

Summary
Endothelial chemokines are instrumental for integrin-mediated lymphocyte adhesion and transendothelial migration (TEM). By dissecting how chemokines trigger lymphocyte integrins to support shear-resistant motility on and across cytokine-stimulated endothelial barriers, we found a critical role for high-affinity (HA) LFA-1 integrin in lymphocyte crawling on activated endothelium. Endothelial-presented chemokines triggered HA-LFA-1 and adhesive filopodia at numerous submicron dots scattered underneath crawling lymphocytes. Shear forces applied to endothelial-bound lymphocytes dramatically enhanced filopodia density underneath crawling lymphocytes. A fraction of the adhesive filopodia invaded the endothelial cells prior to and during TEM and extended large subluminal leading edge containing dots of HA-LFA-1 occupied by subluminal ICAM-1. Memory T cells generated more frequent invasive filopodia and transmigrated more rapidly than their naive counterparts. We propose that shear forces exerted on HA-LFA-1 trigger adhesive and invasive filopodia at apical endothelial surfaces and thereby promote lymphocyte crawling and probing for TEM sites.
INTRODUCTION
A key checkpoint in leukocyte recruitment to inflammatory targets is their ability to firmly arrest on vascular endothelial cells (EC) and maintain resistance to detachment by disruptive shear forces while crawling on the luminal surface of the endothelium (Ley et al., 2007). This lateral migration allows leukocytes to search out sites permissive for endothelial crossing (diapedesis) within variable distances from their original arrest site (Auffray et al., 2007; Muller, 2003; Phillipson et al., 2006; Schenkel et al., 2004). Recent high-speed intravital analysis of inflamed vessels revealed that a major fraction of stably adherent leukocytes migrate laterally on the venular surface, scanning the endothelium for potential transendothelial migration (TEM) sites (Auffray et al., 2007; Phillipson et al., 2006). Even when the leukocyte initially arrests at its final TEM site, it must still translocate a substantial part of its body along the apical endothelial surface. Unlike motility in interstitial spaces (Lammermann et al., 2008), leukocytes crawling on vascular endothelium must maintain (through adhesive integrin bonds) persistent resistance to detachment by disruptive shear forces (Ley et al., 2007). Because leukocytes crawl on endothelial barriers at 5–20 μm/min, these bonds must be rapidly displaced while new bonds are continuously generated at the leukocyte's leading edge. Leukocyte integrins must therefore reversibly adjust their strength of binding to endothelial ligands via spatiotemporal changes in affinity and avidity. In lymphocytes, these changes are tightly coordinated by chemokines displayed on the endothelial surface, which bind and signal through cognate G protein-coupled receptors (GPCRs) (Kinashi, 2005; Ley et al., 2007).
In lymphocytes, three major integrins, LFA-1 and the α4 integrins VLA-4 and α4β7, control essentially all shear-resistant lymphocyte interactions with various endothelial beds, yet it is not known how these integrins contribute to lymphocyte crawling and TEM under disruptive shear flow. In most peripheral sites of inflammation, α4 integrins and LFA-1 have been shown to cooperate in promoting lymphocyte extravasation (Luster et al., 2005), but their relative contribution to individual steps after arrest, and preceding TEM, has not been dissected. In vivo dissection of integrin contribution to these processes with blocking mAbs or gene-deficient models is hampered by the early involvement of these integrins in lymphocyte arrest and adhesion strengthening (Luster et al., 2005).
How chemokine signals dynamically activate integrin adhesiveness in crawling leukocytes has been examined in vitro (Schenkel et al., 2004). Subsets of α4 and β2 integrins are thought to undergo rapid conformational changes both prior to, and shortly after, binding their major endothelial ligands (Kinashi, 2005). Nevertheless, it is unclear when and where high-affinity integrin subsets are generated and how they cooperate with intermediate-affinity integrin subsets and with chemokine-triggered actomyosin machineries in supporting rapid shear-resistant lymphocyte crawling over endothelial cells. Shear forces exerted on leukocyte-endothelial contacts are not solely disruptive, as indicated by the fact that they can positively regulate integrin-ligand bonds (Astrof et al., 2006; Alon and Dustin, 2007; Woolf et al., 2007). Lymphocyte TEM is dramatically facilitated in the presence of shear flow (Cinamon et al., 2001), but most in vitro studies on integrin-mediated lymphocyte transendothelial migration have been conducted under shear-free conditions and therefore could not directly dissect this highly relevant role of hydrodynamic forces in integrin-mediated lymphocyte crawling and TEM.
By using an in vitro model of lymphocyte adhesion and TEM across cytokine-activated human umbilical vein endothelial cells (HUVEC), we demonstrate that although both VLA-4 and LFA-1 integrins are similarly activated by initial chemokine signals, LFA-1 is the major mediator of postarrest T lymphocyte crawling and subsequent TEM. High-affinity LFA-1 subsets are necessary for these two processes, and their turnover at numerous focal dots ensures rapid crawling and resistance to detachment by shear forces. Although traditionally believed to move over endothelial cells as flattened bodies (Ley et al., 2007), we suggest that lymphocytes must use ventral adhesive filopodia to rapidly crawl and scan endothelial cells via millipede-like locomotion.
RESULTS
Chemokine-Stimulated Lymphocytes Crawl and Cluster Endothelial ICAM-1
Endothelial-presented chemokines trigger robust lymphocyte crawling over the apical surface of cytokine-stimulated HUVEC under shear stress conditions in vitro (Cinamon et al., 2001) as well as in vivo (Figure S1 and Movie S1 available online). By exploring the role of the two major lymphocyte integrins, LFA-1 and VLA-4, and their key endothelial ligands, in this shear-resistant crawling, we first analyzed ICAM-1 and VCAM-1 redistribution underneath fresh peripheral blood human T cells (herein T cells) interacting with TNF-a-activated HUVEC. T cells crawling under shear flow on HUVEC monolayers overlaid with CXCL12 were fixed minutes after initial arrest prior to their initiation of TEM, and the distribution of endothelial ICAM-1, VCAM-1, or E-selectin was analyzed. Numerous submicron ICAM-1 dots were highly enriched on the HUVEC surface just underneath the crawling T cells whereas VCAM-1 and E-selectin microclusters were not (Figure 1A). Similar enrichment of ICAM-1 dots was also observed underneath T cells crawling over activated HUVEC overlaid with other prototypic T cell chemokines (Figure S2). Live imaging of T cells crawling on activated HUVEC transfected with ICAM-1-GFP detected transient ICAM-1-GFP clusters (mean duration, 50 s) underneath rapidly crawling T cells (Figure S3 and Movie S2), but similar clustering of VCAM-1-GFP was not detected (Figure S4 and Movie S3). Although coblocking both LFA-1 and α4 integrins eliminated the majority of chemokine-stimulated lymphocyte adhesion and crawling, blocking α4 integrins had no effect on these processes (Figure 1B) in spite of high amount of VCAM-1 on the activated HUVEC (Figure 1A). In contrast, preblocking LFA-1 reduced by 50% the fraction of shear-resistant crawling T cells (Figure 1B) and dramatically reduced ICAM-1 dots underneath the remaining lymphocytes (Figure 1C). Thus, when ICAM-1 is encountered by chemokine-triggered LFA-1, VCAM-1 is not utilized for shear-resistant crawling. Similar dominance of LFA-1 over α4 integrins was also observed on TNF-a- and IL-4-activated HUVEC (data not shown). By analyzing the distribution of LFA-1 with a pan LFA-1 mAb, we found numerous LFA-1 dots scattered under the entire T cell-EC contact area whereas α4 integrins were enriched at the rear of crawling T cells (Figure 1D). Thus, chemokine-stimulated LFA-1 plays a dominant role in T cell crawling on endothelial surfaces under shear flow.
Figure 1. Endothelial ICAM-1 Is Clustered by Chemokine-Stimulated LFA-1 in T Cells Crawling under Shear Flow.
