

Abstract
Molecular chaperones are typically either adenosine triphosphate (ATP) dependent or rely heavily on their ATP-dependent chaperone counterparts in order to promote protein folding. This presents a challenge to chaperones that are localized to ATP-deficient cellular compartments. Here we describe a mechanism by which the pH-regulated acid stress chaperone HdeA is capable of independently facilitating the refolding of acid-denatured proteins in the bacterial periplasm, which lacks both ATP and ATP-dependent chaperone machines. Our results are consistent with a model in which HdeA stably binds substrates at low pH, thereby preventing their irreversible aggregation. pH neutralization subsequently triggers the slow release of substrate proteins from HdeA, keeping the concentration of aggregation-sensitive intermediates below the threshold where they begin to aggregate. This provides a straightforward and ATP-independent mechanism that allows HdeA to facilitate protein refolding. Unlike previously characterized chaperones, HdeA appears to facilitate protein folding by using a single substrate binding-release cycle. This cycle is entirely regulated by the external environment and is therefore energy-neutral for the bacteria.
Keywords: ATP-independent chaperone, periplasm, protein folding, stress response
The acidic environment of the mammalian stomach (pH 1–3) plays an important role not only in the digestion of food, but also as a potent natural barrier against bacterial infections (1). Some microbes, such as Escherichia coli, have evolved systems to deal with the potentially lethal effects of acid stress. Response mechanisms that protect the cytosol of E. coli against acid stress include several amino acid decarboxylases which consume protons, thereby raising the intracellular pH (2). In contrast to the well-protected bacterial cytosol, however, the periplasm has a porous outer membrane. This allows the free diffusion of small (< 600 Da) molecules (3), making its contents vulnerable to external pH changes. Acid stress, like heat stress, exerts detrimental effects on organisms by inducing the unfolding and subsequent aggregation of cellular proteins.
E. coli and Shigella protect their periplasmic proteins against acid-induced aggregation by utilizing the pH-regulated chaperone HdeA. Deletion of hdeA renders the bacteria unable to survive extreme acid stress (4, 5). In vitro studies demonstrated that at neutral pH, HdeA is a well-folded dimer with no chaperone activity (4). Upon exposure to low pH, HdeA, like many other proteins, loses structure (6, 7). However, in contrast to most other proteins, which are inactivated upon pH-induced unfolding, partial unfolding and dissociation of HdeA into monomers causes the activation of its chaperone function (4, 7). We recently reported that low pH induces the exposure of structurally plastic, high-affinity binding sites for unfolding proteins on HdeA, and enables it to effectively prevent protein aggregation in vitro (8). The partially unfolded and therefore highly flexible architecture of active HdeA monomers seems to contribute to HdeA’s ability to adaptively bind to a wide range of different substrate proteins, some of which are much larger than the 9.7 kDa chaperone itself (8). Inactivation of HdeA is triggered by pH neutralization, which induces its refolding and dimerization (7).
While it is well established that HdeA binds substrates at low pH, thereby suppressing protein aggregation (4, 7,8–9), the fate of these substrates following pH neutralization and subsequent HdeA inactivation has not been elucidated. In vivo results suggested HdeA’s involvement in the refolding of periplasmic proteins following recovery from acid stress (10). In vitro studies using the model substrate alcohol dehydrogenase revealed, however, that pH neutralization triggered the release of alcohol dehydrogenase from HdeA in an aggregation-sensitive conformation, such that the substrate protein was unable to fold to its native state (7). Therefore, the fate of HdeA substrates, and particularly the question of whether HdeA is able to independently facilitate their refolding, remained unresolved. This is of particular interest because previously characterized ATP-independent chaperones can prevent substrate aggregation, but substrates typically then require downstream processing by ATP-dependent chaperones (11).
In addition to testing the role of HdeA in assisting the refolding of acid-denatured proteins, HdeA provides us with an opportunity to study how a chaperone has adapted to cope with the absence of ATP and ATP-dependent chaperone machines in the periplasm. To address these issues, we examined the fate of bound-substrate proteins upon shifting from low to neutral pH (i.e., upon returning to nonstress conditions). In this report, we demonstrate that HdeA can independently facilitate the refolding of acid-denatured protein substrates, including the physiological substrate alkaline phosphatase, to their enzymatically active state without assistance from ATP, ATP-dependent chaperones or cochaperones. We find that HdeA-substrate complexes spontaneously but slowly dissociate upon pH neutralization, and the substrates are released in a folding-competent state. To fulfill its function as a chaperone, HdeA appears to utilize the natural host physiology in which external changes in pH, rather than ATP, provides the energy to trigger chaperone activation, inactivation and substrate-protein refolding.
Results
HdeA Suppresses Substrate Aggregation at Low pH and Following pH Neutralization.
