

Abstract
The frequency of intrinsic pulsatile GnRH secretion from endogenous GnRH neurons and GT1 GnRH cell lines is stimulated by increased intracellular cAMP levels. The downstream molecules comprising the cAMP signaling pathway are organized in microdomains by a family of scaffolding proteins, A-kinase anchoring proteins (AKAPs). These molecules tether protein kinase A, cAMP-specific phosphodiesterases, phosphatases to known substrates. In neurons AKAP150 organizes many of the signaling molecules known to regulate the excitability and intrinsic pulsatile activity of GnRH neurons. AKAP150 was expressed in both the GT1-1 and GT1-7 cells. We determined the role of AKAP150 in coordinating GT1-1 cell excitability and intrinsic GnRH pulsatile secretion by lowering AKAP150 levels with a small interfering RNA (siRNA) adenovirus construct to AKAP150 (Ad-AKAP150-siRNA). Infection with Ad-AKAP150-siRNA specifically decreased AKAP150 mRNA levels by 74% and protein levels by 53% relative to uninfected cells or cells infected with a luciferase control adenovirus siRNA vector. In GT1 cells, spontaneous Ca2+ oscillations, an index of neuron excitability, are stimulated by increased levels of intracellular cAMP and lowered by decreased levels. The frequency of spontaneous Ca2+ oscillations in Ad-AKAP150-siRNA-treated GT1-1 cells decreased by 47.2% relative to controls. A dramatic decrease in the number of spontaneous GnRH pulses was also observed after infection with Ad-AKAP150-siRNA. The interpulse interval increased to 143 ± 20.25 min in Ad-AKAP150-siRNA infected cells from 32.2 ± 7.3 min in luciferase control adenovirus siRNA vector-infected cells. These data demonstrate an important role of AKAP150 in coordinating signaling events regulating the frequency of intrinsic pulsatile GnRH secretion.
The macromolecular complexes important for maintaining pulses of GnRH hormone secretion vital for fertility are discussed.
GnRH secretion from primary cultures of olfactory placode GnRH neurons of the Rhesus monkey sheep and rat (1,2,3) and cultures of GT1 cells was pulsatile (4,5,6). The interpulse frequency of GnRH pulses in these experiments was in close accord with that of castrated animals of the respective species. In GT1 cells the interpulse frequency was 24 min, similar to that observed in castrated rats. These findings support the hypothesis that pulsatile release of GnRH is an intrinsic property of GnRH neurons. GT1 cells have provided an excellent model to address the signaling mechanism regulating the initiation and timing of intrinsic GnRH pulsatility.
GnRH secretion from GT1 cells is increased by both dopamine and norepinephrine (5,7,8). The dopamine response is mediated via D1 dopamine receptors, whereas the norepinephrine response is mediated via β1-adrenergic receptors. Consistent with the fact that both receptors are positively coupled to adenylyl cyclase (AC), treatment with norepinephrine or dopamine resulted in a dose-dependent increase in the intracellular concentrations of cAMP through the activation of AC (9). Pharmacologically increasing cAMP concentrations by treatment with forskolin or 8-Bromo-cAMP mimicked the stimulation of GnRH release by norepinephrine or dopamine.
GnRH neurons are spontaneously excitable as shown by the appearance of spontaneous Ca2+ oscillations seen in fura-2-loaded cells (10). Because each Ca2+ oscillation was preceded by an action potential, the frequency of Ca2+ oscillations is an index of neuron excitability. Increases in intracellular cAMP levels resulted in an increase in Ca2+ oscillations and neuron excitability (10). Genetically decreasing the intracellular cAMP concentration by the expression of a constitutively active cAMP-dependent phosphodiesterase (PDE4D1) lowered both the frequency of spontaneous Ca2+ oscillations and the frequency of intrinsic pulsatile GnRH release (11). The frequency of pulsatile GnRH release was also decreased in castrated GPR4 transgenic rats in which PDE4D1 expression was genetically targeted to GnRH neurons (12). cAMP gated Ca2+ channels (CNG) are a downstream component of the cAMP signaling pathway, regulating GT1 cell excitability and intrinsic GnRH pulse frequency. GT1 cells and rat GnRH neurons express all three subunits of the CNG channel, and GT1 cells have electrophysiologically characteristic CNG channels (13,14). Decreasing CNG channel expression in GT1 cells by infection with an adenovirus expressing a specific CNG channel small interfering RNA (siRNA) resulted in a similar decrease in cell excitability and intrinsic GnRH pulse frequency as seen with overexpressing PDE4D1.