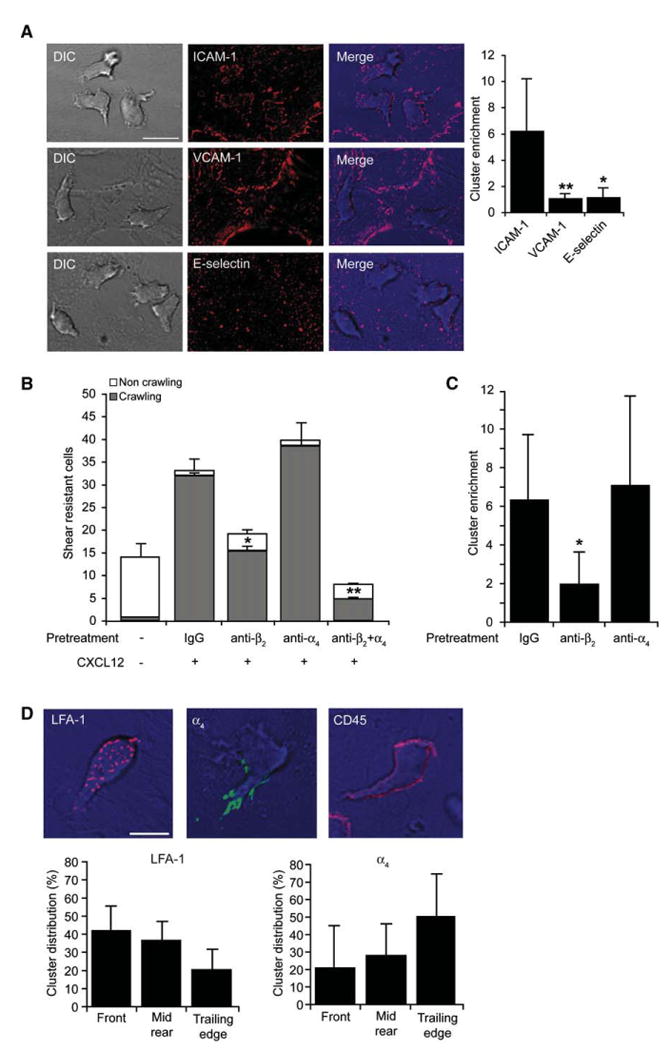
(A) Human peripheral blood T cells accumulated for 1 min on activated HUVEC overlaid with CXCL12 were subjected to shear stress of 5 dyn/cm2. Actively crawling lymphocytes (>90% of originally accumulated T cells) were fixed 7 min after accumulation and stained with mAbs to ICAM-1, VCAM-1, or E-selectin. Scale bar represents 10 μm. Right graph: cluster enrichment in areas underneath crawling T cells versus areas proximal to these cells. Background staining is shown in Figure S2B. Values are the mean ± SD of 20 crawling T cells in each experimental group. *p < 0.00001; **p < 0.00001.
(B) Effects of blocking T cell LFA-1 and α4 integrins (with TS1.18 and HP1/2, respectively) on T cell arrest and crawling on activated HUVEC overlaid with CXCL12. Values represent the mean ± range of two fields in each experimental group and results are representative of three independent experiments. *p < 0.035; **p < 0.006 for the indicated crawling fractions compared to control mAb-treated T cells.
(C) Enrichment of ICAM-1 clusters under T cells treated with control IgG-, β2-, or α4-blocking mAbs measured as in (A). *p < 0.0001.
(D) Crawling T cells were fixed and stained for LFA-1 (red), β4 integrins (green), or CD45 (red). Scale bar represents 6 μm. Lower panels depict the distribution of LFA-1 or β4 integrin clusters in different compartments of crawling T cells. Values are the mean ± SD of 20 crawling T cells in each experimental group.
High-Affinity LFA-1 Is Confined to Focal ICAM-1 Dots underneath Crawling Lymphocytes
T cell LFA-1 is activated in situ by chemokine signals through a rapid acquisition of an open headpiece conformation, identified by the b I domain-specific mAb, 327C (Beals et al., 2001; Shamri et al., 2005). Because the only β2 integrin expressed on resting T cells is LFA-1, this mAb was used to probe high-affinity LFA-1 (herein HA-LFA-1). Among several other conformational epitopes tested, this epitope is insensitive to mild fixation and, being distal to the ICAM-1 binding site on the a I domain, remains accessible even in ICAM-1-occupied LFA-1 (Beals et al., 2001). Fluorescence analysis revealed numerous submicron dots of HA-LFA-1 at the immediate contact between the crawling T cell and the endothelial surface (Figures 2A and 2B). In contrast, pan LFA-1 staining was detected within 2 μm above the T cell-HUVEC contact, and this LFA-1 was negative for 327C, suggesting that it comprises a low-affinity pool (Figure 2A, graph). Similar HA-LFA-1 dots were enriched underneath T cells crawling over CCL19- or CXCL9-bearing HUVEC (Figure S5). Notably, when the reporter 327C mAb was perfused over actively crawling lymphocytes in real time, it artificially entrapped HA-LFA-1 subsets at their rear (Figure S6). Collectively, our data suggest that HA-LFA-1 serially induced by chemokine signals during crawling cluster endothelial ICAM-1 within the direct T cell-EC contact zone (Figures 2A and 2B). We therefore termed these HA-LFA-1:ICAM-1 dots “focal dots.” Substantial levels of focal dots formed within the first 1 min after initial T cell arrest and increased by 2- to 2.5-fold within the next 3 to 6 min, reaching a peak density of 28 ± 10 focal dots, most of which were <0.5 μm2 (Figure 2C). T cells covered during 60 s of crawling a mean area slightly larger than their own (Movie S4 and Figure S7), so all HA-LFA-1 dots underneath crawling T cells got successively replaced by de novo generated dots within this time frame.
Figure 2. Crawling T Cells Rearrange Chemokine-Stimulated High-Affinity LFA-1 in Scattered Dots Enriched with Endothelial ICAM-1.
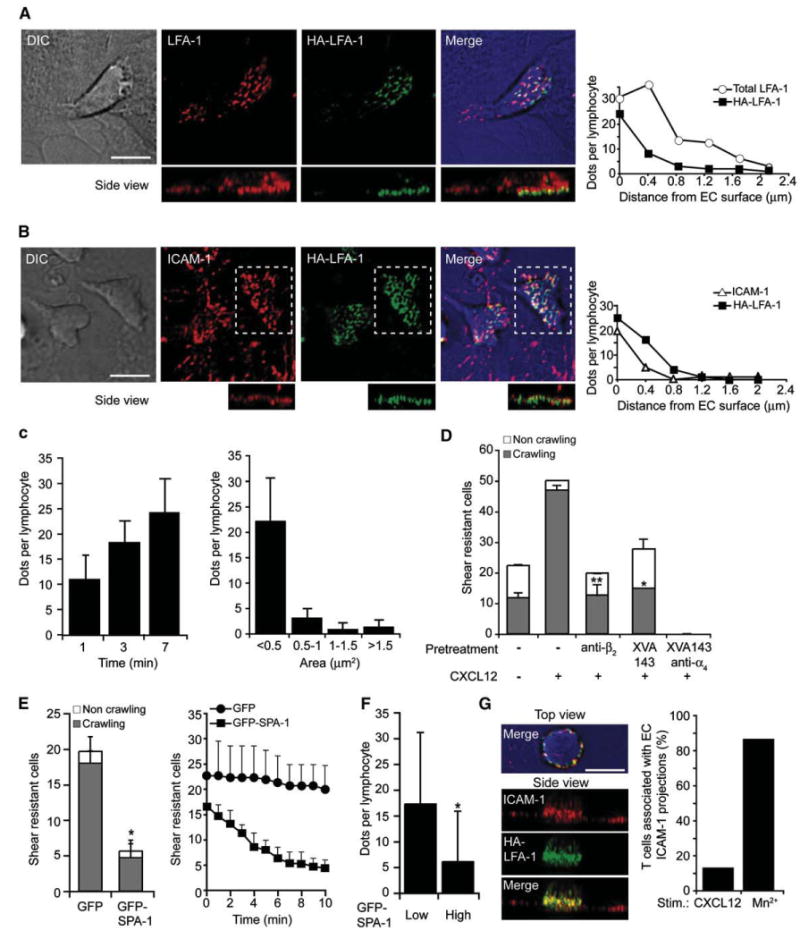
(A) T cells crawling for 7 min on activated HUVEC overlaid with CXCL12 under shear flow were fixed and stained for HA-LFA-1 (via the 327C reporter, green) and total LFA-1 (via the anti-αL mAb TS2/4, red). Right graph: analysis of LFA-1 and HA-LFA-1 dot density per individual Z-sections constituting the indicated side view projections.
(B) T cells crawling on a similar HUVEC monolayer were fixed as in (A) and stained with mAbs to HA-LFA-1 (green) and ICAM-1 (red). In the bottom panels, the side view projections correspond to the dashed rectangles. Right graph: analysis of ICAM-1 and HA-LFA-1 dots density in individual Z-sections as in (A). Scale bar represents 6 μm.
(C) Left: density of HA-LFA-1 dots underneath T cells fixed at various time points on the activated HUVEC. Mean density ± SD of 10 crawling T cells in each group. Right: size distribution of HA-LFA-1 dots in T cells crawling for 7 min. Mean ± SD of 30 crawling T cells from independent experiments is shown.
(D) Effects of XVA143 (1 μm) and integrin-blocking mAbs (20 mg/ml) on shear-resistant adhesion and crawling of T cells analyzed over a 7 min period. Values are the mean ± range of two fields in each experimental group. *p < 0.02, **p < 0.003 for control versus mAb and XVA143-treated crawling T cells. The experiment is a representative of three.
(E) Left: shear-resistant adhesion and crawling of T cells transiently transfected with either GFP-SPA-1 or GFP on CXCL12-bearing HUVEC analyzed as in (D). Right: detachment over time of initially arrested lymphocytes. The experiment is a representative of three.
(F) Density of HA-LFA-1 dots underneath T cells expressing low or high GFP-SPA-1 fixed 3 min after arrest on CXCL12-bearing HUVEC.
(G) T cells were perfused over activated HUVEC in binding medium containing 2 μm Mn2+, fixed after 7 min and stained for ICAM-1 (red) and HA-LFA-1 (green). Right graph: fractions of CXCL12- versus Mn2+-stimulated T cells engulfed by ICAM-1 clusters extended at least 1.2 μm above the EC surface.
