

Structure determination was successfully carried out using single Laue exposures from a group of lysozyme crystals. The Laue method may be a viable option for collection of one-shot-per-crystal data from microcrystals.
Keywords: Laue diffraction, microcrystallography, X-ray optics
Abstract
Crystal size is an important factor in determining the number of diffraction patterns which may be obtained from a protein crystal before severe radiation damage sets in. As crystal dimensions decrease this number is reduced, eventually falling to one, at which point a complete data set must be assembled using data from multiple crystals. When only a single exposure is to be collected from each crystal, the polychromatic Laue technique may be preferable to monochromatic methods owing to its simultaneous recording of a large number of fully recorded reflections per image. To assess the feasibility of solving structures using single Laue images from multiple crystals, data were collected using a ‘pink’ beam at the CHESS D1 station from groups of lysozyme crystals with dimensions of the order of 20–30 µm mounted on MicroMesh grids. Single-shot Laue data were used for structure determination by molecular replacement and correct solutions were obtained even when as few as five crystals were used.
1. Introduction
The determination of macromolecular structures by X-ray crystallography requires the recording of diffraction data that provide a good coverage of reciprocal space out to a limiting resolution determined by crystal quality. These data typically comprise a series of images obtained from a single cryocooled crystal by the monochromatic oscillation technique. In some cases, e.g. when a suitable cryoprotectant cannot be found, multiple room-temperature crystals are used; the full data set is constructed by combining short series of images from each crystal. In either case, each diffraction image is taken as the crystal is rotated (or oscillated) through a small angle (∼1°). Each spot on the image is recorded over an angular range whose width is typically a few tenths of a degree, dependent on the mosaicity of the crystal; the start of this range depends on the orientation of the crystal and the identity of the reflection. Hence, different reflections are excited at different times during the exposure. Moreover, many of them are only partially recorded on a single image and data from multiple images must be combined to obtain the corresponding full intensities. When the properties of the crystal and the X-ray source vary only slowly with time (i.e. are effectively constant over the course of an exposure), such data can be processed with excellent results (Drenth, 1999 ▶) and most of the >50 000 structures in the Protein Data Bank (Berman et al., 2003 ▶) were obtained using the monochromatic oscillation technique.
The useful lifetime of a crystal is limited by radiation damage, which causes increased mosaicity, decreased resolution, altered cell dimensions and the breakage of specific susceptible bonds, e.g. some disulfides (Nave & Garman, 2005 ▶). At room temperature, radiation damage depends on the absorbed X-ray dose and on the rate at which the dose is received (Southworth-Davies et al., 2007 ▶). When the temperature is lowered to ∼100 K, the dependence on dose rate becomes negligible and damage (as measured by the increase in overall B factor) is simply proportional to dose (Kmetko et al., 2006 ▶). When the dimensions of a crystal are reduced, the intensity of diffraction can be maintained by increasing the X-ray flux density striking the crystal; however, this also increases the dose, resulting in a decreased lifetime for smaller crystals (Holton, 2009 ▶). For sufficiently small crystals, the useful lifetime is of the order of the exposure time required to obtain a single diffraction pattern. Cryocooled protein crystals with dimensions of 1–10 µm are predicted to fall into this category (Holton, 2009 ▶). A few structures have been obtained from crystals in the 5 µm size range using multiple monochromatic oscillation exposures per crystal (Standfuss et al., 2007 ▶; Coulibaly et al., 2007 ▶), but severe radiation damage was observed.
Microcrystals are commonly obtained in initial screening trials (Luft et al., 2007 ▶) and increasing their size can sometimes be difficult. The microfluidics-based devices that are being developed for large-scale automated combinatorial studies of crystallization conditions use smaller and smaller volumes of protein, limiting the size of crystals that may be grown in them (Shim et al., 2007 ▶). A few X-ray beamlines exist, and more are being developed, with sufficiently powerful <10 µm diameter beams to produce observable diffraction from <10 µm crystals (Moukhametzianov et al., 2008 ▶; Sanishvili et al., 2008 ▶; Bilderback et al., 2006 ▶; Dierker, 2008 ▶). Hence, it appears that the single-shot-per-crystal regime is of more than theoretical interest. Single oscillation images can be indexed and integrated and partial reflections scaled up to give full intensities, provided that adequate estimates of crystal orientation and mosaicity can be made (Rossmann & van Beek, 1999 ▶; Diprose et al., 2001 ▶). However, there is another possibility for single-shot data collection.
The Laue technique, utilizing a wide-bandwidth X-ray beam and a stationary crystal, is an alternative to the monochromatic method; in fact, it was invented first. Laue experiments on protein crystals were first performed at synchrotron sources over 20 years ago (Moffat et al., 1984 ▶) and diffraction was obtained from crystals as small as ∼20 µm shortly thereafter (Hedman et al., 1985 ▶). When the first condensing monocapillary X-ray microbeams were created, they were used to produce Laue diffraction images (Bilderback et al., 1994 ▶). The Laue method is capable of solving protein structures using a series of images taken from one or a few crystals (Helliwell et al., 1989 ▶; Ren & Moffat, 1995 ▶), but it is not commonly used owing to the greater ease of processing monochromatic data.
Most modern Laue experiments deal with time-resolved changes in known structures (Bourgeois et al., 2007 ▶). For time-resolved studies, there are distinct advantages to the distinguishing characteristics of the Laue technique: the crystal need not be rotated during an exposure, there are no partial reflections and there are more spots per image than for a typical monochromatic image (Moffat, 1997 ▶). With a stationary crystal, all reflections are recorded simultaneously and very short exposures can be recorded without placing unrealistic demands on a rotation stage; hence, high time resolution is possible. The large number of fully recorded reflections per image allows the collection of complete data from a relatively small number of images. The same Laue characteristics offer advantages in the collection of single-shot data: simultaneous recording of all reflections means that they all arise from a crystal experiencing the same dose of radiation, the lack of partials removes a source of error in processing single images and the large number of diffraction spots per image reduces the number of crystals required.