From a variety of studies, it is clear that the cAMP signaling events do not occur globally throughout cells. Rather signaling molecules are organized into microdomains within neurons by molecules that tether signaling molecules to relevant substrates. A large family of more than 50 A-kinase anchoring proteins (AKAPs) was identified. Multivalent AKAPs bring protein kinase A (PKA) along with other regulatory molecules in close molecular proximity to relevant substrates (for reviews see Refs. 15,16,17,18). The restriction of PKA to specific domains in the cell also permits the enzyme to be activated by locally fluctuating pools of cAMP. AKAP79/150 (79 human/150 rodent) is expressed predominantly in the brain and central nervous system. It tethers PKA to AC 5 and AC 6 (19), β2-adrenergic receptor (20,21,22), protein kinase C (PKC), α-amino-3-hydroxyl-5-methyl-4-isoxazole propionic acid receptor (23), KCNQ K+ channels (24), L-type Ca2+ channel (Cav1.2) (25,26), and the calcium/calmodulin-dependent phosphatase (PP2B)/calcineurin (27,28). In AKAP150 knockout mice, the localization of PKA holoenzyme was excluded from dendritic spines in hippocampal neurons (29). This was associated with a loss in bidirectional regulation of 2-amino-3-hydroxy-5-methyl-4-isoxazol propionic acid receptors in the hippocampus. Alterations in complex behaviors including memory retention, motor function and anxiety were observed in knockout mice consistent with AKAP150 playing an important role in the spatial and temporal organization of signaling molecules throughout the brain.
AC 5 and AC 6, CNG channels, and L-type Ca2+ channels were shown to be involved in cAMP signaling regulating GnRH secretion from GT1 cells. Therefore, AKAP150 was an excellent candidate for the spatial and temporal organization of the cAMP signaling pathways involved in regulating intrinsic GnRH pulsatility. We first show that AKAP150 is expressed in GT1 cells and that decreasing its expression with a specific siRNA resulted in decreased cell excitability and the frequency of spontaneous GnRH pulses.
Materials and Methods
Cell culture
GT1-1 cells (passages 14–23) were cultured on 10-cm culture plates in DMEM (Invitrogen Corp., Carlsbad, CA) supplemented with 5% fetal bovine serum (HyClone Laboratories, Inc., Logan, UT), 5% horse serum (HyClone Laboratories), 100 U/ml penicillin, and 100 μg/ml streptomycin.
Adenoviral siRNA constructs
Two pairs of oligos (AKAP150 no. 1 and AKAP150 no. 2) were synthesized from sequences corresponding to AKAP150 cDNA sequence (NP_001094941) (Table 1). These oligos were annealed and ligated into linearized pSIREN-Shuttle (CLONTECH, Palo Alto, CA). The negative control luciferase (Luc) siRNA supplied with the vector was also cloned into pSIREN-Shuttle. Both the AKAP150 siRNA (Ad-AKAP150-siRNA) and the Luc siRNA (Ad-Luc-siRNA) were transferred from the shuttle vector into pAdenoX (CLONTECH) using the recommended protocol. The recombinant adenoviral DNA was transfected into HEK 293 cells to produce viral particles. Cell lysates were then used to reinfect HEK 293 cells for large-scale viral production. The virus was purified on two consecutive cesium chloride gradients, dialyzed in formulation buffer [2.5% glycerol, 25 mm NaCl, and 20 mm Tris-HCl (pH8.0)] and titered using the adenoviral rapid titer kit (CLONTECH). Viral titers were 2.8 × 109 pfu/ml for Ad-AKAP150-siRNA and 2.3 × 109 pfu/ml for Ad-Luc-siRNA.
Table 1.
