosphorylation is known to regulate single channel conductance both negatively and positively. PKC activation by phorbol esters is associated with a reduced single channel conductance (17, 18), and can be prevented by substitution of S368 to alanine (18). The opposite was found in a study of serines 325/328/330, where substitution of these sites to alanine reduced single channel conductance (12). The latter result indicates that dephosphorylation can change single channel conductance.
S306A channels also exhibited altered gating compared to WT channels. In WT channels the most frequent events occurred between the closed and main state, whereas fluctuations between the main and the residual state were less frequent. Although closure of WT channels from the residual state was observed, these events were relatively uncommon. In contrast, in S306A channels openings and closings between the closed and the residual state occurred frequently as revealed in the frequency histogram, and the most frequent event was the fluctuation between the residual and the main state. We could not assess the open probability directly since single channel recordings are only possible upon addition of the uncoupler octanol that alters Po. However, it is conceivable that S306A channels spend more time in the residual state due to altered gating, and this could add to the reduced macroscopic conductance.
Changes in phosphorylation at different sites in Cx43 are not necessarily independent. Lampe and coworkers showed that dephosphorylation of S365 plays a permissive role for the phosphorylation of S368 (13), and it could be speculated that the S306A mutation causes phosphorylation or dephosphorylation of other sites. As mentioned above, dephosphorylation of S325/328/330 results in a shift to smaller single channel conductances (12), similar to that observed in the present study. Therefore, any effect of a changed phosphorylation status of S306 on these sites could potentially reduce channel conductance indirectly. However, the S325/328/330 triple alanine-mutant was associated with intracellular retention (12), which was not the case in S306A (Fig. 3). We therefore find it unlikely that the S306A mutation induces a dephosphorylation of the S325/328/330 sites.
S306 was originally discovered as a phosphorylated site by mass spectrometry in rat hearts. When hearts were subsequently exposed to global ischemia for 7 minutes, phosphorylation at S306 was no longer detectable (11). Notably, no studies have been published that examine the phosphorylation of S306 in human cardiac myocytes.. However, as the amino acid sequences of Cx43 from rats and humans are identical, and since the effects of these alanine substitutions have been observed in a cell line of human origin, it is likely that the observed changes in unitary conductance also are relevant to the regulation of human Cx43. A previous study has shown that most of the Cx43 exists in a hemi- or dephosphorylated form in HeLa cells (19), whereas cardiomyocytes have been shown to contain mainly phosphorylated Cx43 (20). Nonetheless, the Cx43 residing in plaques exhibit phosphorylation at S306 in both HeLa cells and in rat cardiac tissue (Fig. 4A and C), consistent with our previous findings using mass spectrometry (11). We therefore conclude that HeLa cells are capable of phosphorylating S306 and resembles, with regard to this phosphorylation site, cardiomyocytes under normal conditions.
During ischemia, coupling is partly reduced by internalization of gap junctions and probably also by reduced activity of the channels remaining at the intercalated disc. Dephosphorylation at S306 will contribute to the latter process by reducing the conductance of active channels and possibly by increasing the time spent in the residual and closed state. This will in turn reduce electrical coupling, and increase electrical heterogeneity and the susceptibility to re-entry arrhythmia.
Conclusions
The present study shows that the exchange of serine 306 for alanine results in a reduction of single channel conductance and a change in channel kinetics compared to WT-Cx43. We therefore conclude that the S306 is a regulatory phosphorylation site that may play an important role in ischemic uncoupling.