The effectiveness of the Laue technique in time-resolved experiments has been demonstrated. However, these studies have examined changes to systems whose initial structures are very well known, a somewhat different case from the determination of an unknown structure from a collection of microcrystals. As a first step in applying the Laue method to the latter task, we evaluated the success of the method in determining a known structure from crystals in the 20–30 µm range.
2. Choosing the X-ray spectrum
Laue data have been obtained using a broad range of X-ray sources, including the Bremsstrahlung radiation of an X-ray tube (Rabinovich & Lourie, 1987 ▶), the full white beam from a synchrotron bending magnet (Ren & Moffat, 1995 ▶) or wiggler (Helliwell et al., 1989 ▶), the output from wide-bandpass multilayer optics (Kazimirov, 2005 ▶) and the highly peaked spectrum from an undulator (Key et al., 2007 ▶). Wider bandwidth sources produce more spots per image, with a concomitant increase in both spatially overlapped spots and spots containing contributions from multiple orders of diffraction (harmonic overlaps). Many of the overlaps of both types can be deconvoluted (Campbell, 1995 ▶; Arzt et al., 1999 ▶), but the process introduces error. Widening the bandwidth also increases the level of background scatter. A smooth relatively broad spectrum, as opposed to a ‘peaky’ one, has the advantage of being easier to model with Chebyshev polynomials and introduces less noise arising from inaccurate assignment of wavelength to individual reflections.
As a reasonable compromise between increasing the data per image and reducing overlaps and background, we chose to use an X-ray beam with a 30% bandwidth, from 9.5 keV (λ = 1.30 Å) to 13.5 keV (λ = 0.92 Å), with the peak at 12 keV (λ = 1.03 Å). This spectrum avoids the low-energy range where absorption, e.g. by the sample mount, could become significant, as well as higher energies at which the CCD detector has reduced sensitivity (Ren et al., 1999 ▶). Experiments were carried out at the CHESS D-line (at the 5.2 GeV Cornell storage ring), where a hard bend dipole magnet produces a spectrum containing energies from a few keV to beyond 50 keV. Traditionally, a restricted bandwidth from such a source has been selected using either a wide-bandwidth multilayer or a reflection mirror plus absorption filter combination. The desired 30% bandwidth is too large for multilayers, whose upper limit is presently ΔE/E ≃ 10% (Kazimirov et al., 2006 ▶). Total external reflection mirrors can provide a relatively sharp high-energy cutoff, which can be tuned over a range of about 10–20 keV by changing the angle of the mirrors in the incident beam (Fig. 1 ▶); such mirrors are commonly used for suppression of higher harmonics. For an absorption filter, a material with no absorption edges in the desired region of the spectrum is required; aluminium would be suitable in the present case. However, using an Al absorption filter to remove low-energy radiation has a major drawback in that the energy cutoff is very broad, of the order of several keV in the 5–20 keV range.
Figure 1.

Diagram of the setup for the reflection and transmission mirrors used to create a tunable large-bandwidth beam.
In order to achieve a spectrum with a sharper low-energy cutoff, we chose a third option (Figs. 1 ▶ and 2 ▶): a reflection mirror and transmission mirror combination (Lairson & Bilderback, 1982 ▶). Like a reflection mirror, a transmission mirror has a sharp energy cutoff, but it is a high-pass filter rather than a low-pass filter (Cornaby & Bilderback, 2008 ▶). The low-energy cutoff can be tuned over a range of about 8–12 keV by changing the angle of the mirror with respect to the X-ray beam (Fig. 1 ▶). With this new combination of optics, the desired 30% bandwidth was readily achieved.
Figure 2.

Predicted and actual spectrum of the 30% bandwidth (FWHM) X-ray beam. Red, calculated reflection from a Rh-coated reflection mirror at 0.28° (4.9 mrad). Blue, calculated transmission through an Si3N4 transmission mirror at 0.24° (4.2 mrad). Magenta, calculated spectrum produced by two reflections and one transmission. Black, spectrum measured using Compton scattering from a Kapton foil. The sharp peaks arise from fluorescent scattering from impurities in the foil and are not present in the incident X-ray beam. Green, spectrum determined from Laue data modeled with 15 Chebyshev polynomials. The heights of the black and green curves have been scaled to match the magenta curve.
The first transmission X-ray mirror was created more than 20 years ago from a soap-bubble film and was used for Laue experiments (Bilderback et al., 1984 ▶). Such mirrors had a very limited lifetime of a few hours, making for nonstable and nonrepeatable X-ray optics; hence, they have not been used beyond the proof-of-principle demonstration. A 0.5 µm Mylar film stretched on a frame was also tried (Iida et al., 1985 ▶), but it too had a short lifetime and produced an inhomogeneous beam owing to non-uniformity of the film thickness. For the current experiment, we designed and constructed a novel X-ray transmission mirror from a 300 nm thick silicon nitride membrane using the Cornell NanoScale Science and Technology Facility (CNF; http://www.cnf.cornell.edu). Although fragile, mirrors made with the new design were stable over a period of weeks to months. Mirrors exposed to white beam at the CHESS B1 station for 24 h continuously and to pink beam at D1 intermittently for two weeks showed no sign of degradation of their optical properties.