Sequences of the siRNA oligonucleotides designed to the mouse AKAP150 mRNA
| Oligo | Sequence (5′–3′) |
|---|---|
| AKAP150 no. 1F | gatccGGCAAAGAGAAGTCGTCATTTCAAGAGAATGACGACTTCTCTTTGCCTTTTTTACGCGTg |
| AKAP150 no. 1R | aattcACGCGTAAAAAAGGCAAAGAGAAGTCGTCATTCTCTTGAAATGACGACTTCTCTTTGCcg |
| AKAP150 no. 2F | gatccGAAAAAGGCAAAATCCAGACTTTCAAGAGAAGTCTGGATTTTGCCTTTTTCTTTTTTACGCGTg |
| AKAP150 no. 2R | aattcACGCGTAAAAAAGAAAAAGGCAAAATCCAGACTTCTCTTGAAAGTCTGGATTTTGCCTTTTTcg |
Sequences corresponding to the AKAP150 mRNA are shown in bold.
RT-PCR
GT1-1 and GT1-7 cells were grown on 10-cm dishes until 70% confluency. Total RNA was isolated using 1 ml Trizol (Invitrogen). Total RNA was isolated from mouse cortex. cDNA was synthesized using 2 μg total RNA. Primer sequences were as follows: forward, 5′-ATGGAGACCAGCGTTTCTGA-3′ and reverse primer, 5′-GTGGGTGTTTTTCTCTCCTG-3′. The expected product size was 97 bp.
Real-time quantitative RT-PCR analysis
GT1-1 cells plated in six-well plates were infected with either Ad-AKAP150-siRNA or Ad-Luc-siRNA at 5 multiplicity of infection (MOI; the average number of viral particles per cell) were washed with PBS and lysed with 0.5 ml Trizol (Invitrogen). Total RNA was prepared according to the recommended protocol. cDNA was synthesized from 1 μg of total RNA using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) Real-time quantitative PCR was performed using SYBR Advantage qPCR mix (CLONTECH) with an iCycler thermal cycler (Bio-Rad). We used the following primers: AKAP150 forward, 5′-AGGATGGGGCTCTTCCTAAG-3′ and reverse, 5′-GGGTCTGGGCTTTTATCTCC-3′; β-actin forward, 5′-GTCCACACCCGCCACCAGTTC-3′ and reverse, 5′-GACCCATTCCCACCATCACACC-3′. The data were collected and analyzed using the comparative threshold cycle method using β-actin expression as the reference gene. Data were collected in triplicate from three separate experiments analyzed using GraphPad Prism (GraphPad, San Diego, CA). Statistically significant differences were determined by t test.
Western blotting
COS-7 cells were plated into six-well plates at a density of 3 × 105 cells/well. Cells were transfected with 1 μg of AKAP150 cDNA into a mammalian expression vector using Lipofectamine (Invitrogen). After 24 h the cells were transfected with 1 μg pSiren-AKAP150 no. 1/no. 2 cDNA. Cells were incubated for 24 h and then washed twice in cold PBS and total cell lysate was collected using membrane protein extraction kit containing a complete protease inhibitor cocktail (Roche, Indianapolis, IN). For Western blots on GT1-1 cells, cells were cultured on 10-cm dishes and infected with 5 MOI Ad-AKAP150-siRNA or Ad-Luc-siRNA. Protein was extracted 48 h after infection using the membrane protein extraction kit (Pierce, Rockford, IL). Protein yield was determined by BCA assay (Pierce), and 50 μg of protein in Laemmli sample buffer were resolved by SDS-PAGE and transferred to a polyvinyl difluoride membrane (Bio-Rad). The immunoblots were probed with a goat polyclonal antibody to the carboxy terminus of rat AKAP150 (Santa Cruz Biotechnology, Santa Cruz, CA) in 10 mm Tris (pH 7.4), 150 mm NaCl, and 0.05% Tween 20 containing 5% nonfat milk powder and 10% bovine calf serum. Immunoreactive proteins were visualized with the ECL plus Western blotting detection system (Amersham Biosciences, GE Healthcare, Indianapolis, IN).
Perifusion studies
GT1-1 cells (300,000 cells/well) were plated on Matrigel (BD Biosciences, Discovery Labware, Franklin Lakes, NJ)-coated, 25-mm plastic coverslips (Thermanox plastics, Thermo Fisher Scientific, Rochester, NY) in six-well plates. When the cells reached 50–60% confluency, they were infected with 5 MOI of Ad-AKAP150-siRNA or Ad-Luc-siRNA. Cells were incubated for 24 h after infection. The media were then replaced with Opti-MEM (Invitrogen), and incubated for an additional 24 h before sampling. The coverslips were placed in a modified Sykes-Moore chamber (Bellco Glass, Inc. Vineland NJ), and the cells were perifused with oxygenated Locke’s medium (154 mm NaCl, 5.6 mm KCl, 2.2 mm CaCl2, 1 mm MgCl2, 6 mm NaHCO3, 10 mm glucose, 2 mm HEPES) supplemented with 0.1% BSA and 20 μm bacitracin at a flow rate of 70–100 μl/min. Chambers were washed for 60 min and then samples were collected every 2 min for 3 h. Each sample was boiled for 3 min cooled on ice and then stored at −80 C for RIA.