Supplementary Material
Acknowledgements
This work was supported by The Danish National Research Foundation; The John and Birthe Meyer Foundation; The Danish Health Sciences Research Councils; The Velux Foundation; The Danish Heart Association; The Novo Nordisk Foundation; The A.P. Møller Foundation. We thank lab technician Ninna Buch for her skillful help and Rune Berg for valuable help and discussion of data filtering.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: None
Reference List
- 1.Peters NS, Coromilas J, Severs NJ, et al. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95:988–996. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- 2.Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. 1989;69:1049–1169. doi: 10.1152/physrev.1989.69.4.1049. [DOI] [PubMed] [Google Scholar]
- 3.Gutstein DE, Morley GE, Tamaddon H, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–339. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kleber AG, Riegger CB, Janse MJ. Electrical uncoupling and increase of extracellular resistance after induction of ischemia in isolated, arterially perfused rabbit papillary muscle. Circ Res. 1987;61:271–279. doi: 10.1161/01.res.61.2.271. [DOI] [PubMed] [Google Scholar]
- 5.Beardslee MA, Lerner DL, Tadros PN, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:656–662. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- 6.Burt JM. Block of intercellular communication: interaction of intracellular H+ and Ca2+ Am J Physiol. 1987;253:C607–C612. doi: 10.1152/ajpcell.1987.253.4.C607. [DOI] [PubMed] [Google Scholar]
- 7.Morley GE, Taffet SM, Delmar M. Intramolecular interactions mediate pH regulation of connexin43 channels. Biophys J. 1996;70:1294–1302. doi: 10.1016/S0006-3495(96)79686-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fluri GS, Rudisuli A, Willi M, et al. Effects of arachidonic acid on the gap junctions of neonatal rat heart cells. Pflugers Arch. 1990;417:149–156. doi: 10.1007/BF00370692. [DOI] [PubMed] [Google Scholar]
- 9.Massey KD, Minnich BN, Burt JM. Arachidonic acid and lipoxygenase metabolites uncouple neonatal rat cardiac myocyte pairs. Am J Physiol. 1992;263:C494–C501. doi: 10.1152/ajpcell.1992.263.2.C494. [DOI] [PubMed] [Google Scholar]
- 10.Hofgaard JP, Banach K, Mollerup S, et al. Phosphatidylinositol-bisphosphate regulates intercellular coupling in cardiac myocytes. Pflugers Arch. 2008;457:303–313. doi: 10.1007/s00424-008-0538-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Axelsen LN, Stahlhut M, Mohammed S, et al. Identification of ischemia-regulated phosphorylation sites in connexin43: A possible target for the antiarrhythmic peptide analogue rotigaptide (ZP123) J Mol Cell Cardiol. 2006;40:790–798. doi: 10.1016/j.yjmcc.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Lampe PD, Cooper CD, King TJ, et al. Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci. 2006;119:3435–3442. doi: 10.1242/jcs.03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solan JL, Marquez-Rosado L, Sorgen PL, et al. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol. 2007;179:1301–1309. doi: 10.1083/jcb.200707060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xing D, Kjølbye AL, Nielsen MS, et al. ZP123 increases gap junctional conductance and prevents reentrant ventricular tachycardia during myocardial ischemia in open chest dogs. J Cardiovasc Electrophysiol. 2003;14:510–520. doi: 10.1046/j.1540-8167.2003.02329.x. [DOI] [PubMed] [Google Scholar]
- 15.Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.VanSlyke JK, Musil LS. Analysis of connexin intracellular transport and assembly. Methods. 2000;20:156–164. doi: 10.1006/meth.1999.0933. [DOI] [PubMed] [Google Scholar]
- 17.Kwak BR, van Veen TA, Analbers LJS, et al. TPA increases conductance but decreases permeability in neonatal rat cardiomyocyte gap junction channels. Exp Cell Res. 1995;220:456–463. doi: 10.1006/excr.1995.1337. [DOI] [PubMed] [Google Scholar]
- 18.Lampe PD, TenBroek EM, Burt JM, et al. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;149:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Solan JL, Fry MD, TenBroek EM, Lampe PD. Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J Cell Sci. 2003;116:2203–2211. doi: 10.1242/jcs.00428. [DOI] [PubMed] [Google Scholar]
- 20.Laird DW, Puranam KL, Revel JP. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem J. 1991;273:67–72. doi: 10.1042/bj2730067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.