Two reflection mirrors and one transmission mirror were installed in the D1 optics enclosure and the resulting X-ray spectrum was characterized using an energy-resolving X-Flash detector to record the Compton scattering from a strip of Kapton tape placed in the beam just upstream of the capillary used to focus the X-rays onto the samples. The bandwidth was readily tuned by adjusting the angles of the mirrors. The spectrum used for the Laue experiment is shown in Fig. 2 ▶. The lambda curve (spectrum) determined in the course of processing Laue images recorded from crystals using the focused beam corresponded closely to the Compton spectrum. The spectrum was peaked at 12 keV (λ = 1.03 Å), with half-height values of 9.6 keV (λ = 1.29 Å) and 13.4 keV (λ = 0.93 Å).
Hence, the reflection/transmission mirror combination was successful in setting the desired bandwidth of the beam, although the low-energy cutoff was not quite as sharp as predicted. Measurements taken previously on a similar transmission mirror at the B1 station showed the same low-energy tail. Calculations indicated that this was the consequence of a deviation from flatness of the mirror surface of ∼120 µrad (Cornaby & Bilderback, 2008 ▶). The spectrum measured at D1 was very similar to that from the B1 experiment, indicating good reproducibility in fabrication of the mirrors. No instabilities were observed in the optics over the course of the week of Laue experiments.
3. Focusing the beam
The X-ray beam was focused to a size comparable to that of the crystals used (∼20 µm) in order to maximize signal to noise in the diffraction images. A single-bounce monocapillary was used; such capillaries are achromatic and so are able to focus over the entire spectral range of the incoming X-rays (Huang & Bilderback, 2006 ▶; Cornaby, 2008 ▶). The capillary (designated f1b-mr9f20-01) was a 7.5 mrad optic with a 22 mm focal length mounted in a housing compatible with the protein crystallography goniostat used for the experiment. Slits upstream of the capillary could be used to adjust the divergence of the output beam from ∼2 to 7.5 mrad and a small upstream beamstop could be used to block the straight-through unfocused beam (Cornaby, 2008 ▶; Lamb et al., 2007 ▶).
A highly focused X-ray beam maximizes the flux density at the crystal but also produces relatively divergent diffracted rays. Lowering the divergence limits the flux but also reduces the number of spots which cannot be integrated owing to excessive overlap or overly elongated shapes. Fig. 3 ▶ (overlapping spots) and Fig. 4 ▶ (isolated spots) show examples of Laue patterns obtained using different divergence settings of the capillary–slits combination. The shapes of the diffraction spots in the Laue images differed from simple ellipses because of the beam’s angular profile (revealed by the far-field pattern used to align the optics; see Fig. 5 ▶) and divergence, which was significantly higher than that produced by a typical collimator. The shapes of diffraction spots per se are not critical; oddly-shaped spots are acceptable for intensity integration as long as they have reasonably consistent spot shapes within a limited area of the image. However, overly elongated spots produce a large number of spatial overlaps whose presence degrades data quality.
Figure 3.

Laue diffraction image taken from a small lysozyme crystal, with magnified views of two small regions of the image. The divergence from the capillary was too large, resulting in many overlapping diffraction spots. The odd shapes seen in these particular spots represent approximately half of the full far-field pattern (Fig. 5 ▶), with a small portion of the direct beam (owing to pre-capillary beamstop misalignment). If the full divergence from the capillary were used, the diffraction spots would have elliptical shapes.
Figure 4.

Laue diffraction from a lysozyme crystal. Left: crystal on a MicroMesh mount. The circle around the crystal is ∼100 µm in diameter; the crystal is about 30 µm across. Right: diffraction pattern from the crystal with a 10 s exposure time. The inset shows well separated acceptably shaped spots.
Figure 5.

Images of the capillary-focused beam observed on a fluorescent screen attached to a small video camera located 300–400 mm downstream of the capillary, i.e. well beyond the sample position. These images reveal the angular profile of the beam passing through the ∼13 µm focal spot. (a) Full far-field image with no upstream stop for the central beam. The central spot is from the direct beam passing straight through the capillary; the outer ring is from X-rays that have been focused by a single reflection off the inner wall of the capillary. (b) Slit-down far-field image of a beam suitable for collecting Laue images with well separated spots. The grids in the images have 1 × 1 mm squares.
Without slitting down the beam entering the monocapillary, i.e. with a full 7.5 mrad (0.43°) divergence in both the horizontal (H) and vertical (V) directions, the total flux was ∼1.9 × 1011 photons s−1 in a 16 (H) × 10 (V) µm2 spot (∼1 × 109 photons s−1 µm−2). In order to reduce the overlap of diffraction spots, slits were used to reduce the divergence of the beam to ∼5 (H) × 2 (V) mrad2 [0.3° (H) × 0.12° (V)] (Fig. 5 ▶). The resulting total flux was 4.4 × 1010 photons s−1 in a 10 (H) × 13 (V) µm2 spot (∼3.4 × 108 photons s−1 µm−2). This beam was used to collect the Laue images used for processing; an example is shown in Fig. 4 ▶.
4. Collection of Laue data
Hen (chicken) egg-white lysozyme (HEWL; Hampton Research HR7-108) was dissolved to 100 mg ml−1 in 20 mM sodium acetate pH 4.6. Sitting-drop trials were set up over 1 ml of a reservoir solution consisting of 100 mM sodium acetate pH 4.8, 0.625–1.0 M sodium chloride and 25% ethylene glycol. 4 µl protein solution was mixed with 4 µl reservoir solution for each trial. An abundance of small cryo-ready crystals grew overnight.