GnRH RIA
Levels of GnRH in the media from the perifusion studies were determined by a RIA using rabbit antibody R1245 (obtained from T. Nett, College of Veterinary Medicine and Biomedical Sciences, Colorado State University, Fort Collins, CO). This antiserum was specific for intact GnRH. Synthetic human GnRH (Sigma, St. Louis, MO) was used for the standard and 125I-[d-Trp6]GnRH was purchased from PerkinElmer (Norwalk, CT). All samples from an experiment were analyzed in the same assay. The limit of detection of the assay was 0.25–25 pg/tube, and the intraassay coefficient of variation was 7.2%. The limit of detection of the assay was defined as 90% of maximal binding. Analysis of the GnRH pulse data was performed using the hormone pulse analysis software Cluster 8 (Michael L. Johnson, University of Virginia, Charlottesville, VA). Cluster analysis was performed on measurements done in singlicate. The coefficient of variation was determined from intraassay controls. Cluster size or nadir was defined by two points that significantly increased or decreased with a t statistic of 4.
Ca2+ assays
Intracellular Ca2+ concentration was measured using fluorescence ratio imaging with MetaFluor imaging software (Universal Imaging Corp., Westchester, PA) as previously described (30). Briefly, GT1-1 cells cultured on Matrigel-coated, 25-mm glass coverslips were loaded with fura-2 (Molecular Probes, Eugene, OR) by incubation in 5 μm fura-2-AM for 30 min at 37 C in oxygenated Locke’s medium supplemented with 0.1% BSA. The cells were then washed in fresh Locke’s medium for 15 min. Coverslips were placed in a temperature-controlled modified Sykes-Moore chamber mounted on a TE2000 inverted fluorescence microscope (Nikon, Tokyo, Japan). Cells were perifused with Locke’s medium or Locke’s medium containing 10 μm forskolin at a flow rate of 50 μl/min. Fura-2 fluorescence at 510 nm was measured at five second intervals for 18 min and a further 2 min after the addition of forskolin at excitation settings of 340 and 380 nm. Approximately 40–60 cells/coverslip were imaged and four coverslips were studied for the control GT1-1 cells as well as the Ad-AKAP150-siRNA- or Ad-Luc-siRNA-infected cells.
Quantification of Ca2+ concentration and Ca2+ oscillations
To distinguish living cells from dead cells, only cells that showed a 20% increase in Ca2+ concentration after treatment with forskolin for 5 min were used for analysis. The intracellular Ca2+ concentration was estimated from the ratio of the fluorescence intensities and comparison with fura-2 standards using the fura-2 Ca2+ calibration kit (Invitrogen) (31). These values were plotted in Raster plots using Transform (Fortner Software, Sterling, VA). Ca2+ oscillations were quantified using JPULSAR1 (written by Dr. Xiaoyu Pan, Department of Molecular and Cellular Biology, University of California, Davis, CA), a program modified from PULSAR (32), to detect peaks in intracellular Ca2+ concentration ([Ca2+]i) oscillations. The sd was determined by using the mean of the sds identified by time series analysis using a 5-sec window. The coefficient of variation for calcium imaging was described by the formula: Y = 4.1827X + 0.4826. The peak detection parameters for JPULSAR, G(1)–G(5), were 1.77, 1.05, 0.73, 0.53, and 0.39. A smoothing window of 25 sec was used to determine segmented baseline values.
Results
Expression of AKAP150 in GT1-1 and GT1-7 GnRH neurons
AKAP150 expression was shown to be high in the forebrain regions including the cerebral cortex (33). We determined whether AKAP150 was expressed in GnRH neurons using RT-PCR. Specific primers designed to the AKAP150 cDNA sequences yielded a single predicted band of 97 bp with mRNA from both GT1-1 and GT1-7 cells (Fig. 1A). An identical size band was observed with mRNA extracted from mouse cerebral cortex. A strong fluorescence signal associated with the plasma membrane was observed in GT1 cells transfected with AKAP150-red fluorescent protein (RFP) (Fig. 1C).