Because T cell crawling on ICAM-1 can be mediated by both high- and intermediate-affinity LFA-1 subsets (Morin et al., 2008; Stanley et al., 2008), we next tested whether CXCL12-triggered T cell crawling on activated HUVEC can be mediated by both LFA-1 subsets. XVA143 is an α/β I-like allosteric antagonist that blocks the acquisition of high-affinity LFA-1:ICAM-1 binding (Salas et al., 2004; Yang et al., 2006) (Figure S8). Notably, XVA143 reduced CXCL12-triggered T cell crawling over activated HUVEC to an identical extent as that caused by total LFA-1 blockade, and this residual crawling was all α4 dependent (Figure 2D). Because XVA143 stabilizes LFA-1 in an extended intermediate-affinity state (Salas et al., 2004) (Figure S8), these results taken together with the HA-LFA-1 staining with the 327C reporter indicate that functional HA-LFA-1 is indispensable for T cell crawling on endothelial ICAM-1 under shear flow.
A key regulator of chemokine-stimulated HA-LFA-1 in T cells is the Rap1 GTPase (Shimonaka et al., 2003; Ghandour et al., 2007). Overexpression of the Rap1 GAP, SPA-1 inhibited up to 50% of the chemokine-triggered HA-LFA-1 reporter epitope, 327C (data not shown), and dramatically reduced the number of T cells initially arrested on CXCL12-bearing HUVEC that could resist detachment by high shear over time (Figure 2E; Movies S5 and S6). Interestingly, although SPA-1 overexpression did not affect T cell motility on CXCL12 under shear-free conditions (data not shown), shear-resistant lymphocyte crawling was impaired, along with reduced density of HA-LFA-1 focal dots under crawling T cells (Figures 2E and 2F).
Previous in vitro reports suggested that endothelial ICAM-1 clusters within proactive microvilli-like membrane projections engulf lymphoblasts in ring-like docking structures enriched with VCAM-1 (Barreiro et al., 2002; Carman et al., 2003). Nevertheless, the ICAM-1 microclusters generated by the primary T cell crawling on CXCL12 bearing TNF-a-activated HUVEC were not associated with elongated engulfing projections unless Mn2+, a strong artificial integrin-activating cation, was included (Figures 2B and 2G). Mn2+-induced HA-LFA-1 clustering interfered, however, with T cell crawling and TEM (Figure S9). Thus, the chemokine-triggered HA-LFA-1 dots generated by crawling primary T cells are dynamic submicron focal assemblies rather than long-lived micron-scale EC projections engulfing stationary lymphocytes.
Chemokine-Triggered HA-LFA-1 Focal Dots Promote Lymphocyte Crawling Independently of Endothelial Machineries
We next wished to explore whether any endothelial machinery plays an active role in HA-LFA-1 focal dot formation and function. Most of the LFA-1 on intact T cells was in low-affinity state and evenly distributed on the T cell surface (Figure 3A). Isolated ICAM-1 and coimmobilized CXCL12 were both necessary and sufficient to support robust crawling of T cells under high shear stress (Figure 3B). Notably, HA-LFA-1 was induced underneath the entire surface of T cells crawling over the chemokine-bearing substrate (Figure S10) and ICAM-1 was critical for HA-LFA-1 focal dots, because many fewer dots were formed underneath T cells crawling on VCAM-1 and CXCL12 surface (Figures 3B and 3C). Both CXCL12- and Mn2+-induced firm lymphocyte arrest on ICAM-1 but not on VCAM-1 were eliminated by the HA-LFA-1 blocker, XVA143 (Figure S8; Figure 3D, right; and not shown). In contrast, perfusing XVA143 over adherent T cells actively crawling on ICAM-1 and CXCL12 surface resulted in their rapid detachment (Figure 3D), whereas irreversible Mn2+-induced HA-LFA-1 adhesions were resistant to the XVA143 blocker (Figure 3D; Figure S9). Interestingly, suboptimal doses of XVA143 perfused over actively crawling T cells caused only partial lymphocyte detachment but still halted crawling (Figure S11). These findings collectively suggest that maximal turnover of chemokine-stimulated HA-LFA-1 is mandatory for continuous LFA-1-mediated crawling but not for stationary LFA-1-dependent firm adhesion. Notably, CXCL12-triggered HA-LFA-1 dots colocalized with the key LFA-1 affinity regulator, talin, but not with ERM adaptors (Figure 3E). Ultrastructural analysis of T cells revealed numerous adhesive filopodia underneath T cells crawling on ICAM-1 and CXCL12 surface and to lesser extent on VCAM-1 and CXCL12 surface (Figures 3F and 3G, right). Nevertheless, T cells forced to attach and crawl on an ICAM-1-free VCAM-1 and CXCL12 surface did not form LFA-1 dots and their LFA-1 was polarized to the leading edge (Figures 3F and 3G). Thus, the chemokine-induced HA-LFA-1 is stabilized by immobile ICAM-1. HA-LFA-1 dots form independently of active endothelial machineries and their high turnover allows rapid shear-resistant lymphocyte crawling under shear flow.
Figure 3. T Cells Crawling under Shear Flow Generate Short-Lived HA-LFA-1 Dots Independently of Endothelial Machineries.
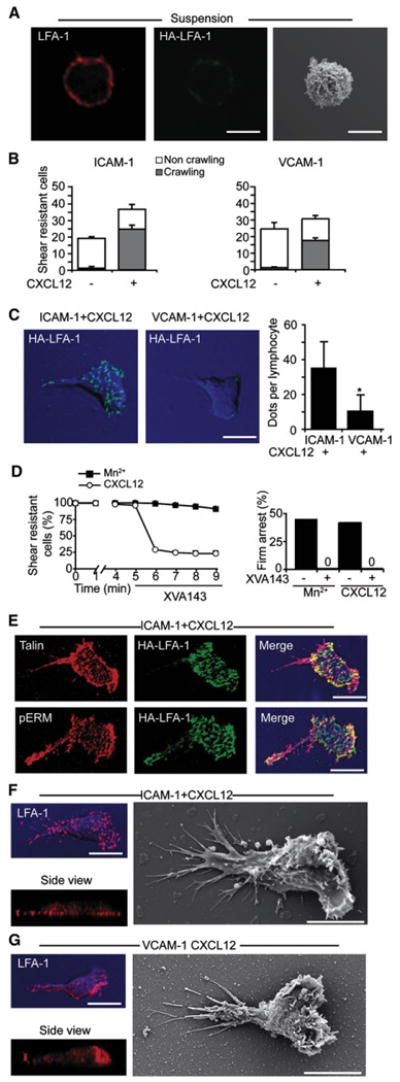
(A) T cells were fixed in suspension and stained for LFA-1 (red) and HA-LFA-1 (green). Right: SEM image of a representative resting T cell.
(B) Equal number of T cells accumulated on purified ICAM-1 or VCAM-1 coimmobilized without or with CXCL12 were subjected to a shear stress of 5 dyn/ cm2 for 7 min. Values are the mean ± range of two fields in each experimental group.
(C) T cells crawling on the indicated substrates under shear flow as in (B) were fixed and stained for HA-LFA-1 (green). DIC images (blue) overlaid with fluorescence images are depicted. Right graph: the density of HA-LFA-1 dots underneath T cells crawling on ICAM-1 or VCAM-1, each coimmobilized with CXCL12. Values are the mean ± SD of 20 crawling T cells in each experimental group. *p < 0.00001.
(D) Left: T cells crawling on ICAM-1+CXCL12 or arrested on ICAM-1 in presence of Mn2+ (2 μm) were exposed to XVA143 (1 μm) 5 min after arrest. The fractions of T cells remaining resistant to detachment thereafter are shown. Right: effect of XVA143 on the frequency of T cells arrested on ICAM-1 in the presence of the indicated LFA-1 stimuli.
(E) T cells crawling on ICAM-1+CXCL12 as in (B) were costained for HA-LFA-1 (green) and talin (red) or pERM (red).
(F and G) T cells crawling on ICAM-1 (F) or on VCAM-1 (G), each coimmobilized with CXCL12, were fixed and stained for LFA-1 (red). Side view projections are shown at the bottom. SEM images of T cells crawling on identical surfaces are shown in the right panels. Scale bars represent 6 μm.
T Cell Crawling and TEM Are Independent of Src and DAG PKCs
The focal dots generated by ICAM-1-occupied HA-LFA-1 resemble, in size and distribution, podosomes, specialized integrin-enriched cell-matrix and cell-EC contacts detected mainly in myeloid cells (Linder and Aepfelbacher, 2003). In monocytes spread on ICAM-1 and CXCL12 surfaces, we identified podosome-like structures with high-affinity β2 staining colocalized with dense F-actin and pTyr staining (Figure S12). In contrast, HA-LFA-1 focal dots generated by crawling T cells were largely devoid of F-actin or pTyr (Figure 4A; Figure S12). Furthermore, whereas Src activity is critical for podosome formation (Carman et al., 2007; Linder and Aepfelbacher, 2003), Src inhibition in T cells did not affect chemokine-triggered lymphocyte crawling on activated HUVEC under shear flow prior to TEM and did not reduce TEM (Figure 4B). Src inhibition also did not reduce the ability of chemokine-triggered LFA-1 to support T cell adhesion and crawling on isolated ICAM-1 (Figure 4C) and did not affect the density of HA-LFA-1 dots underneath crawling lymphocytes (Figure 4D), but completely blocked T cell receptor-induced LFA-1 activation (Figure 4C, right).