To examine the fate of substrate proteins upon pH neutralization and inactivation of HdeA, we studied the interaction of HdeA with the model substrate porcine mitochondrial malate dehydrogenase (MDH). Light-scattering measurements indicate that HdeA suppresses MDH aggregation at low pH (Fig. 1A), as previously reported (8). In the absence of HdeA, pH neutralization causes MDH to rapidly aggregate, as indicated by the increase in the light-scattering signal immediately after the pH shift (Fig. 1A, trace 1). In sharp contrast, in the presence of stoichiometric amounts of HdeA, pH neutralization results in a very small increase in the light-scattering signal, indicating a strong suppression of MDH aggregation by HdeA (Fig. 1A, traces 2–4). SDS-PAGE analysis of the insoluble pellet fraction (P) and the soluble supernatant fraction (S) following centrifugation of the samples supports the light-scattering results. In the presence of increasing HdeA concentrations, increasing amounts of MDH remained soluble during incubation at pH 2 (Fig. 1B) and following pH neutralization (Fig. 1C). The two most likely explanations for these observations are: (1) HdeA remains bound to MDH and continues to prevent aggregation even upon return to neutral pH; or (2) HdeA releases MDH in a less aggregation-prone conformation.
Fig. 1.

HdeA suppresses aggregation of MDH at low pH and following pH neutralization. (a) MDH (8 μM) was incubated in buffer A (pH 2) in the (1) absence or presence of (2) 4 μM, (3) 8 μM, or (4) 16 μM HdeA for 30 min before the pH was neutralized (indicated by the arrow). The gap represents the mixing time during which data were not acquired. Apparent increases in absorbance at 320 nm due to light scattering were recorded to monitor protein aggregation. (b and c) SDS-PAGE analysis of the soluble supernatant (S) and aggregated pellet (P) protein in each sample (b) after 30 min at pH 2 or (c) 30 min after pH neutralization at the indicated HdeA:MDH ratios.
While light-scattering measurements are a useful tool to monitor the relative amount of protein aggregation, they do not provide any information about the size, structure, or heterogeneity of the aggregated molecules. Nor can they distinguish whether or not the chaperone-substrate complex dissociates following pH neutralization. To gain structure and molecular mass information about the conformational states of HdeA during pH-induced activation and inactivation and upon association with MDH, we analyzed the populated species by sedimentation velocity analytical ultracentrifugation.
Ultracentrifugation data confirmed that HdeA forms ∼20 kDa dimers at neutral pH (Fig. S1A), dissociates into ∼9 kDa monomers upon incubation at pH 2 (Fig. S1B), and reassociates into ∼20 kDa dimers upon subsequent neutralization (Fig. S1C). The sedimentation of the HdeA monomer is not consistent with a completely unfolded polypeptide as has been proposed (7, 12), but rather is indicative of a fairly compact structure, consistent with our previous circular dichroism and fluorescence data suggesting that HdeA is only partially unfolded at low pH (8).
The behavior of MDH as monitored by sedimentation velocity agrees well with our light-scattering measurements. MDH forms a number of high molecular weight species (∼150–800 kDa) upon incubation at pH 2.0 (Fig. S1D). The presence of HdeA during incubation at low pH leads to the formation of smaller complexes (Fig. S1E). pH neutralization in the absence of HdeA induces MDH to form very large, heterogeneous particles ranging from ∼170 to 2200 kDa (Fig. S1F). Strikingly, neutralization of MDH solutions that had been incubated in the presence of HdeA at low pH produces two major species that sediment like native MDH and HdeA dimers (see Fig. S1G, and also Table S1 for a summary of ultracentrifugation data). This suggests that HdeA releases MDH upon pH neutralization, and in agreement with light-scattering measurements, the released MDH does not show a high aggregation propensity. Rather, it is in a soluble and possibly native form.
HdeA Supports Refolding of Acid-Unfolded MDH to the Native State.