Laue data were collected from a total of 52 lysozyme crystals in 13 groups (‘spreads’). Groups of crystals were scooped up with MicroMesh (Mitegen) mounts and positioned on a single-axis goniometer. They were flash-cooled in the cold stream and maintained at 100 K using an Oxford Cryostream device. For one group of nine crystals, a custom helium-filled enclosure surrounded the capillary, beamstop, crystals on their mount and cryostream nozzle; the enclosure was flushed and maintained at a slight positive pressure of He by using helium at 100 K in the cryostream in place of the usual nitrogen. Crystal dimensions ranged from ∼20 to ∼120 µm. Data were recorded from one crystal at a time using an ADSC Quantum-4 CCD detector with an exposure time of 1–15 s. For each crystal, a series of still images was recorded, with a ϕ (spindle) rotation of typically 10° between images (Fig. 4 ▶).
5. Processing of Laue data
5.1. General procedure
The goal of the experiment was to evaluate single-shot-per-crystal data, but various other data sets were also processed for comparison. Images were evaluated visually (eliminating extremely weak patterns and those with very elongated spots owing to high mosaicity) and by preliminary processing; several sets of images, all with 10 s exposure times, were selected for further processing. Owing to radiation damage, only the first 4–5 frames in each set were useful, i.e. the survival time of a crystal in the beam was about 45 s. Frames taken after this point had a visible increase in the streakiness of the spots and scaled poorly with earlier frames. The wavelength range was set to 0.84–1.58 Å (7.8–14.8 keV) based on the points at which the measured Compton scattering curve fell to background levels.
The unit-cell parameters of HEWL previously determined from a monochromatic experiment were used initially: space group P43212, unit-cell parameters a = b = 78.56, c = 36.98 Å. Refinement of the c unit-cell parameter based on the Laue data gave a final value of 36.61 Å. Data were indexed, refined and integrated to 2.0 Å resolution with the program LAUEGEN (Campbell, 1995 ▶) and scaled with LSCALE (Arzt et al., 1999 ▶), both from the Daresbury Laue processing suite, with local modifications by D. M. Szebenyi. Integration was by profile-fitting, using a circular spot area with a diameter of 1.0 mm and a minimum spot separation for non-overlapped spots of 1.0 mm. Spatially overlapped spots were deconvoluted by LAUEGEN if their centers were at least 0.25 mm apart; closer spots were rejected. Multiple orders of reflections were deconvoluted by LSCALE when possible, i.e. when sufficient symmetry-related reflections were available to solve the equations involved. Scale factors and relative B factors for each frame were refined. The X-ray spectrum was modeled with 15 Chebyshev polynomials, whose coefficients were refined simultaneously with the per-frame parameters. At the scaling stage, outlier observations (with |I(j) − I mean|/I mean > 1.0) were excluded, only observations with I/σ(I) > 2 were used for scaling and only observations with I/σ(I) > 3 were written to the output file. For one set only (‘best’, see below), the wavelength range of observations used for scaling and merging was restricted to 0.92–1.25 Å, omitting weak data from the tails of the spectral peak, and just ten Chebyshev polynomials were used.
Scaled but unmerged data were written in MTZ format by LSCALE. Subsequent processing used programs in the CCP4 suite v.6.1.0 (Collaborative Computational Project, Number 4, 1994 ▶) via the CCP4i graphical interface (Potterton et al., 2003 ▶). Data were merged, with unit scale factors, by SCALA (Evans, 2006 ▶) and converted to Fs with TRUNCATE. 5% of the reflections were reserved for calculation of a free R factor; the same set of reflections was used for all data sets. Molecular replacement was carried out using MOLREP (Vagin & Teplyakov, 1997 ▶), with hexagonal turkey lysozyme (PDB code 1tew) as the (similar but non-identical) model. Following one cycle of rigid-body refinement and one cycle of restrained coordinate and B-factor refinement using REFMAC5 (Murshudov et al., 1997 ▶), Fourier maps were calculated and used to evaluate the quality of the solution. Coot (Emsley & Cowtan, 2004 ▶) was used with these maps to mutate the seven residues which differed between turkey and chicken lysozyme and one more round of restrained refinement was carried out. A monochromatic HEWL data set was subjected to the same molecular-replacement procedure for comparison. This data set comprised 60 1° oscillation images collected at the CHESS F2 station on an ADSC Q-210 detector from a single cryocooled crystal of approximately 200 µm dimensions. There was no evidence of radiation damage occurring during the course of collection of the monochromatic data.
5.2. Data sets used
Four different sets of Laue data were analyzed.
(i) A ‘best’ set comprising the first 3–7 frames recorded from each of seven crystals from a single ‘spread’ (i.e. a group of crystals mounted on the same MicroMesh mount). In scaling this set, reflections in the tails of the wavelength range, i.e. below 0.92 Å or above 1.25 Å, were excluded; these are on average weaker than other reflections and also suffer more from any inaccuracy in wavelength determination (owing to the steepness of the lambda curve in these regions). Because of the large number of frames used (36 in total), the data had high completeness and multiplicity even after the exclusions.
(ii) A ‘firsts’ set comprising the initial images from 18 different crystals (from several different spreads), i.e. a single-shot data set.
(iii) A ‘small firsts’ set including only the initial images from five crystals, all from the same spread, as an example of single-shot data that could be obtained if only a small number of crystals were available.
(iv) A ‘single-crystal’ set comprising five frames from a single crystal; the completeness was about the same as for ‘small firsts’ but inter-frame discrepancies reflected radiation damage rather than inter-crystal variability.
Two monochromatic data sets were used for comparison.
(i) A ‘complete’ set comprising all 60 frames from a single crystal.
(ii) A ‘small’ set comprising the first 22 frames of ‘complete’, with approximately the same completeness and multiplicity as the Laue sets ‘small firsts’ and ‘single-crystal’.
Statistics for the six data sets are given in Table 1 ▶. Molecular replacement gave a single clear solution in every case and refinement improved the initial model. Refinements were preliminary only: no solvent was added, no manual adjustments were made to residues other than the seven mutations and default values were used for refinement parameters such as X-ray versus geometry weighting.