Figure 4. High-Affinity LFA-1 Dots Are Devoid of F-actin and pTyr and Support T Cell Crawling Independent of Src Signaling.
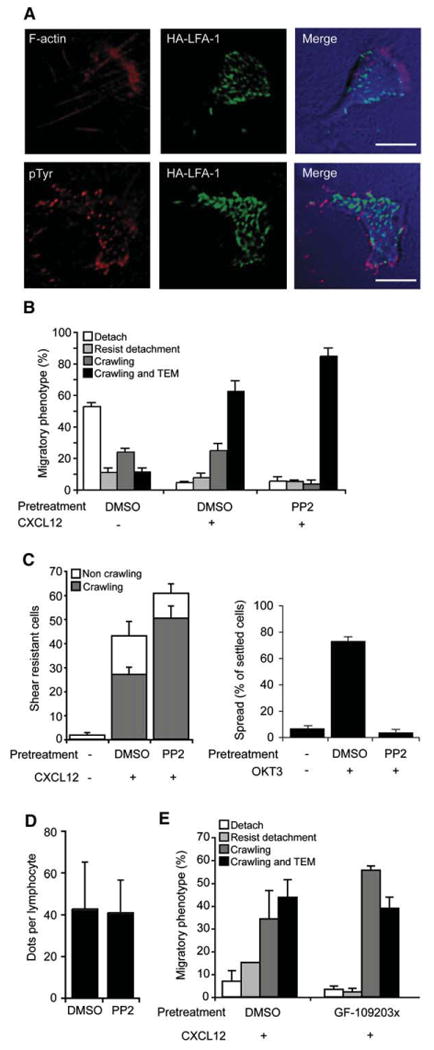
(A) T cells crawling on activated HUVEC were fixed and costained for HA-LFA-1 (green) and F-actin (red, top) or pTyr (red, bottom). Images are representative of 30 crawling T cells. Scale bars represent 6 μm.
(B) Migratory phenotypes of T cells pretreated with PP2 (10 μm) or a carrier interacting with activated HUVEC.
(C) Effects of PP2 pretreatment on T cell adhesion and crawling on ICAM-1+CXCL12 analyzed as in Figure 3B. Right: effects of PP2 on TCR-induced (anti-CD3-stimulated) lymphocyte spreading on ICAM-1.
(D) HA-LFA-1 dot density in PP2 and control treated T cells adhered and crawling on ICAM-1+CXCL12. Values are the mean ± SD of 15 cells in each group.
(E) Migratory phenotypes of T cells pretreated with the DAG-PKC inhibitor, GF-109203x (5 μm) or a carrier interacting with activated HUVEC overlaid with CXCL12.
Values in experiments shown in (B), (C), and (E) represent the mean ± range of two fields.
Diacyl glycerol (DAG)-dependent protein kinase C (PKC) iso-forms can be directly activated by LFA-1:ICAM-1 occupancy to promote lymphoblast motility on ICAM-1 (Volkov et al., 2001). DAG-dependent PKCs also function in CXCL12 triggering of VLA-4 adhesions to VCAM-1 (Ghandour et al., 2007). Nevertheless, pretreatment of resting T cells with a general inhibitor of DAG-dependant PKCs had no effect on T cell arrest, crawling on CXCL12-bearing activated HUVEC, or on ICAM-1 and CXCL12 surfaces (Figure 4E and data not shown). Thus, neither Src nor PKCs are required for the formation and function of HA-LFA-1 focal dots in shear-resistant T cell crawling over and through activated endothelium under shear flow. Additional LFA-1 outside-in signals were previously shown to promote myosin contractility in lymphoblasts migrating over ICAM-1 (Smith et al., 2003). Indeed, myosin light chain (MLC) activation took place preferentially in the rear of resting T cells crawling over activated HUVEC presenting CXCL12, as indicated by staining for phosphorylated MLC (pMLC), which controls myosin-mediated contractility (Figure S13A). pMLC was excluded, however, from HA-LFA-1 focal dots (Figure S13A), and inhibition of MLC kinase while inhibiting crawling did not affect chemokine-triggered HA-LFA-1 dot formation (Figures S13B and S13C). Thus, HA-LFA-1 dot formation is necessary but insufficient for myosin-dependent lymphocyte crawling on EC (Figure S13C).
Shear-Promoted TEM Is Associated with Invasive Filopodia underneath Crawling Lymphocytes
We next performed ultrastructural analysis of T cells accumulated on CXCL12-bearing activated HUVEC fixed shortly after initial arrest but much earlier to TEM initiation (t = 6–10 min after initial arrest). More than 90% of polarized T cells crawled at this stage (Figure 1B), so all amoeboid-shaped T cells were assumed as actively crawling. Interestingly, in the presence of shear flow, crawling T cells generated a 3-fold higher number of filopodia than under shear-free conditions (Figure 5A; Figure S14), and this augmenting effect was independent of endothelial machineries as it was also observed in T cells crawling on EC-free ICAM-1 and CXCL12 (Figure 5A, right graph; Figure S15) surface. Strikingly, in two-thirds of all crawling T cells (Figure S14), a fraction of the adhesive filopodia invaded into the HUVEC surface and the mean density of these filopodia was increased by 3-fold under shear flow conditions (Figure 5A). Similar invasive filopodia were observed in T cells crawling on microvascular EC presenting CXCL12 (Figure S16), where T cell TEM was also LFA-1, chemokine, and shear stress dependent (data not shown). Transmission EM of middle cross sections of crawling T cells confirmed the generation of invasive filopodia underneath T cells, in addition to the lymphocyte periphery (Figure 5B). The depth of individual invasive filopodia ranged from 0.2 to 0.5 μm and immunostaining revealed enrichment of HA-LFA-1 at the base of individual invasive filopodia (Figure 5C). Furthermore, HA-LFA-1 staining surrounded clusters of activated phophorylated ERMs, markers of filopodia (Figure 3E). Importantly, blocking HA-LFA-1 with XVA143 dramatically reduced the number of both adhesive and invasive filopodia underneath crawling T cells (Figure 5D) concomitantly with more than 70% reduction in lymphocyte TEM (data not shown). Notably, the majority of invasive filopodia were formed well before T cells reached paracellular EC junctions (Figure S17), the predominant sites of TEM in this setting (Yang et al., 2005).
Figure 5. T Cells Crawling on Activated EC Generate Adhesive and Invasive Filopodia Preferentially under Shear Flow.
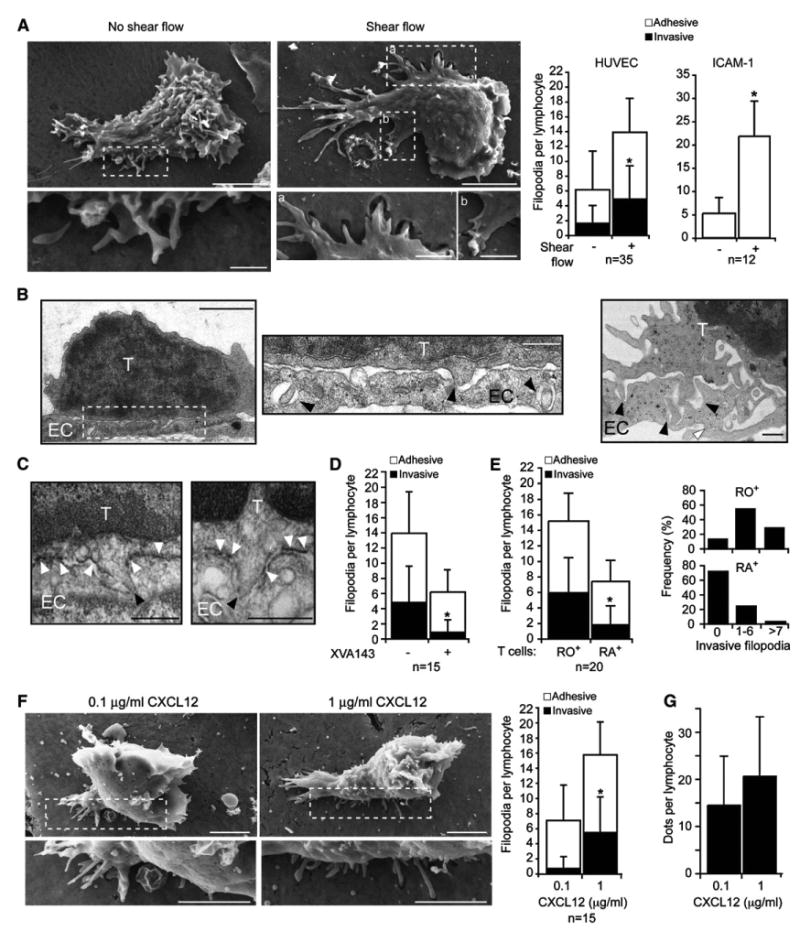
(A) T cells settled on activated HUVEC overlaid with CXCL12 (1 mg/ml) were left in shear-free conditions (left) or subjected to shear stress for 3 min (right) prior to fixation. SEM images of representative T cells (scale bars represent 3 μm) with enlarged segments (scale bars represent 0.4 μm) are depicted. Left graph depicts average densities (per lymphocyte) of adhesive and invasive filopodia underneath T cells crawling on HUVEC. Right graph depicts the average densities of adhesive filopodia underneath T cells crawling on ICAM-1+CXCL12. Values are mean ± SD. *p < 0.0003.