Figure 1.

Specific primers were designed to the sequence of AKAP150 and used in RT-PCR to determine expression (A). PCR on cDNA from GT1-1, GT1-7 cells, and mouse cortex show expression of AKAP150. B, Whole-cell extracts from untransfected COS 7 cells or cells transiently transfected with AKAP150-RFP cDNA alone or with the control luciferase (Luc) siRNA, AKAP150 siRNA no. 1, or AKAP150 siRNA no. 2, respectively, were immunoblotted with anti-AKAP150 antibody. Both AKAP-siRNA no. 1 and AKAP150-siRNA no. 2 but not Luc-siRNA inhibited the expression of AKAP150 in transiently transfected COS 7 cells. WB, Western blot; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. C, AKAP150-RFP transfected into GT1-1 cells is expressed at the plasma membrane.
Development of AKAP150 siRNA
We designed two siRNAs specific to the mouse AKAP150 cDNA (AKAP150 no. 1; AKAP150 no. 2; Table 1). Before cloning these oligonucleotides into the Adenovirus expression vector, we tested their ability to knockdown expression of AKAP150-RFP cDNA. We performed these experiments in COS cells transiently expressing the AKAP150 cDNA. The oligonucleotides were cloned into pSIREN-Shuttle and expressed in COS cells transiently cotransfected with a AKAP150 expression construct. Western blot analysis showed that both AKAP150 no. 1 and AKAP150 no. 2 were equally effective in decreasing the expression of the AKAP150 cDNA (Fig. 1B). Because only a small portion of GT1 cells are transiently transfected by available techniques, for subsequent experiments in GT1 cells, we developed an adenovirus vector to direct expression of the AKAP150 no. 1 siRNA. The AKAP150 no. 1 siRNA (Ad-AKAP150-siRNA) as well as the control luciferase siRNA (Ad-Luc-siRNA) were cloned into an adenovirus vector for all further studies.
Inhibition of AKAP150 mRNA and protein by Ad-AKAP150-siRNA treatment
We measured AKAP150 mRNA levels in GT1-1 cells 48 h after infection by real-time quantitative RT-PCR. At 5 MOI, Ad-AKAP150-siRNA caused a 74% average decrease in mRNA levels of AKAP150 relative to uninfected cells (Fig. 2). A small nonsignificant increase was seen after infection with 5 MOI of Ad-Luc-siRNA. The Ad-AKAP150-siRNA appeared specific because no affect was observed on the mRNA levels of β-actin or GnRH (data not shown). All subsequent experiments were done with 5 MOI of the siRNA vectors because infection of GT1 cells with a MOI above 5 was shown to have a nonspecific effect on spontaneous GnRH pulses and Ca2+ oscillations (11). Furthermore, infection with 5 MOI of the same adenovirus vector expressing green fluorescent protein (GFP) resulted in GFP expression in 90–95% of GT1 cells (data not shown).
Figure 2.
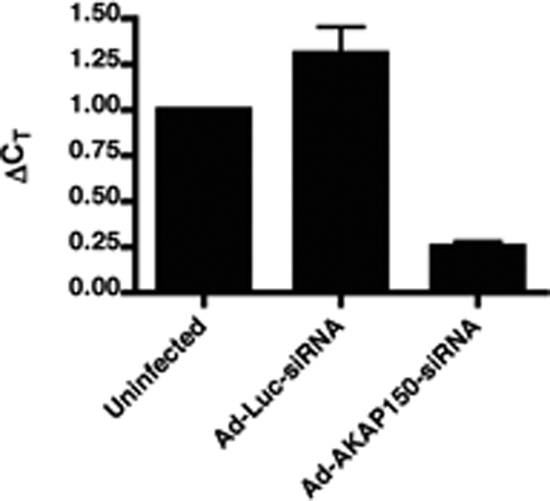
Quantitative RT-PCR showed that Ad-AKAP150-siRNA decreased AKAP150 mRNA expression by approximately 74%. GT1-1 cells were infected with Ad-AKAP150-siRNA (5 MOI) or Ad-Luc-siRNA (5 MOI) and RNA was isolated 48 h after infection. The data were analyzed using the comparative threshold cycle (Ct) method with β-actin as the reference gene. The data, expressed as a ratio of the control AKAP150 mRNA in uninfected cells, represent the mean ± sem from three separate experiments.