Our ultracentrifugation experiments suggested that the presence of HdeA during low pH incubation not only prevents protein aggregation, but might also support the refolding of MDH upon neutralization and complex dissociation. This was a somewhat surprising result given that other ATP-independent chaperones, such as the small heat shock proteins, are capable of suppressing protein aggregation but unable to release their substrate proteins unless ATP-dependent chaperone systems are present to support substrate refolding (11). To test whether the presence of HdeA is indeed sufficient to support the refolding of acid-denatured MDH, we incubated MDH at pH 2 in the absence or presence of HdeA, neutralized the pH to initiate refolding, and assayed for MDH activity at regular intervals. As shown in Fig. 2A, in the absence of HdeA, acid-denatured MDH has a low propensity for spontaneous refolding under our experimental conditions, with < 5% activity recovered after ∼3 h. In contrast, the presence of HdeA during the low pH incubation period greatly enhanced the recovery of MDH activity. Nearly 50% of the original enzymatic activity was restored in the presence of a 2-fold excess of HdeA. In addition, we tested the periplasmic protein alkaline phosphatase (AP), which has the potential to interact with HdeA in vivo. AP also showed a low propensity for spontaneous refolding (< 10%), but refolded to > 50% in the presence of a 2-fold excess of HdeA (Fig 2B). Thus, HdeA appears to be well suited to protect proteins against pH-induced aggregation and to support their reactivation. In contrast, the presence of the same amount of BSA had minimal impact on MDH or AP refolding (Fig. 2, open squares). Strikingly, the homologous protein HdeB, which has been reported to have HdeA-like function (9, 10), was also completely incapable of assisting in MDH or AP refolding (Fig. 2, closed squares). This may be indicative of nonoverlapping substrate specificities between HdeA and HdeB.
Fig. 2.

HdeA facilitates the refolding of acid-denatured substrates to an enzymatically active state. (A) 1 μM MDH or (B) 3 μM AP were incubated in the absence or in the presence of increasing concentrations of HdeA in buffer A (pH 2) for 1 h at 37 °C prior to equilibration at 20 °C and pH neutralization. Aliquots were taken at time points following pH neutralization and assayed in triplicate for MDH or AP activity. Error bars are ± 1 s.d. The effect of a twofold molar excess of BSA (□) and HdeB (▪) on MDH and AP refolding were also tested.
To determine at what point HdeA acts in suppressing aggregation and facilitating substrate refolding, we conducted order-of-addition experiments in which we added HdeA either immediately before or various times after the start of the low pH incubation of MDH. HdeA was most effective both in aggregation suppression and assisting in MDH refolding when present from the beginning of the low pH incubation period (Fig. S2A, B). These observations underscore the importance of the rapid activation of HdeA for its optimal function (8). Binding of HdeA to MDH at low pH likely prevents the formation of off-pathway MDH intermediates or small aggregates, which rapidly form upon exposure to low pH, and have a strong tendency to aggregate rather than refold upon neutralization.
HdeA Releases Substrates in a Nonnative but Folding-Competent State.
Although our analytical ultracentrifugation experiments provided us with useful insights about the fate of HdeA-MDH complexes under equilibrium conditions, they do not provide us with any kinetic information. To investigate the rates of substrate binding and release, we used a fluorescence anisotropy-based approach. This technique monitors the relative rate of rotational diffusion of a fluorophore (13), and should allow us to distinguish between free and substrate-bound HdeA. To label HdeA, we made use of the previously characterized HdeA(S27C) variant, which is fully chaperone-active at pH 2 and can be modified with the pH-insensitive fluorophore bimane at Cys27 (8). As shown in Fig. 3, bimane-labeled HdeA(S27C) exhibits an anisotropy of ∼0.05 at pH 2. Upon addition of MDH (Fig. 3A) or AP (Fig. 3B), the anisotropy rapidly increased approximately 4-fold, reflecting the decreased rotational diffusion rate of labeled HdeA that accompanies binding of acid-unfolded substrate.
Fig. 3.

Substrate binding and release kinetics as monitored by fluorescence anisotropy. Anisotropy of 0.5 μM HdeA (S27C)-bimane at pH 2 in the absence or presence of 0.5 μM (A) MDH or (B) AP (added at t = 1 min) was monitored for 60 min at 37 °C. The temperature was then adjusted to 20 °C for 5 min, during which time data acquisition was paused. HdeA inactivation and substrate release were then triggered by pH neutralization (as indicated by the arrows). The traces corresponding to substrate release after neutralization were fit to double exponential functions; fit parameters: (A) A1 = 0.055, k1 = 0.30 min-1, A2 = 0.066, k2 = 0.020 min-1 and (B) A1 = 0.14, k1 = 4.3 min-1, A2 = 0.03, k2 = 0.29 min-1.
To test the kinetic stability of these complexes at pH 2, we added a large excess of unlabeled HdeA to preformed complexes of MDH and labeled HdeA. If the HdeA-MDH complexes are dynamic, the labeled HdeA should exchange with unlabeled HdeA, resulting in a decrease in anisotropy back to the starting value of ∼0.05. We observed very little change in anisotropy upon addition of a 10-fold excess of unlabeled HdeA over the course of the experiment (Fig. S2C). This suggests that HdeA-substrate complexes are kinetically very stable at low pH, with an estimated t1/2 of roughly 200 min. Thus, on a physiological timescale, HdeA may essentially irreversibly bind substrates at low pH.