(B) T cells crawling on activated HUVEC were fixed as in (A) and · 80 nm cross sections in the middle of T cells were viewed by transmission EM. Scale bars represent 2 μm (left) and 0.4 μm (enlarged segment and right). Black arrowheads depict tips of invasive filopodia; white arrowhead marks EC junction.
(C) Immunoelectron microscopy of crawling T cells labeled after fixation with the HA-LFA-1-specific mAb and stained with HRP-labeled secondary antibody. White arrowheads mark dense HA-LFA-1 staining around invasive filopodia and black arrowheads mark tips of filopodia. Scale bars represent 0.5 μm.
(D) Average densities of adhesive and invasive filopodia underneath crawling T cells treated with or without XVA143. *p < 0.005.
(E) Average densities of adhesive and invasive filopodia underneath either crawling CD45RO+ or CD45RA+T cells. *p < 0.01. Inset graphs depict the fractions of crawling T cells with the indicated range of invasive filopodia per lymphocyte.
(F) Representative SEM images of T cells crawling on identically activated HUVEC overlaid with either 0.1 or 1 mg/ml CXCL12. Scale bars represent 3 μm. Graph, average densities of adhesive and invasive filopodia underneath crawling T cells. *p < 0.002.
(G) Density of HA-LFA-1 dots underneath crawling T cells on activated HUVEC overlaid with either 0.1 or 1 mg/ml CXCL12. Values represent the mean ± SD of 30 crawling T cells in each group.
To examine whether the ability to generate invasive filopodia correlates with successful TEM, we next compared the number of these submicron structures in memory (CD45RO+) T cells, which transmigrate at much higher rates than their naive (CD45RA+) counterparts (Figure S18). Notably, the density of both adhesive and invasive filopodia detected underneath crawling memory T cells was 2- and 3-fold higher, respectively, than under naive T cells (Figure 5E). To further verify that the ability to generate invasive filopodia correlates with successful TEM, HUVEC were overlaid with lower levels of CXCL12, a concentration insufficient to promote any T cell TEM (Figure S19). Under these limiting conditions, no invasive filopodia could be detected (Figure 5F). Nevertheless, a high number of HA-LFA-1 dots were still generated (Figure 5G), indicating that the threshold of chemokine signals required for the generation of invasive filopodia and TEM is higher than for HA-LFA-1 focal dots and crawling on the apical EC surface under flow.
Cdc42 Is Required for Chemokine-Triggered Lymphocyte Crawling under Flow
Because chemokine-stimulated T cell crawling on HUVEC and on isolated ICAM-1 involved adhesive filopodia, we next wished to address the role of the Cdc42 GTPase, a master regulator of filopodia formation, in lymphocyte crawling. Complete suppression of Cdc42 by siRNA was not achieved (data not shown), so we used a selective Cdc42 inhibitor, Secramine A (SecA), recently shown to inhibit Cdc42 binding to membranes, GTP, and effectors in a RhoGDI-dependent manner (Pelish et al., 2006). We found that SecA inhibits the activation of Cdc42 but not of Rac1 in T cells treated with CXCL12 (Figure 6A). Notably, SecA did not interfere with CXCL12-dependent induction of HA-LFA-1 (Figure 6B), or with shear-resistant LFA-1-mediated T cell adhesion to ICAM-1 or to activated EC (Figures 6C and 6D), nor with the density of HA-LFA-1 focal dots (Figure 6E). Nevertheless, SecA-treated T cells arrested on ICAM-1 or to activated HUVEC under shear flow could not crawl away from their arrest site (Figures 6C and 6D) and failed to initiate TEM (data not shown). In addition, their ability to generate both adhesive and invasive filopodia was dramatically reduced (Figure 6F). Importantly, SecA did not affect random integrin-independent T cell motility on immobilized CXCL12 under shear-free conditions (Figure 6G), a process tightly regulated by chemokine-activated Rac (Shulman et al., 2006). Furthermore, TCR-induced LFA-1-mediated T cell spreading on ICAM-1, another actin-dependent process, was also not affected by the Cdc42 inhibitor (Figure 6H). These results collectively suggest that although Cdc42 activation by chemokine has no role in LFA-1 activation as previously shown (Bolomini-Vittori et al., 2009), it has a major role in the ability of HA-LFA-1 to generate filopodia and support millipede-like crawling under shear flow.
Figure 6. LFA-1-Mediated T Cell Crawling and Filopodia Formation under Shear Flow, but Not Chemokine-Triggered Adhesiveness, Require Activated Cdc42.
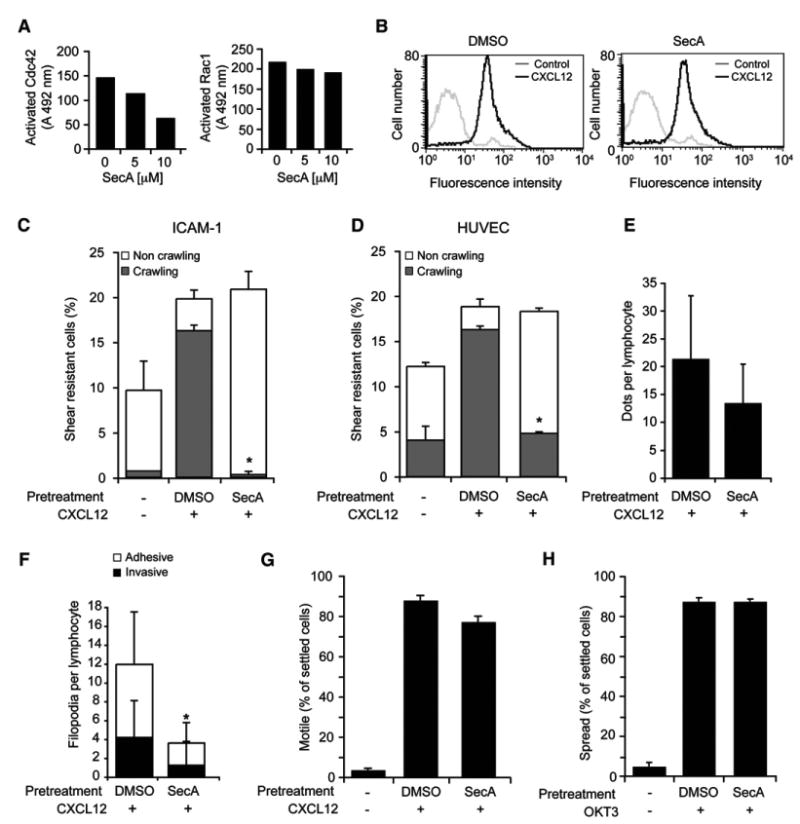
(A) T cells were treated with the indicated doses of SecA and then stimulated for 30 s with 250 nM CXCL12. Cdc42 (left) or Rac1 (right) activation was assessed by ELISA as described in the Supplemental Experimental Procedures section. Values are the average OD units of six samples with background subtracted.
(B) T cells pretreated with the Cdc42 inhibitor SecA or a carrier were stimulated with CXCL12, and induction of HA-LFA-1 was analyzed by flow cytometry with the HA-LFA-1 reporter, 327C.
(C) Effects of SecA (present throughout the assay) on shear-resistant T cell adhesion and crawling on ICAM-1 coimmobilized with CXCL12. *p < 0.003.
(D) Effects of SecA on T cell adhesion and crawling on activated HUVEC overlaid with CXCL12. *p < 0.002. Flow conditions and analysis in (C) and (D) were preformed as described in Figures 2 and 3. Results are representative of two independent experiments.
(E) Effects of SecA on HA-LFA-1 dot density under T cells crawling on activated HUVEC. Average dot densities were determined for 30 crawling T cells in each experimental group.
(F) The effects of SecA on the average density of adhesive and invasive filopodia underneath T cells crawling on activated HUVEC bearing CXCL12. Values are the mean ± SD of 15 crawling T cells in each experimental group. *p < 0.0002.
(G) Effects of SecA on random T cell motility over immobilized CXCL12 under shear-free conditions.
(H) Effects of SecA on TCR-triggered LFA-1-mediated T cell spreading on ICAM-1.
In (G) and (H), at least 40 T cells were analyzed in each experimental group and values represent the mean ± range of two fields.