Table 1. Summary of 2.0 Å lysozyme data sets.
Values in parentheses are for lowest resolution (∞–6.32 Å) and highest resolution (2.11–2.00 Å) bins. The actual low-resolution limit of the data was 15–20 Å.
| Name | Laue best | Laue firsts | Laue small firsts | Laue single-crystal | Mono complete | Mono small |
|---|---|---|---|---|---|---|
| Crystals/frames | 7/36 | 18/18 | 5/5 | 1/5 | 1/60 | 1/22 |
| Completeness (%) | 89.6 (83.3, 74.9) | 90.2 (86.9, 81.3) | 70.4 (61.3, 49.1) | 70.9 (52.7, 50.8) | 98.3 (89.2, 99.4) | 70.7 (54.0, 71.4) |
| Multiplicity | 7.7 (4.5, 4.1) | 5.5 (4.0, 3.0) | 2.2 (1.8, 1.6) | 2.3 (1.9, 1.6) | 4.5 (3.5, 4.6) | 2.3 (2.3, 2.3) |
| Rmerge† | 0.174 (0.098, 0.300) | 0.200 (0.150, 0.217) | 0.144 (0.125, 0.151) | 0.127 (0.069, 0.206) | 0.052 (0.046, 0.171) | 0.049 (0.032, 0.118) |
| Rp.i.m.‡ | 0.057 (0.048, 0.139) | 0.072 (0.074, 0.112) | 0.093 (0.099, 0.119) | 0.080 (0.051, 0.157) | 0.027 (0.024, 0.083) | 0.036 (0.023, 0.083) |
| MOLREP score | 0.524 | 0.528 | 0.485 | 0.494 | 0.551 | 0.494 |
| R after rigid-body refinement | 0.392 | 0.394 | 0.390 | 0.384 | 0.399 | 0.394 |
| R/Rfree after all refinement | 0.266/0.318 | 0.259/0.319 | 0.256/0.331 | 0.264/0.320 | 0.241/0.265 | 0.228/0.266 |
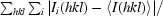

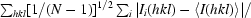
The utility of a molecular-replacement solution depends on how well it can reveal features that are not present in the starting model. Hence, we calculated difference Fourier maps from the six data sets after one cycle of rigid-body refinement and one cycle of restrained positional and B-factor refinement and examined the seven residues which differed between the model (turkey lysozyme) and the true structure (chicken lysozyme). Fig. 6 ▶ shows the result for residue 41, which is histidine in turkey and glutamine in chicken lysozyme. Other residues are shown in Supplementary Figs. 1–3.1
Figure 6.

Difference Fourier maps around residue 41, which is His in turkey lysozyme and Gln in chicken lysozyme. The contour level is 3σ, with green for positive and red for negative peaks. The data sets used were (a) monochromatic ‘complete’, (b) Laue ‘best’, (c) Laue ‘small firsts’, (d) monochromatic ‘small’, (e) Laue ‘firsts’, (f) Laue ‘single-crystal’; see the text for the definitions of these designations. The structure of turkey lysozyme after refinement but before mutations is shown as multicolored sticks; the corresponding part of chicken lysozyme from PDB entry 1bwh is shown as thin blue sticks. Maps and model were displayed using Coot.
5.3. Evaluation of data
Based on the refinement statistics and Fourier maps, the ‘best’ Laue data set was clearly adequate for structure determination, although not quite up to the standard of the monochromatic reference set. The relatively high R merge was primarily a consequence of (i) the necessity of combining data from multiple crystals and (ii) the asymmetry and variation across the image of spot shapes. The high multiplicity was able to produce good merged intensities in spite of these difficulties. The overall completeness of 90% was satisfactory. Laue data often suffer from incompleteness at low resolution owing to (i) the closeness of the Ewald spheres for the minimum and maximum λ values at low resolution and (ii) the fact that many spots recording low-index reflections contain multiple harmonics, which can not always be successfully deconvoluted. In the present experiment, using a 30% X-ray spectral bandwidth, the ‘best’ set was 83% complete in the low-resolution (∞–6.3 Å) range, which is an acceptable value, although certainly less than the 96% completeness observed in the most populous mid-resolution (3.6–3.2 Å) bin. At high resolution, a gradual fall-off in completeness was observed (Fig. 7 ▶). This was the consequence of the rejection of reflections which were either (i) poorly measured because they were weak and located in the outer regions of the image where spot shapes were more elongated or (ii) produced by longer wavelength X-rays and outside the active area of the detector. In spite of this effect, completeness was over 90% in the range from about 5 to 2.25 Å. The 5% difference between R and R free after refinement was a little high, perhaps indicating a need to adjust refinement parameters such as the relative weighting of X-ray and geometrical terms.
Figure 7.

Completeness (blue) and R p.i.m. (red) as a function of resolution for the six data sets described in the text. ×s, monochromatic ‘complete’; crosses, monochromatic ‘small’; open circles, Laue ‘best’; filled circles, Laue ‘firsts’; open squares, Laue ‘single-crystal’; filled squares, Laue ‘small firsts’.
The single-shot-per-crystal ‘firsts’ set, judging from Fourier maps and R/R free values, was of comparable quality to the ‘best’ set, i.e. it could be used to produce a correct structure. A plot of R p.i.m. versus resolution (Fig. 7 ▶) hinted that the high-resolution data in ‘firsts’ were of better quality than those in ‘bests’, which is consistent with less radiation damage in the single-shot set. The somewhat higher overall R p.i.m. was a consequence of (i) the use of data from more crystals and (ii) the inclusion of reflections at the tails of the X-ray spectrum, which are generally weaker than those in the middle of the range. Inclusion of these reflections appeared to be beneficial overall, as omitting them from ‘firsts’ decreased the completeness of the data, increased the overall R p.i.m. and raised the R free after refinement (data not shown).