To determine the kinetics of HdeA inactivation and substrate release, we tested the effects of pH neutralization on the anisotropy of HdeA(S27C) in the absence or presence of bound MDH. In the substrate-free state, the fluorescence anisotropy of bimane-labeled HdeA(S27C) rapidly jumped from 0.05 to 0.08 following pH neutralization (Fig. 3). As shown by bis-ANS fluorescence and FRET experiments, this increase in fluorescence anisotropy upon a shift to neutral pH occurs on a timescale that is similar to the rapid refolding of HdeA (Fig. S3A) and association into chaperone-inactive dimers (Fig. S3B). A fluorescence anisotropy signal of 0.08 was therefore considered to be the expected end point of our anisotropy measurements upon complete substrate-protein dissociation. pH neutralization of the HdeA-MDH (Fig. 3A) and HdeA-AP (Fig. 3B) complexes caused a decrease in anisotropy, which eventually approached the signal expected for substrate-free HdeA dimers. These results indicate that both substrates are being released from HdeA. AP is released via a rather rapid two-step process, with half times of ∼0.2 and 2 min (Fig. 3B). MDH is also released in an apparent two-step process with half times of 2 and 35 min (Fig. 3A), which is significantly slower than the inactivation of substrate-free HdeA, indicating that the presence of bound substrate can substantially decelerate the formation of the inactive HdeA dimer. The slower kinetic phase appears to correlate very well with the kinetics of MDH reactivation (t1/2 ∼ 35 min, Fig. 2A). While we can not say with certainty what causes these two phases, our analytical ultracentrifugation data (Fig. S1E) revealed that HdeA and MDH form multimeric complexes at low pH. It is possible that fast dissociation of these oligomers might underlie the first observed exponential phase, and the slower phase may be due to the complete dissociation of 1∶1 HdeA-MDH complexes.
The observation that the rate of MDH release from HdeA and the rate of MDH refolding are apparently identical, raised the intriguing possibility that MDH might actually refold while bound to HdeA. In this case, substrate release could simply be triggered by the refolding of the bound protein. A second possible explanation is that slow release of substrate proteins from HdeA might function to greatly reduce the concentration of free nonnative substrate at any given time, thereby suppressing aggregation and favoring rapid on-pathway refolding upon release. Alternatively, the slow kinetic phase may be a result of “iterative annealing” (14) or repetitive cycles of chaperone binding and release until MDH reaches the native state.
To distinguish between these possibilities, we used the GroEL/ES system as an in vitro controllable molecular trap for nonnative MDH molecules. GroEL, in the absence of GroES and ATP, has been shown to tightly bind substrates, including MDH, and to prevent them from folding; refolding of the trapped substrate molecules can then be initiated by addition of GroES and ATP (15, 16). To analyze whether the presence of GroEL affects the apparent rate of MDH dissociation from HdeA, we formed bimane-labeled HdeA(S27C)-MDH complexes at low pH. We then shifted the pH to neutral, and immediately added an equimolar amount of GroEL-14-mers. As shown in Fig. 4A, GroEL addition did not significantly impact the kinetics of MDH dissociation from HdeA. This result suggests that HdeA and MDH do not undergo iterative cycles of binding and release, because addition of GroEL would then be expected to compete with HdeA for rebinding and should, therefore, increase the apparent rate of anisotropy decay. To further exclude that HdeA and MDH undergo iterative cycles of release and rebinding, we performed similar experiments but added an excess of unlabeled HdeA following pH neutralization instead of GroEL (Fig. S4). Like GroEL, unlabeled HdeA did not significantly affect the rate of anisotropy decay, which is consistent with the conclusion that HdeA functions via a single binding and release event.
Fig. 4.

GroEL does not significantly impact the apparent rate of MDH release from HdeA but traps nonnative MDH following its release from HdeA. (A) 0.5 μM HdeA(S27C)-bimane was preincubated with 0.5 μM MDH for 1 h at pH 2, 37 °C, equilibrated to 20 °C for 5 min, and then neutralized, at which point data acquisition began. One sample was supplemented with 0.5 μM GroEL (14-mer) immediately following neutralization (+GroEL), and the other sample was not (-GroEL). The data were fit to double exponential functions (smooth lines) with fit parameters: A1 = 0.051, k1 = 0.31 min-1, A2 = 0.054, k2 = 0.025 min-1 (-GroEL), and A1 = 0.053, k1 = 0.35 min-1, A2 = 0.058, k2 = 0.034 min-1 (+GroEL). (B and C) Recovery of enzymatic activity of MDH that was incubated at low pH in the (B) absence or (c) presence of HdeA following pH neutralization. Duplicate samples were prepared at low pH, which were then allowed to refold after pH neutralization in the absence (□) or presence (▪) of GroEL. For samples containing GroEL, GroES and ATP were added where indicated by the arrows (to 0.5 μM and 5 mM, respectively) to test for possible refolding of MDH that had been trapped by GroEL.