Transmigrating T Cells Engage Subendothelial ICAM-1 via HA-LFA-1 Dots
ICAM-1 is expressed on both the apical and subluminal (basal) sides of activated endothelial cells (Oppenheimer-Marks et al., 1991). We therefore next asked whether HA-LFA-1 is continuously generated during TEM. Once reaching paracellular EC junctions, lymphocytes send invasive filopodia into both the EC body and through the EC junction to the subluminal EC side (Figure 5B; Figure S20). Once TEM was initiated, it took the transmigrating T cell 10–20 s to fully extend its leading edge underneath the endothelial layer, and an additional 20 s period to insert its upper lobe and complete TEM (Figure 7A; Movie S7). HA-LFA-1 was detected in numerous submicron focal dots in the leading edge of transmigrating T cells (Figure 7B; Movie S8). Interestingly, these HA LFA-1 dots were segregated to the apical side of the T cell's leading edge, in direct contact with basal endothelial ICAM-1 (Figure 7B, side views). Ultrastructural analysis further indicated that the leading edge remained in close contact with the subluminal EC membrane (Figure 7B, right). Shortly after TEM was completed, T cells still engaged the subluminal ICAM-1 via HA-LFA-1 dots (Figure 7C), yet these lymphocytes did not send invasive filopodia into the basal endothelial surface (Figure 7C, right). Thus, focal dots fail to generate invasive filopodia in lymphocyte-endothelial contacts that do not experience shear forces. These findings collectively suggest that HA-LFA-1 guides the transmigrating T cells through serial engagements with basal endothelial ICAM-1, a novel modality of inverted motility underneath the basal endothelial surface.
Figure 7. High-Affinity LFA-1:ICAM-1 Dots Are Stabilized on the Apical Side of the Leading Edge of Actively Transmigrating T Cells.
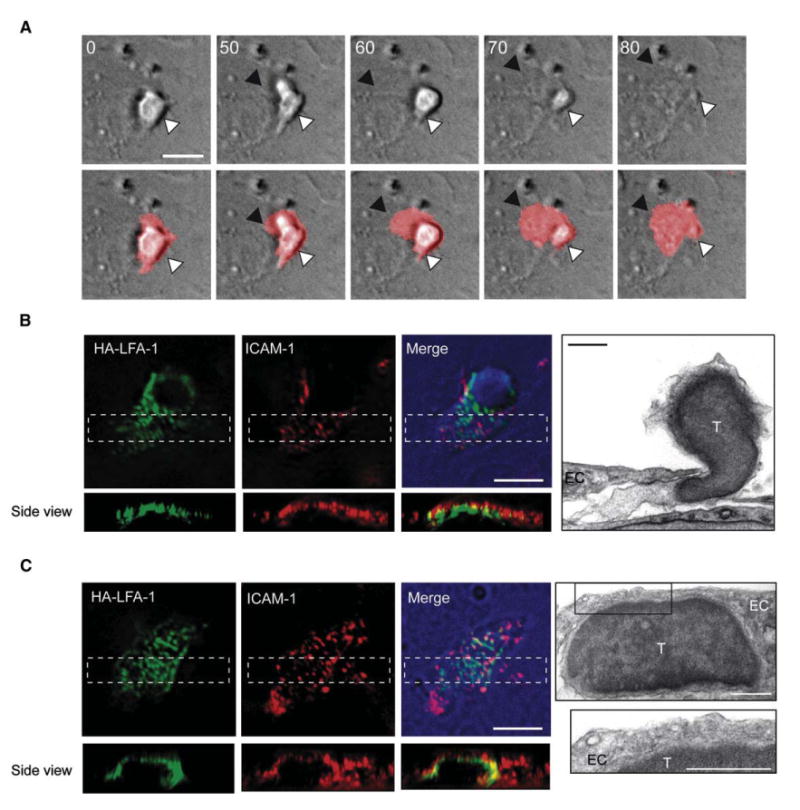
(A) T cells prelabeled with the membranal dye, DiD, were recorded during crawling and TEM across activated-HUVEC bearing CXCL12. Time-lapse images taken from Movie S7. Top and lower panels depict DIC images alone or merged with DiD fluorescence, respectively. The white arrowheads point to the T cell rear adhered to the apical side of the EC, and the black arrowheads mark the leading edge of the transmigrating T cell that engages the subluminal aspect of the EC. The time indicated is in seconds. Scale bar represents 10 μm.
(B and C) T cells migrating through and below activated HUVEC displaying CXCL12 were fixed and stained for HA-LFA-1 and ICAM-1. Fluorescence microscopy images of at least 10 cells (scale bars represent 6 μm) and transmission EM image (scale bars represent 3 μm) are depicted. Images of a T cell undergoing TEM are shown in (B) and Movie S8. Images of a T cell crawling under the EC are shown in (C).
DISCUSSION
Unlike interstitial leukocyte motility, leukocyte crossing of endothelial barriers is an integrin-dependent process tightly regulated by shear forces (Cinamon et al., 2001, 2004; Cuvelier and Patel, 2001; Phillipson et al., 2006). To productively crawl on endothelial cells, leukocytes must integrate three external signals: inside-out integrin activation signals from chemokine-occupied GPCRs, outside-in integrin activation signals, and chemokine-GPCR-triggered signals to actomyosin-remodeling Rho GTPases. The recent realization that chemokine-stimulated T cell integrins are further activated by shear forces (Astrof et al., 2006; Woolf et al., 2007) prompted us to assess whether and how these forces alter the contribution of specific integrins to T cells crawling over and through defined endothelial barriers. We utilized an in vitro model of resting T cells interacting with primary cytokine-activated HUVEC coexpressing the two major integrin ligands, VCAM-1 and ICAM-1, a classical setting for TEM studies. Although initial T cell arrest on activated endothelium can be mediated by both VLA-4 and LFA-1 integrins, T cell crawling under shear flow is mostly LFA-1 dependent. Signals from chemokines displayed on the endothelium stabilize HA-LFA-1 in transient focal dots at the immediate T cell-EC interface without which adhesiveness and crawling on ICAM-1 under shear flow are abolished. We found numerous dynamic HA-LFA-1:ICAM-1 dots scattered below crawling T cells, which nucleate the formation of submicron ventral filopodia, particularly in the presence of shear forces. These small and transient LFA-1 focal dots allow T cells to establish fast motility in the X-Y plane while serially generating confined adhesive filopodia in the perpendicular direction. Interestingly, a subset of these ventral filopodia constantly invades the endothelial surface not only during TEM but also at time points well before initiation of TEM and its density is closely correlated with subsequent TEM efficiency.
We suggest that T cell crawling on endothelial integrin ligands involves numerous evenly scattered traction sites with local high resistance to detachment by disruptive shear forces, and rapid local turnover, maintaining very fast millipede-like T cell translocation over the endothelial surface. The crawling T cell encounters chemokine signals that serially induce HA-LFA-1 stabilized by ICAM-1. Individual LFA-1:ICAM-1 bonds may be further stabilized by application of shear force (Astrof et al., 2006). In contrast, lymphoblasts migrating over ICAM-1 concentrate their HA-LFA-1 in a large mid-cell focal zone (Smith et al., 2005). Four alternative models could account for the high turnover of HA-LFA-1 focal contacts in lymphocytes crawling on endothelial ICAM-1. (1) Rapid localized downregulation of HA-LFA-1 via GTPase deactivation, without which T cells would remain stuck to the endothelial surface, as recently found for T cells with LFA-1 artificially locked in activated state (Semmrich et al., 2005; Yang et al., 2006). (2) Accelerated dissociation of HA-LFA-1:ICAM-1 bonds by disruptive shear forces (Alon and Dustin, 2007). (3) Chemokine induction of intermediate-affinity LFA-1 with high bond dissociation rates (Stanley et al., 2008). (4) Shedding of the ectodomain of LFA-1 occupied by ICAM-1 (Evans et al., 2006). Our results are consistent with possibilities (1) and (2). Freezing chemokine-stimulated LFA-1 at intermediate states abolished LFA-1-dependent T cell crawling on activated endothelium under shear stress, suggesting that only HA-LFA-1:ICAM-1 bonds can promote shear-resistant crawling under physiological shear flow. As for possibility (4), shedding is unlikely to be a major player in T cell motility on ICAM-1, because LFA-1 ectodomain shedding takes place in neutrophils but not in T cells (Evans et al., 2006). We thus favor the possibility that chemokine stimulation of HA-LFA-1 within submicron focal dots generates high, reversible, and confined adhesiveness that allows rapid T cell translocation to the next endothelial contact. Such adhesive contacts may take place on preformed endothelial nanostructures enriched with ICAM-1 and tetraspanins, recently suggested to support and regulate leukocyte-EC interactions (Barreiro et al., 2008).
A variety of endothelial regulatory GTPases, including RhoA and RhoG, are triggered by microclustered ICAM-1 (Hordijk, 2003; van Buul et al., 2007) and may translate HA-LFA-1:ICAM-1 focal contacts into endothelial invaginations surrounding invasive T cell filopodia. ICAM-1 is expressed on both sides of activated HUVEC (Oppenheimer-Marks et al., 1991), so both apical and basolateral ICAM-1 may assemble distinct TEM-guiding structures. Indeed, we observed that basal endothelial ICAM-1 clusters with HA-LFA-1 dots in T cell-EC contacts at the leading edge of actively transmigrating T cells. Thus, the ability of HA-LFA-1 to engage with and cluster ICAM-1 is utilized beyond the crawling phase, to guide subluminal protrusions sent by the transmigrating T cell through paracellular and transcellular routes.