The ‘small firsts’ set was clearly of poorer quality than the ‘best’ and ‘firsts’ sets, largely owing to its lower completeness and multiplicity. The monochromatic ‘small’ set, with similar completeness and multiplicity, showed a similar deficiency in difference Fourier quality relative to the more complete monochromatic set. Nevertheless, even this five-crystal single-shot Laue set was adequate to produce a molecular-replacement solution which refined sensibly and showed some evidence for differences between the model and the true structure. The Laue ‘single-crystal’ data set was of roughly the same size as the ‘small firsts’ set but was derived from a single crystal. It exhibited somewhat better statistics and maps, which is consistent with the expectation that a group of cryocooled crystals, even within a single spread, would vary in properties such as unit-cell parameters (Murray & Garman, 2002 ▶). As with the larger sets, the single-shot set appeared slightly better at high resolution than the multi-shot set (Fig. 7 ▶).
All the crystals used in the Laue experiment came from the same batch of protein, but those used for the ‘best’, ‘small firsts’ and ‘single-crystal’ sets also came from a single crystallization drop, while those in ‘firsts’ came from several different drops. We attempted to evaluate the variation in unit-cell parameters within and between drops. Determination of accurate absolute values of unit-cell parameters from Laue data requires special measures such as a filter which produces a sharp edge in the X-ray spectrum (Carr et al., 1993 ▶) or an energy-resolving detector (Hanley et al., 1997 ▶), owing to uncertainty in the exact X-ray wavelength producing each spot in ‘simple’ data (Ravelli et al., 1996 ▶). However, ratios between unit-cell parameters can routinely be refined. In the present case, refinement (by LAUEGEN) of the a/c ratio for different crystals gave similar results for most crystals, with the exception of those mounted in a helium enclosure (see below), which gave a value about 0.5% lower. Merging statistics were little affected by whether the He-box data were included or not, indicating that the difference in a/c was either insignificant or an artifact of the different backgrounds on images taken in He and in air. Overall, variation between crystals and drops did not appear to cause significant problems; note that the multiplicity-independent merging statistic R p.i.m. (Diederichs & Karplus, 1997 ▶) is actually better for ‘firsts’ (18 crystals) than for ‘small firsts’ (five crystals) or ‘single-crystal’ (one crystal).
5.4. Effects of experimental details
For weak diffraction signals, such as those from microcrystals, background scatter by air in the path of the direct beam can potentially reduce the signal-to-noise ratio. To evaluate the effect of a lower background, a helium-filled enclosure around the crystals was used for one spread. The helium was effective in reducing background at low to medium scattering angles, as is clearly visible on the diffraction images (Supplementary Fig. 4 ▶ 1); however, there was no apparent effect on the quality of the resultant Laue data and we conclude that air scatter was not a significant factor in this experiment.
Crystals from different locations on the MicroMesh mount were used: some on the mesh area, some on the solid part of the mount and some in the keyhole region where there is no mount and the crystal is supported solely by solidified crystallization solution. No effect of location on data quality was observed.
A few crystals were examined at room temperature and produced only very streaky Laue patterns indicative of high mosaic spread. This is an area for future investigation: to determine whether careful crystal handling and rapid data collection can result in good room-temperature data. If so, the one-shot Laue technique could potentially be applied to microcrystals for which cryocooling has not been successful.
6. Limitations and future work
Crystals exhibited a wide range in diffraction quality. Only about 35% of potential data sets were actually useful. For example, in a spread of 20 crystals three were too weak, six were too mosaic, four were oriented such that indexing was not successful and seven were good. Optimization of cryoconditions and crystal-handling techniques could potentially reduce the number of high-mosaicity crystals and would clearly be worthwhile. There is also room for improvement in the LAUEGEN indexing procedure, perhaps by allowing it to use multiple frames for indexing.
The crystals used in this work were 20–30 µm in dimensions, but the anticipated need for single-shot structure solution is for microcrystals with dimensions of ∼1 µm. In order to minimize background scatter from a crystal’s surroundings, it is desirable to match the X-ray beam size to the crystal size. Collecting good data from a 1 µm crystal with a 1 µm beam is not a trivial task. Using a combination of slits, apertures and Kirkpatrick–Baez (K-B) mirrors with an undulator source a polychromatic beam as small as 95 × 80 nm has been produced (Liu et al., 2005 ▶). With similar optics, protein structures have been obtained using monochromatic data from beams and crystals in the 5 µm range (Coulibaly et al., 2007 ▶; Sanishvili et al., 2008 ▶) and from a 1 µm beam used to select a small volume of a larger crystal (Moukhametzianov et al., 2008 ▶). Laue data could perhaps be collected using a similar combination of undulator source, apertures and K-B mirrors (which are achromatic).
The beam from such a source would have a considerably narrower bandpass than the 30% used in the present experiment. For the lysozyme crystals used, the Laue patterns contained approximately 5000 spots on a typical image, of which approximately 200 were multiples, 100 were too close to integrate and the remainder were processable singles (roughly 3400 of which were non-overlapped and 1300 were overlapped but could be deconvoluted). The overall I/σ(I) calculated by LAUEGEN was in the range 5–8. Reducing the bandwidth would have the advantage of reducing the background and hence increasing the signal-to-noise ratio at the expense of reducing the number of spots per image. Experimentation will be required to determine the optimum bandwidth; moderate reduction from 30% can be obtained by adjusting the mirror angles in the current setup and smaller values can be obtained using multilayer optics or an undulator source.