Whereas GroEL had no effect on MDH release from HdeA, the recovery of MDH activity was substantially influenced by GroEL. We incubated duplicate samples of MDH either alone (Fig. 4B) or in the presence of HdeA (Fig. 4C) at pH 2 and supplemented one of each of the samples with GroEL immediately after neutralization. As expected, the presence of HdeA during the low pH incubation significantly increased the reactivation of MDH upon pH neutralization (Fig. 4B–C). The presence of GroEL during the neutral pH incubation, however, completely suppressed this reactivation (Fig. 4C, compare closed and open squares), until the point of GroES/ATP addition. These results suggest that MDH is not released from HdeA in a fully folded conformation, but in a state that can still be recognized by GroEL. GroEL apparently binds these inactive MDH molecules upon release from HdeA and remains in complex until addition of GroES and ATP triggers MDH folding. MDH that was incubated at low pH in the absence of HdeA served as a poor substrate for the GroEL/ES system upon neutralization (compare closed squares in Fig. 4B with closed squares in Fig. 4C). This result agrees with the observation that in the absence of HdeA, most MDH molecules partition to off-pathway aggregates. These results provide further evidence that HdeA’s rapid interaction with acid-unfolding proteins maintains them in a nonnative but folding-competent form. Because we have excluded the possibility that MDH refolds completely while bound to HdeA, the observation that the rate of MDH release closely mimics the rate of MDH refolding suggests that dissociation from HdeA may be rate-determining in MDH reactivation.
To determine whether the slow release from HdeA and the correlation between rate of release and refolding rate observed for MDH may also apply to other substrates, we measured the release and refolding kinetics of two additional model substrate proteins, GAPDH and aldolase. By using fluorescence anisotropy, we found that HdeA(S27C) associates with both proteins at pH 2 and releases them upon neutralization (Fig. S5 and S6). Importantly, HdeA supports the refolding of both substrates to an enzymatically active state. Analysis of the kinetics of release and refolding revealed that aldolase behaves similarly to MDH by following apparent biphasic release kinetics, with the overall half-time of dissociation corresponding roughly to the rate of refolding (Fig. S5). GAPDH (Fig. S6) behaved more similarly to alkaline phosphatase, where the apparent rate of release is faster than the appearance of recovered enzymatic activity, suggesting that these substrates are first released and then spontaneously refold.
The observation that at least some substrates appear to be released significantly faster than they acquire activity would seem to suggest that release from HdeA is not always rate limiting for the complete refolding of all substrate proteins. However, it is important to note that the release of all tested substrates is slow (i.e., on the minute timescale) when compared to the typical rate of early protein folding events such as hydrophobic collapse, which occurs on the microsecond or millisecond timescale (17). To investigate the possibility that the substrates we tested refold via an intermediate state under our experimental conditions, we used bis-ANS as a conformational probe. This dye binds hydrophobic regions that are exposed in unfolded proteins but has a lower propensity to bind native proteins (18). Each of the substrates we tested bind bis-ANS at pH 2; however, following neutralization they each very rapidly collapse to a state that is apparently less hydrophobic (Fig. S7). For all four substrate proteins, this occurs on a timescale that is 2–4 orders of magnitude faster than the observed rate of release from HdeA. Therefore, the relatively slow release of substrates may serve to decrease the concentration of aggregation-sensitive intermediates following pH neutralization. This would effectively suppress substrate aggregation and thereby facilitate refolding to the native state. Further support for this model comes from experiments in which we varied the starting concentration of HdeA:MDH complexes and tested refolding efficiency following pH neutralization. We found an inverse relationship between MDH refolding yield and the HdeA:MDH complex concentration (Fig. S8). This is likely because increased concentrations of partially unfolded MDH as it is released from HdeA leads to increased aggregation and less refolding.
Discussion
We have provided evidence for a molecular chaperone that is capable of assisting in the refolding of acid-denatured proteins independent of ATP. This capability makes HdeA well suited to protect proteins against acid-induced unfolding and aggregation in the ATP-free environment of the bacterial periplasm.