Filopodia are narrow protrusive structures enriched with F-actin bundles that serve various exploratory functions (Gupton and Gertler, 2007). The HA-LFA-1:ICAM-1 dots serve as nucleating sites for adhesive filopodia generated underneath T cells crawling under shear flow. A subset of these adhesive filopodia become invasive under shear flow and a critical density of these invasive filopodia correlates with successful TEM. Below a critical amount of apical chemokine signals, T cells do not form invasive filopodia and fail to transmigrate once reaching paracellular EC junctions. The density of invasive filopodia formation by crawling T cells and TEM are therefore highly dependent on the magnitude of the apical chemokine signal. Notably, the density of invasive filopodia was particularly high in memory rather than in naive T cells, corresponding to the higher transmigratory capacity of memory lymphocytes across inflamed endothelial barriers.
Recently, Src-dependent LFA-1-enriched podosome-like assemblies were shown to organize submicron ICAM-1 rings and drive transcellular migration of lymphoblasts (Carman et al., 2007). In contrast to blasts, the resting T cells studied by us crawled on activated EC via HA-LFA-1 dots generated independently of Src signaling. Invasive protrusions underneath our T cells did not coincide with transcellular TEM, so the majority of the invasive filopodia observed in vivo (Anderson and Anderson, 1976) are likely to be associated with T cell crawling and general probing for TEM sites rather than with obligatory transcellular diapedesis.
Our findings establish a paradigm for the role of chemokine and integrin signals in millipede-like T cell crawling and probing for potential TEM sites under shear flow. We propose that ventral LFA-1-dependent filopodia serve as nucleating sites for either paracellular TEM (representing the majority of events) or transcellular TEM (in a minority of events). Within the paracellular junctions, lymphocytes may use their filopodia and LFA-1 to guide their leading edge along basolateral endothelial ICAM-1. Key open questions are whether millipede-like crawling and invasive filopodia are similarly used by other types of leukocytes crossing endothelial barriers and how GTPases on these leukocytes translate chemokine and integrin signals into shear-resistant crawling and TEM.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
See Supplemental Experimental Procedures.
Cells
Human T cells were isolated from citrate-anticoagulated whole blood of healthy donors (approved by the Institutional Review Board of the Rambam Medical Center, in accordance with the provisions of the Declaration of Helsinki) by dextran sedimentation and density separation over Ficoll-Hypaque as described (Grabovsky et al., 2000). The resulting PB T cells (>90% CD3+ T cells) were cultured for 16–18 hr before experiments. HUVEC were isolated from umbilical cord veins and established as primary cultures as described (Larrivee and Karsan, 2005) or purchased from Promocell (Heidelberg, Germany). Human dermal microvascular endothelial cells (HDMVEC, Promocell) were cultured according to the supplier's instructions.
Analysis of T Cell Migration under Shear Flow in Real Time
Primary HUVEC were plated at confluence on glass-bottom dishes spotted with fibronectin (20 mg/ml). EC were stimulated with 200 U/ml TNF-α for 20–28 hr. CXCL12, CCL19, or CXCL9 (1 mg/ml) were overlaid for 5 min on an EC monolayer assembled in the flow chamber and washed extensively. All flow experiments were performed at 37° C. T cells were perfused over the EC monolayer at 0.75 dyn/cm2 for 1 min (accumulation phase) and then left under constant shear (5 dyn/cm2) throughout the assay. Images were video recorded through a 203 phase contrast objective at 2 frame/s. For migratory phenotype analysis, T cells accumulated in multiple fields (0.11 μm2 in area) were individually tracked throughout the assay and categorized as described (Cinamon et al., 2001). Crawling lymphocytes moved at least 3 cell diameters over the EC surface without crossing through the monolayer whereas transmigrating lymphocytes crawled variable distances before crossing the endothelium (crawling and TEM). Crawling categories were normalized to the T cell flux (Grabovsky et al., 2000) whereas migratory phenotypes were calculated as fractions of T cells originally accumulated during the first 1 min phase. For blocking integrin function, T cells were preincubated with appropriate blocking mAbs (20 mg/ml, 5 min) or with XVA143 (1 μm) and perfused through the flow chamber in binding medium containing 2 mg/ml of the mAbs or 1 μm XVA143, respectively. For analysis of crawling T cells on purified integrin ligands, ICAM-1-Fc (5 mg/ml) or VCAM-1-Fc (2 mg/ml) were cocoated with CXCL12 (2 mg/ml) on glass-bottom plates precoated with Protein A (20 mg/ml), as described (Grabovsky et al., 2000). Where indicated, T cells were pretreated with either PP2, GF-109203x, ML-7, or their DMSO carrier for 30 min. For inhibition of Cdc42, T cells were pretreated with Secramine A (SecA, 5 μm/ 5 × 105 cells, 15 min). All inhibitors were included in the perfused medium throughout the experiments. Real-time imaging of fluorescently labeled or transiently transfected cells is described in the Supplemental Experimental Procedures section.
Immunostaining of Fixed Cells, Fluorescence Images Acquisition, and Processing
For immunofluorescence staining, PBS containing 4% (w/v) paraformaldehyde and 2% sucrose was perfused into the flow chamber for 5 min at indicated time points. All experiments were video-recorded for parallel analysis of arrest and crawling. Fixed cells were extensively washed with PBS and blocked with TBS (25 μm Tris [pH 7.4], 150 μm NaCl) supplemented with either 2% HSA or serum. Cells were incubated with either primary directly labeled mAb (45 min, 37° C) or with unlabeled mAb followed by secondary fluorescence antibody staining. For double staining of intracellular molecules and HA-LFA-1, cells were permeabilized with saponin (0.1% w/v, 5 min) before blocking with goat serum. Subsequently, cells were incubated with primary antibody for 45 min and washed and incubated with secondary fluorescence antibodies for 30 min in the presence of 0.05% saponin. Mouse serum was added to quench saponin and secondary antibodies. For intracellular staining of F-actin, TRITC- or coumarin-phalloidin was added after permeabilization of the cells. Fluorescence microscopy was carried out with the DeltaVision system (Applied Precision, Issaquah, WA) with an oil 603/1.4 PlanApo (DIC) objective. All fixed cell images were acquired as serial Z-stacks (0.2 μm apart) and subjected to digital deconvolution and 3D reconstructions with the SoftWoRx software (Applied Precision). Endothelial ligand enrichment underneath T cells crawling on ECs was determined with SoftWoRx software and was expressed as the number of ICAM-1, VCAM-1, or E-selectin clusters in the T cell-EC contact section divided by the number of clusters in lymphocyte-proximal EC areas of identical dimensions. Integrin dot density was defined as the number of microclusters per lymphocyte within the EC contact plane. Integrin and integrin ligand clusters were always compared at fixed fluorescence thresholds.
Electron Microscopy and Filopodia Analysis
Cells were fixed under shear flow as described for fluorescence microscopy followed by additional post fixation (3% glutaraldehyde, 1% sucrose). Fixed cells were processed as described in the Supplemental Experimental Procedures. Adhesive filopodium was defined as a protrusion extended from the lymphocyte body in direct contact with the substrate. Invasive filopodium was defined as lymphocyte protrusion that invaded into the EC body.
Statistical Analysis
All data are reported as mean values ± SD or range (as indicated) and were analyzed by Student's t test. Group comparisons were deemed significant for 2-tailed p values below 0.05.
Supplementary Material
Acknowledgments
We thank S. Schwarzbaum for editorial assistance and O. Barreiro (University Autonoma, Madrid), P. Goichberg, and C. Luxenburg for helpful suggestions. We also thank Ishai Sher and the Weizmann Institute graphic department for graphic assistance. R.A. is the Incumbent of The Linda Jacobs Chair in Immune and Stem Cell Research. R.A. is supported by the Israel Science Foundation, by the Minerva Foundation, Germany, and by MAIN, the EU6 Program for Migration and Inflammation.
Footnotes
SUPPLEMENTAL DATA: Supplemental Data include Supplemental Experimental Procedures, 20 figures, and 8 movies and can be found with this article online at http://www.immunity.com/supplemental/S1074-7613(09)00102-2.