A focusing capillary–slits combination similar to that used in the present experiment could also be used to produce a beam in the 1 µm size range. The relatively high divergence of the beam from a strongly focusing capillary presents a problem; in the work reported here much of the variation in spot shape across an image arose from the properties of the capillary-focused beam. A capillary with a lower divergence and a longer focal length is one possible way to address the problem (capillaries with different properties are readily produced as needed by the Bilderback group at CHESS), but other options such as toroidal mini-mirrors (Cornaby et al., 2008 ▶) are also being considered.
Additional considerations which will present challenges for future investigations include mounting techniques for crystals in the 1 µm range (is there a way to remove the MicroMesh-plus-solvent-film source of background scatter?), visualization of microcrystals for centering purposes (or would it be better to simply scan the sample mount, recording images on a fine grid and sorting out the good ones afterwards?), vibration of samples and heating effects in an extremely bright X-ray beam.
Structure solution for crystals of known unit cell and symmetry using the molecular-replacement technique was successful. For a new protein form with unknown cell and symmetry it would be necessary to collect some monochromatic data to determine these quantities. Luckily, only a few frames would be needed to make this determination and radiation damage during their collection would not matter; only spot positions are needed to determine unit-cell parameters. Structure determination using isomorphous replacement should be possible but needs to be tried. A typical MAD (multiple-wavelength anomalous diffraction) experiment (using data collected at wavelengths on and near an absorption edge) is probably not feasible with Laue data owing to the small number of reflections excited by any particular wavelength and inaccuracies in determining the wavelength for each reflection. However, SAD (single-wavelength anomalous diffraction) is a possibility using only reflections produced by X-rays of energy comfortably above the relevant absorption edge, where there is little variation in f′′ with wavelength. With accurate wavelength calibration, even reflections recorded near an absorption edge might become useful. Such calibration could be provided by comparison of data collected with and without a filter in the X-ray beam, where the filter material exhibits a sharp absorption edge within the appropriate wavelength range. Further experiments will be needed in order to investigate these options.
7. Conclusions
We have demonstrated that structure solution is possible using single-shot-per-crystal Laue data. The Mitegen MicroMesh grid proved an excellent tool to mount a group of small crystals, cool them in a cryostream and position each crystal in turn in the X-ray beam. The choice of a 30% bandwidth beam peaked at 12 keV was appropriate for producing Laue data for structure solution (although it is not necessarily optimal in all cases) and the combination of reflection and transmission mirrors used, readily produced the desired spectrum. A single-bounce focusing monocapillary produced a beam at the crystal of ∼13 µm diameter, which was well matched to the crystal size and had a flux density from a bending-magnet line comparable to those from wiggler and undulator beamlines (J. Holton; http://bl831.als.lbl.gov/damage_rates.pdf).
Laue data were collected from multiple small (dimensions of ∼20 µm) crystals and used for structure determination by molecular replacement; a successful solution could be obtained using as few as five diffraction images. The quality of multi-crystal single-shot data sets was comparable to that of single-crystal or few-crystal multi-shot sets; crystal-to-crystal variation did not pose a serious problem. The Laue technique appears to hold potential for structure determination in the case where radiation damage precludes the collection of more than one exposure per crystal; further experiments will be needed in order to determine the extent of this potential.
Supplementary Material
Supplementary material file. DOI: 10.1107/S0907444909037731/gm5005sup1.pdf
Acknowledgments
This work is based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation and the National Institutes of Health/National Institute of General Medical Sciences under NSF award DMR-0225180, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by award RR-01646 from the National Institutes of Health through its National Center for Research Resources. We are very grateful to the staffs of CHESS and MacCHESS, particularly Bill Miller, Mike Cook and Scott Smith, for assistance in setting up the complex equipment needed to carry out the Laue experiment. We also wish to thank the anonymous reviewers of this paper for their very detailed and constructive comments, which led to a much improved final version.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: GM5005). Services for accessing this material are described at the back of the journal.
References
- Arzt, S., Campbell, J. W., Harding, M. M., Hao, Q. & Helliwell, J. R. (1999). J. Appl. Cryst.32, 554–562.
- Berman, H., Henrick, K. & Nakamura, H. (2003). Nature Struct. Mol. Biol.10, 980. [DOI] [PubMed]
- Bilderback, D. H., Hoffman, S. A. & Thiel, D. J. (1994). Science, 263, 201–203. [DOI] [PubMed]
- Bilderback, D. H., Moffat, K. & Szebenyi, D. M. E. (1984). Nucl. Instrum. Methods Phys. Res. A, 222, 245–251.
- Bilderback, D. H., Sinclair, C. & Gruner, S. M. (2006). Synchrotron Rad. News, 19(6), 30–35.
- Bourgeois, D., Schotte, F., Brunori, M. & Vallone, B. (2007). Photochem. Photobiol. Sci.6, 1047–1056. [DOI] [PubMed]
- Campbell, J. W. (1995). J. Appl. Cryst.28, 228–236.
- Carr, P. D., Dodd, I. M. & Harding, M. M. (1993). J. Appl. Cryst.26, 384–387.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Cornaby, S. (2008). Dissertation. Cornell University.
- Cornaby, S. & Bilderback, D. H. (2008). J. Synchrotron Rad.15, 371–373. [DOI] [PMC free article] [PubMed]
- Cornaby, S., Smilgies, D. M. & Bilderback, D. (2008). Adv. X-Ray Anal.52
- Coulibaly, F., Chiu, E., Ikeda, K., Gutmann, S., Haebel, P., Schulze-Briese, C., Mori, H. & Metcalf, P. (2007). Nature Struct. Mol. Biol.446, 97–101. [DOI] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Mol. Biol.4, 269–275. [DOI] [PubMed]
- Dierker, S. et al. (2008). NSLS-II Conceptual Design Report. Brookhaven National Laboratory, Upton, USA.