The direct activation of a molecular chaperone by stress conditions at the protein level enables presynthesized proteins to be activated by stress conditions that even strongly inhibit transcription and translation. One such stress condition is the shift from neutral pH to the pH of stomach acid (pH ∼ 2) that accompanies oral ingestion of bacteria. This drop in pH has been shown to rapidly activate the periplasmic chaperone HdeA (4, 7, 8). Other chaperones that are directly activated by stress conditions at the protein level are Hsp26, a member of the small Hsp family, which is activated by elevated temperatures (19), and Hsp33, which is activated by oxidative stress conditions that lead to protein unfolding (20). As with HdeA, these proteins appear to be efficiently adapted to be active only when the bacteria encounter stresses that necessitate their activity. One feature of HdeA that stands in stark contrast to these other examples is that return to nonstress conditions is sufficient to trigger substrate release and refolding. Hsp26 and Hsp33, on the other hand, require the assistance of the ATP-consuming chaperone systems to facilitate substrate release and support refolding (20, 21). HdeA, however, by virtue of its periplasmic localization, must function in the absence of these chaperone systems and ATP. It seems to have therefore evolved to take efficient advantage of the natural host physiology. HdeA uses external pH shifts provided by the host to trigger chaperone activation and inactivation. Unlike previously characterized chaperones, return to nonstress conditions (i.e., neutral pH) is sufficient to trigger substrate release and refolding of substrate proteins. We should note that HdeA is not the only protein known to facilitate protein folding in an ATP-independent manner. The ribosome-associated peptidyl proline isomerase, trigger factor, is capable of independently facilitating protein folding in vitro, although it is involved in the folding of nascently synthesized polypeptides in the cytosol and is also thought to function in concert with ATP-dependent chaperone systems (22). HdeA on the other hand, is likely not involved in the folding of newly translocated proteins, but rather only with the refolding of proteins following acid stress.
One tentative model that is consistent with our experimental observations, is that HdeA binds substrates stably at low pH and slowly releases them following pH neutralization (Fig. 5). The stable binding at low pH is undoubtedly important, as this maintains substrates in a soluble and therefore folding-competent form. The slow release of substrate proteins from HdeA upon pH neutralization may help explain why one single binding and release cycle appears to be sufficient to support substrate-protein refolding. Slow release may provide a convenient way to keep the concentration of aggregation-sensitive folding intermediates low at any given time. Because aggregation is strongly dependent on the concentration of aggregation-sensitive folding intermediates (23), any process that decreases the concentration of these intermediates should greatly decrease the extent of aggregation. In contrast, in the absence of HdeA, pH neutralization may quickly generate a large population of aggregation-sensitive intermediates, which favors their partitioning into the aggregation pathway instead of the refolding pathway. Thus, slow release in concert with preventing off-pathway aggregation at low pH provides an elegant, mechanistically simple and ATP-independent way of allowing HdeA to suppress aggregation and thereby promote refolding.
Fig. 5.

Model for HdeA-mediated substrate refolding. At low pH, substrate proteins populate an ensemble of acid-unfolded states (SU), which are aggregation-sensitive. The aggregation propensity of SU is even more pronounced following pH neutralization, causing the rapid formation of inactive protein aggregates. A small population appears to escape aggregation and fold into a relatively aggregation-resistant intermediate state (SI), which is able to refold to the native state. Acid-activated HdeA is able to prevent aggregation of unfolded substrates at low pH by directly binding to them and sequestering otherwise aggregation-prone substrate. Upon pH neutralization, the substrates are released from HdeA at a rate which is very slow compared to the rate of folding to productive, aggregation-resistant intermediates and/or the native state. Because the rate of aggregation (kagg) is a second or higher order process (23), the decrease in SU concentration dictated by slow release from HdeA following neutralization effectively suppresses aggregation and favors on-pathway substrate-protein refolding.
Other chaperone systems may also work through a kinetic partitioning mechanism that keeps the free concentration of substrates sufficiently low to prevent aggregation while passively allowing them to fold to the native state (24,25–26). One aspect of the HdeA mechanism that seems strikingly different compared to other chaperones capable of facilitating protein folding, is that its chaperone “cycle” appears to involve single binding and release events. This is not true of Hsp60 or Hsp70 systems, both of which undergo repetitive cycles of ATP binding and hydrolysis, which in turn drive repetitive cycles of protein substrate binding and release (27, 28). This iterative annealing process has been proposed to be an underlying mechanism of chaperone function (14). In the case of HdeA, we have provided evidence that iterative binding does not occur. Rather, HdeA appears to rapidly refold into the chaperone-inactive state upon substrate-protein release. In our in vitro system, substrate proteins then proceed to refold without requiring additional assistance from energy cofactors, chaperones, or cochaperones. That HdeA can work in vitro in the absence of additional chaperones in no way excludes the possibility that other periplasmic chaperones may assist HdeA in vivo, much like cytoplasmic chaperones often work as part of chaperone networks (20, 26, 27). HdeA appears to be capable of kinetic partitioning through a single binding and release cycle, which is governed by the unidirectional flow of food and the accompanying bacteria through the mammalian digestive system where the pH drops a single time upon entering the stomach and then increases a single time when the bacteria reach the small intestine.