References
- Alon R, Dustin ML. Force as a facilitator of integrin conformational changes during leukocyte arrest on blood vessels and antigen-presenting cells. Immunity. 2007;26:17–27. doi: 10.1016/j.immuni.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Anderson AO, Anderson ND. Lymphocyte emigration from high endothelial venules in rat lymph nodes. Immunology. 1976;31:731–748. [PMC free article] [PubMed] [Google Scholar]
- Astrof NS, Salas A, Shimaoka M, Chen J, Springer TA. Importance of force linkage in mechanochemistry of adhesion receptors. Biochemistry. 2006;45:15020–15028. doi: 10.1021/bi061566o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, Furthmayr H, Sanchez-Madrid F. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol. 2002;157:1233–1245. doi: 10.1083/jcb.200112126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro O, Zamai M, Yanez-Mo M, Tejera E, Lopez-Romero P, Monk PN, Gratton E, Caiolfa VR, Sanchez-Madrid F. Endothelial adhesion receptors are recruited to adherent leukocytes by inclusion in preformed tetraspanin nanoplatforms. J Cell Biol. 2008;183:527–542. doi: 10.1083/jcb.200805076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beals CR, Edwards AC, Gottschalk RJ, Kuijpers TW, Staunton DE. CD18 activation epitopes induced by leukocyte activation. J Immunol. 2001;167:6113–6122. doi: 10.4049/jimmunol.167.11.6113. [DOI] [PubMed] [Google Scholar]
- Bolomini-Vittori M, Montresor A, Giagulli C, Staunton D, Rossi B, Martinello M, Constantin G, Laudanna C. Regulation of conformer-specific activation of the integrin LFA-1 by a chemokine-triggered Rho signaling module. Nat Immunol. 2009;10:185–194. doi: 10.1038/ni.1691. [DOI] [PubMed] [Google Scholar]
- Carman CV, Jun CD, Salas A, Springer TA. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J Immunol. 2003;171:6135–6144. doi: 10.4049/jimmunol.171.11.6135. [DOI] [PubMed] [Google Scholar]
- Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, Dvorak HF, Dvorak AM, Springer TA. Transcellular diapedesis is initiated by invasive podosomes. Immunity. 2007;26:784–797. doi: 10.1016/j.immuni.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinamon G, Shinder V, Alon R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat Immunol. 2001;2:515–522. doi: 10.1038/88710. [DOI] [PubMed] [Google Scholar]
- Cinamon G, Shinder V, Shamri R, Alon R. Chemoattractant signals and β2 integrin occupancy at apical endothelial contacts combine with shear stress signals to promote transendothelial neutrophil migration. J Immunol. 2004;173:7282–7291. doi: 10.4049/jimmunol.173.12.7282. [DOI] [PubMed] [Google Scholar]
- Cuvelier SL, Patel KD. Shear-dependent eosinophil transmigration on interleukin 4-stimulated endothelial cells: a role for endothelium-associated eotaxin-3. J Exp Med. 2001;194:1699–1709. doi: 10.1084/jem.194.12.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans BJ, McDowall A, Taylor PC, Hogg N, Haskard DO, Landis RC. Shedding of lymphocyte function-associated antigen-1 (LFA-1) in a human inflammatory response. Blood. 2006;107:3593–3599. doi: 10.1182/blood-2005-09-3695. [DOI] [PubMed] [Google Scholar]
- Ghandour H, Cullere X, Alvarez A, Luscinskas FW, Mayadas TN. Essential role for Rap1 GTPase and its guanine exchange factor Cal-DAG-GEFI in LFA-1 but not VLA-4 integrin mediated human T-cell adhesion. Blood. 2007;110:3682–3690. doi: 10.1182/blood-2007-03-077628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabovsky V, Feigelson S, Chen C, Bleijs R, Peled A, Cinamon G, Baleux F, Arenzana-Seisdedos F, Lapidot T, van Kooyk Y, et al. Sub-second induction of α4 integrin clustering by immobilized chemokines enhances leukocyte capture and rolling under flow prior to firm adhesion to endothelium. J Exp Med. 2000;192:495–505. doi: 10.1084/jem.192.4.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupton SL, Gertler FB. Filopodia: the fingers that do the walking. Sci STKE. 20072007 doi: 10.1126/stke.4002007re5. re5. [DOI] [PubMed] [Google Scholar]
- Hordijk P. Endothelial signaling in leukocyte transmigration. Cell Biochem Biophys. 2003;38:305–322. doi: 10.1385/cbb:38:3:305. [DOI] [PubMed] [Google Scholar]
- Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol. 2005;5:546–559. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- Lammermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Soldner R, Hirsch K, Keller M, Förster R, Critchley DR, Fassler R, Sixt MT. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 2008;453:51–55. doi: 10.1038/nature06887. [DOI] [PubMed] [Google Scholar]
- Larrivee B, Karsan A. Isolation and culture of primary endothelial cells. Methods Mol Biol. 2005;290:315–329. doi: 10.1385/1-59259-838-2:315. [DOI] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- Linder S, Aepfelbacher M. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 2003;13:376–385. doi: 10.1016/s0962-8924(03)00128-4. [DOI] [PubMed] [Google Scholar]
- Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- Morin NA, Oakes PW, Hyun YM, Lee D, Chin YE, King MR, Springer TA, Shimaoka M, Tang JX, Reichner JS, Kim M. Nonmuscle myosin heavy chain IIA mediates integrin LFA-1 de-adhesion during T lymphocyte migration. J Exp Med. 2008;205:195–205. doi: 10.1084/jem.20071543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24:327–334. doi: 10.1016/s1471-4906(03)00117-0. [DOI] [PubMed] [Google Scholar]
- Oppenheimer-Marks N, Davis LS, Bogue DT, Ramberg J, Lipsky PE. Differential utilization of ICAM-1 and VCAM-1 during the adhesion and transendothelial migration of human T lymphocytes. J Immunol. 1991;147:2913–2921. [PubMed] [Google Scholar]
- Pelish HE, Peterson JR, Salvarezza SB, Rodriguez-Boulan E, Chen JL, Stamnes M, Macia E, Feng Y, Shair MD, Kirchhausen T. Secramine inhibits Cdc42-dependent functions in cells and Cdc42 activation in vitro. Nat Chem Biol. 2006;2:39–46. doi: 10.1038/nchembio751. [DOI] [PubMed] [Google Scholar]
- Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. 2006;203:2569–2575. doi: 10.1084/jem.20060925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas A, Shimaoka M, Kogan AN, Harwood C, von Andrian UH, Springer TA. Rolling adhesion through an extended conformation of integrin αLβ2 and relation to aI and bI-like domain interaction. Immunity. 2004;20:393–406. doi: 10.1016/s1074-7613(04)00082-2. [DOI] [PubMed] [Google Scholar]
- Schenkel AR, Mamdouh Z, Muller WA. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol. 2004;5:393–400. doi: 10.1038/ni1051. [DOI] [PubMed] [Google Scholar]
- Semmrich M, Smith A, Feterowski C, Beer S, Engelhardt B, Busch DH, Bartsch B, Laschinger M, Hogg N, Pfeffer K, Holzmann B. Importance of integrin LFA-1 deactivation for the generation of immune responses. J Exp Med. 2005;201:1987–1998. doi: 10.1084/jem.20041850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamri R, Grabovsky V, Gauguet JM, Feigelson S, Manevich E, Kolanus W, Robinson MK, Staunton DE, von Andrian UH, Alon R. Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat Immunol. 2005;6:497–506. doi: 10.1038/ni1194. [DOI] [PubMed] [Google Scholar]
- Shimonaka M, Katagiri K, Nakayama T, Fujita N, Tsuruo T, Yoshie O, Kinashi T. Rap1 translates chemokine signals to integrin activation, cell polarization, and motility across vascular endothelium under flow. J Cell Biol. 2003;161:417–427. doi: 10.1083/jcb.200301133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman Z, Pasvolsky R, Woolf E, Grabovsky V, Feigelson SW, Erez N, Fukui Y, Alon R. DOCK2 regulates chemokine-triggered lateral lymphocyte motility but not transendothelial migration. Blood. 2006;108:2150–2158. doi: 10.1182/blood-2006-04-017608. [DOI] [PubMed] [Google Scholar]
- Smith A, Bracke M, Leitinger B, Porter JC, Hogg N. LFA-1-induced T cell migration on ICAM-1 involves regulation of MLCK-mediated attachment and ROCK-dependent detachment. J Cell Sci. 2003;116:3123–3133. doi: 10.1242/jcs.00606. [DOI] [PubMed] [Google Scholar]
- Smith A, Carrasco YR, Stanley P, Kieffer N, Batista FD, Hogg N. A talin-dependent LFA-1 focal zone is formed by rapidly migrating T lymphocytes. J Cell Biol. 2005;170:141–151. doi: 10.1083/jcb.200412032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P, Smith A, McDowall A, Nicol A, Zicha D, Hogg N. Intermediate-affinity LFA-1 binds alpha-actinin-1 to control migration at the leading edge of the T cell. EMBO J. 2008;27:62–75. doi: 10.1038/sj.emboj.7601959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Buul JD, Allingham MJ, Samson T, Meller J, Boulter E, Garcia-Mata R, Burridge K. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol. 2007;178:1279–1293. doi: 10.1083/jcb.200612053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov Y, Long A, McGrath S, Ni Eidhin D, Kelleher D. Crucial importance of PKC-beta(I) in LFA-1-mediated locomotion of activated T cells. Nat Immunol. 2001;2:508–514. doi: 10.1038/88700. [DOI] [PubMed] [Google Scholar]
- Woolf E, Grigorova I, Sagiv A, Grabovsky V, Feigelson SW, Shulman Z, Hartmann T, Sixt M, Cyster JG, Alon R. Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat Immunol. 2007;8:1076–1085. doi: 10.1038/ni1499. [DOI] [PubMed] [Google Scholar]
- Yang L, Froio RM, Sciuto T, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005;106:584–592. doi: 10.1182/blood-2004-12-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Carman CV, Kim M, Salas A, Shimaoka M, Springer TA. A small molecule agonist of an integrin, alphaLbeta2. J Biol Chem. 2006;281:37904–37912. doi: 10.1074/jbc.M606888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.