- Diprose, J., Burroughs, J., Sutton, G., Goldsmith, A., Gouet, P., Malby, R., Overton, I., Zientara, S., Mertens, P., Stuart, D. & Grimes, J. (2001). EMBO J.20, 7229–7239. [DOI] [PMC free article] [PubMed]
- Drenth, J. (1999). Principles of Protein X-ray Crystallography. New York: Springer.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Hanley, Q. S., Campbell, J. W. & Denton, M. B. (1997). J. Synchrotron Rad.4, 214–222. [DOI] [PubMed]
- Hedman, B., Hodgson, K. O., Helliwell, J. R., Liddington, R. & Papiz, M. Z. (1985). Proc. Natl Acad. Sci. USA, 82, 7604–7607. [DOI] [PMC free article] [PubMed]
- Helliwell, J. R., Habash, J., Cruickshank, D. W. J., Harding, M. M., Greenhough, T. J., Campbell, J. W., Clifton, I. J., Elder, M., Machin, P. A., Papiz, M. Z. & Zurek, S. (1989). J. Appl. Cryst.22, 483–497.
- Holton, J. M. (2009). J. Synchrotron Rad.16, 133–142.
- Huang, R. & Bilderback, D. H. (2006). J. Synchrotron Rad.13, 74–84. [DOI] [PubMed]
- Iida, A., Matsushita, T. & Gohshi, Y. (1985). Nucl. Instrum. Methods Phys. Res. A, 235, 597–602.
- Kazimirov, A. (2005). CHESS News Magazine 2005, pp. 24–27.
- Kazimirov, A., Smilgies, D.-M., Shen, Q., Xiao, X., Hao, Q., Fontes, E., Bilderback, D. H., Gruner, S. M., Platonov, Y. & Martynov, V. V. (2006). J. Synchrotron Rad.13, 204–210. [DOI] [PubMed]
- Key, J., Šrajer, V., Pahl, R. & Moffat, K. (2007). Biochemistry, 46, 4706–4715. [DOI] [PubMed]
- Kmetko, J., Husseini, N. S., Naides, M., Kalinin, Y. & Thorne, R. E. (2006). Acta Cryst. D62, 1030–1038. [DOI] [PubMed]
- Lairson, B. & Bilderback, D. (1982). Nucl. Instrum. Methods Phys. Res.195, 79–83.
- Lamb, J. S., Cornaby, S., Andresen, K., Kwok, L., Park, H. Y., Qiu, X., Smilgies, D.-M., Bilderback, D. H. & Pollack, L. (2007). J. Appl. Cryst.40, 193–195.
- Liu, W., Ice, G. E., Tischler, J. Z., Khounsary, A., Liu, C., Assoufid, L. & Macrander, A. T. (2005). Rev. Sci. Instrum.76, 113701.
- Luft, J., Wolfley, J., Said, M., Nagel, R., Lauricella, A., Smith, J., Thayer, M., Veatch, C., Snell, E., Malkowski, M. & DeTitta, G. (2007). Protein Sci.16, 715–722. [DOI] [PMC free article] [PubMed]
- Moffat, K. (1997). Methods Enzymol.277, 433–447. [DOI] [PubMed]
- Moffat, K., Szebenyi, D. & Bilderback, D. (1984). Science, 223, 1423–1425. [DOI] [PubMed]
- Moukhametzianov, R., Burghammer, M., Edwards, P. C., Petitdemange, S., Popov, D., Fransen, M., McMullan, G., Schertler, G. F. X. & Riekel, C. (2008). Acta Cryst. D64, 158–166. [DOI] [PMC free article] [PubMed]
- Murray, J. & Garman, E. (2002). J. Synchrotron Rad.9, 347–354. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nave, C. & Garman, E. F. (2005). J. Synchrotron Rad.12, 257–260. [DOI] [PubMed]
- Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. (2003). Acta Cryst. D59, 1131–1137. [DOI] [PubMed]
- Rabinovich, D. & Lourie, B. (1987). Acta Cryst. A43, 774–780.
- Ravelli, R. B. G., Hezemans, A. M. F., Krabbendam, H. & Kroon, J. (1996). J. Appl. Cryst.29, 270–278.
- Ren, Z., Bourgeois, D., Helliwell, J. R., Moffat, K., Šrajer, V. & Stoddard, B. L. (1999). J. Synchrotron Rad.6, 891–917.
- Ren, Z. & Moffat, K. (1995). J. Appl. Cryst.28, 461–481.
- Rossmann, M. G. & van Beek, C. G. (1999). Acta Cryst. D55, 1631–1640. [DOI] [PubMed]
- Sanishvili, R., Nagarajan, V., Yoder, D., Becker, M., Xu, S., Corcoran, S., Akey, D. L., Smith, J. L. & Fischetti, R. F. (2008). Acta Cryst. D64, 425–435. [DOI] [PMC free article] [PubMed]
- Shim, J., Cristobal, G., Link, D., Thorsen, T. & Fraden, S. (2007). Cryst. Growth Des.7, 2192–2194. [DOI] [PMC free article] [PubMed]
- Southworth-Davies, R. J., Medina, M. A., Carmichael, I. & Garman, E. F. (2007). Structure, 15, 1531–1541. [DOI] [PubMed]
- Standfuss, J., Xie, G., Edwards, P., Burghammer, M., Oprian, D. & Schertler, G. (2007). J. Mol. Biol.372, 1179–1188. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- Weiss, M. S. (2001). J. Appl. Cryst.34, 130–135.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material file. DOI: 10.1107/S0907444909037731/gm5005sup1.pdf
Supplementary material file. DOI: 10.1107/S0907444909037731/gm5005sup1.pdf