Materials and Methods
HdeA Expression and Purification.
Wild-type HdeA and HdeA(S27C) were expressed and purified as described previously (8).
MDH Aggregation Assays.
Porcine mitochondrial MDH (Roche) was incubated at a concentration of 8 μM in 8 mM H3PO4, 150 mM KCl, and 150 mM ammonium sulfate, pH 2 (buffer A) at 37 °C for 30 min in the absence or presence of 4, 8, or 16 μM HdeA. After 30 min, the pH was neutralized by adding 0.133 volumes of 0.5 M sodium phosphate, pH 8. Apparent changes in absorbance due to light-scattering aggregates were monitored at 320 nm by using a Cary100 spectrophotometer equipped with a Peltier temperature control block.
Substrate refolding assays.
Model substrate proteins MDH, aldolase, GAPDH and AP were acid-denatured by incubation in buffer A, pH 2 for 30–60 min in the absence or presence of HdeA. Then the pH was neutralized by addition of 0.133 volumes 0.5 M phosphate pH 8. 10–20 μl aliquots were then removed and assayed for enzymatic activity at various time points as detailed in SI Materials and Methods. Activity is reported relative to an equal amount of native enzyme. HdeA and substrate concentrations are indicated in the appropriate figure legends and in SI Materials and Methods.
Fluorescence Anisotropy.
HdeA(S27C) was labeled with monobromobimane as described previously (8). The labeled HdeA (0.25–0.5 μM) was incubated in buffer A (pH 2) at 37 °C for 1 h in the absence or presence of an equimolar amount of substrate protein, equilibrated to 20 °C, and then neutralized by adding 0.133 volumes of 0.5 M sodium phosphate, pH 8. To some experiments, 0.5 μM GroEL (14-mer) was added immediately after neutralization. Anisotropy (r) was calculated according to the following equations:
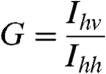 |
[1] |
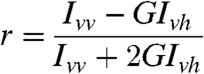 |
[2] |
where G is the instrument correction factor, r is anisotropy, and I is the fluorescence intensity measured with polarizers in the orientations indicated by the subscripts (13). Anisotropy was recorded with a Cary Eclipse Spectrofluorimeter using λex = 390 nm (10 nm bandpass) and λem = 475 nm (10 nm bandpass).
Supplementary Material
Acknowledgments.
The authors thank Dr. Stefan Walter for stimulating discussions and for providing the GroEL and GroES proteins. Calculations to analyze analytical ultracentrifugation data were performed on the UltraScan LIMS cluster at the Bioinformatics Core Facility at the University of Texas Health Science Center at San Antonio, the Lonestar cluster at the Texas Advanced Computing Center (supported by National Science Foundation Teragrid Grant MCB070038 to Borries Demeler), and the National Supercomputer HLRB-II at the Leibnitz-Rechenzentrum, Munich, Germany (supported by Project pr28ci to Johannes Buchner and T. M. F.).This work was supported by National Institutes of Health Grant GM065318 to U.J.; J.C.A.B. is an investigator for the Howard Hughes Medical Institute.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0911610107/DCSupplemental.
References
- 1.Smith JL. The role of gastric acid in preventing foodborne disease and how bacteria overcome acid conditions. J Food Protect. 2003;66:1292–1303. doi: 10.4315/0362-028x-66.7.1292. [DOI] [PubMed] [Google Scholar]
- 2.Richard HT, Foster JW. Acid resistance in Escherichia coli. Adv Appl Microbiol. 2003;52:167–186. doi: 10.1016/s0065-2164(03)01007-4. [DOI] [PubMed] [Google Scholar]
- 3.Koebnik R, Locher KP, Van Gelder P. Structure and function of bacterial outer membrane proteins: Barrels in a nutshell. Mol Microbiol. 2000;37:239–253. doi: 10.1046/j.1365-2958.2000.01983.x. [DOI] [PubMed] [Google Scholar]
- 4.Gajiwala KS, Burley SK. HDEA, a periplasmic protein that supports acid resistance in pathogenic enteric bacteria. J Mol Biol. 2000;295:605–612. doi: 10.1006/jmbi.1999.3347. [DOI] [PubMed] [Google Scholar]
- 5.Waterman SR, Small PL. Identification of sigma S-dependent genes associated with the stationary-phase acid-resistance phenotype of Shigella flexneri. Mol Microbiol. 1996;21:925–940. doi: 10.1046/j.1365-2958.1996.00058.x. [DOI] [PubMed] [Google Scholar]
- 6.Goto Y, Calciano LJ, Fink AL. Acid-induced folding of proteins. Proc Natl Acad Sci USA. 1990;87:573–577. doi: 10.1073/pnas.87.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong W, et al. Periplasmic protein HdeA exhibits chaperone-like activity exclusively within stomach pH range by transforming into disordered conformation. J Biol Chem. 2005;280:27029–27034. doi: 10.1074/jbc.M503934200. [DOI] [PubMed] [Google Scholar]
- 8.Tapley TL, et al. Structural plasticity of an acid-activated chaperone allows promiscuous substrate binding. Proc Natl Acad Sci USA. 2009;106:5557–5562. doi: 10.1073/pnas.0811811106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kern R, Malki A, Abdallah J, Tagourti J, Richarme G. Escherichia coli HdeB is an acid stress chaperone. J Bacteriol. 2007;189:603–610. doi: 10.1128/JB.01522-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malki A, et al. Solubilization of protein aggregates by the acid stress chaperones HdeA and HdeB. J Biol Chem. 2008;283:13679–13687. doi: 10.1074/jbc.M800869200. [DOI] [PubMed] [Google Scholar]
- 11.Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: The structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–846. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- 12.Wu YE, Hong W, Liu C, Zhang L, Chang Z. Conserved amphiphilic feature is essential for periplasmic chaperone HdeA to support acid resistance in enteric bacteria. Biochem J. 2008;412:389–397. doi: 10.1042/BJ20071682. [DOI] [PubMed] [Google Scholar]
- 13.Lakowicz JR. Principles of fluorescence spectroscopy. Vol XXVI. New York: Springer; 2006. p. 954. [Google Scholar]
- 14.Stan G, Thirumalai D, Lorimer GH, Brooks BR. Annealing function of GroEL: structural and bioinformatic analysis. Biophys Chem. 2003;100:453–467. doi: 10.1016/s0301-4622(02)00298-3. [DOI] [PubMed] [Google Scholar]
- 15.Buchner J, et al. GroE facilitates refolding of citrate synthase by suppressing aggregation. Biochemistry. 1991;30:1586–1591. doi: 10.1021/bi00220a020. [DOI] [PubMed] [Google Scholar]
- 16.Miller AD, et al. Escherichia coli chaperonins cpn60 (groEL) and cpn10 (groES) do not catalyse the refolding of mitochondrial malate dehydrogenase. Biochem J. 1993;291:139–144. doi: 10.1042/bj2910139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eaton WA, Thompson PA, Chan CK, Hage SJ, Hofrichter J. Fast events in protein folding. Structure. 1996;4:1133–1139. doi: 10.1016/s0969-2126(96)00121-9. [DOI] [PubMed] [Google Scholar]
- 18.Rosen CG, Weber G. Dimer formation from 1-amino-8-naphthalenesulfonate catalyzed by bovine serum albumin. A new fluorescent molecule with exceptional binding properties. Biochemistry. 1969;8:3915–3920. doi: 10.1021/bi00838a006. [DOI] [PubMed] [Google Scholar]
- 19.Haslbeck M, et al. Hsp26: a temperature-regulated chaperone. EMBO J. 1999;18:6744–6751. doi: 10.1093/emboj/18.23.6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffmann JH, Linke K, Graf PC, Lilie H, Jakob U. Identification of a redox-regulated chaperone network. EMBO J. 2004;23:160–168. doi: 10.1038/sj.emboj.7600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee GJ, Roseman AM, Saibil HR, Vierling E. A small heat shock protein stably binds heat-denatured model substrates and can maintain a substrate in a folding-competent state. EMBO J. 1997;16:659–671. doi: 10.1093/emboj/16.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deuerling E, Schulze-Specking A, Tomoyasu T, Mogk A, Bukau B. Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature. 1999;400:693–696. doi: 10.1038/23301. [DOI] [PubMed] [Google Scholar]
- 23.Zettlmeissl G, Rudolph R, Jaenicke R. Reconstitution of lactic dehydrogenase. Noncovalent aggregation vs. reactivation. 1. Physical properties and kinetics of aggregation. Biochemistry. 1979;18:5567–5571. doi: 10.1021/bi00592a007. [DOI] [PubMed] [Google Scholar]
- 24.Frieden C, Clark AC. Protein folding: how the mechanism of GroEL action is defined by kinetics. Proc Natl Acad Sci USA. 1997;94:5535–5538. doi: 10.1073/pnas.94.11.5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Apetri AC, Horwich AL. Chaperonin chamber accelerates protein folding through passive action of preventing aggregation. Proc Natl Acad Sci USA. 2008;105:17351–17355. doi: 10.1073/pnas.0809794105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 27.Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 28.Weissman JS, Kashi Y, Fenton WA, Horwich AL. GroEL-mediated protein folding proceeds by multiple rounds of binding and release of nonnative forms. Cell. 1994;78:693–702. doi: 10.1016/0092-8674(94)90533-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.