A specific band of approximately 150 kDa, corresponding to the AKAP150 protein, was observed in immunoblots of membrane protein extracted from GT1-1 cells (Fig. 3A). The blots were quantified and band intensity was normalized for loading using an antibody to glyceraldehyde-3-phosphate dehydrogenase. In agreement with findings in transfected COS cells, a 54% decrease in the intensity of the AKAP150 band relative to the uninfected GT1-1 cells was observed after infection with 5 MOI of the Ad-AKAP150-siRNA relative to uninfected controls (Fig 3B). Infection with 5 MOI of Ad-Luc-siRNA had no effect on AKAP protein levels.
Figure 3.
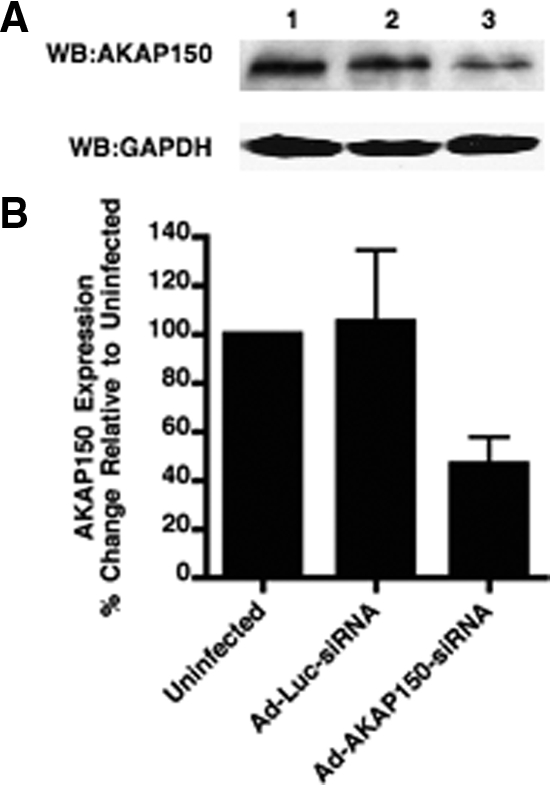
Infection with Ad-AKAP150-siRNA but not Ad-Luc-siRNA inhibited the expression of AKAP150 protein in GT1-1 cells. A, Immunoblot of membrane extracts with antibodies to AKAP150 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in GT1-1 cells. Lane 1, Uninfected GT1-1 cells; lane 2, GT1-1 cells infected with Ad-Luc-siRNA 5 MOI; lane 3, GT1-1 cells infected with Ad-AKAP150-siRNA 5 MOI. WB, Western blot. B, Immunoblot data were quantitated and represented as the percent of the uninfected GT1-1 cells (lane 1 above). Error bars, mean ± sem from different exposures of two separate experiments.
Effect of Ad-AKAP150-siRNA infection on frequency of Ca2+ oscillations
We then asked whether lowering the levels of AKAP150 expression in GT1-1 cells would affect the occurrence of spontaneous Ca2+ oscillations. Calcium was measured using fura-2 imaging in living GT1-1 cells by fluorescence microscopy (Fig. 4). In untreated, fura-2-labeled cells spontaneous Ca2+ oscillations were observed with a similar frequency to those previously reported (10). Quantitation of the Ca2+ oscillations showed a significant 47.2% decrease (from 4.4 to 2.3 oscillations per 18 min experiment, P < 0.0001) in GT1-1 cells infected with the Ad-AKAP150-siRNA relative to the uninfected cells (Fig. 5A). No difference was observed in the Ad-Luc-siRNA infected cells (Fig. 5A). Consistent with these findings, the interpulse interval increased more than 2-fold in Ad-AKAP150-siRNA-treated cells relative to the uninfected or Ad-Luc-siRNA infected cells (Fig. 5B).
Figure 4.

Effect of Ad-AKAP150-siRNA on [Ca2+]i. Fura-2 Ca2+ imaging experiments in uninfected GT1-1 neurons (A) or GT1-1 cells infected with 5 MOI of Ad-AKAP150-siRNA (B) and Ad-Luc-siRNA (C). The tracings at the top of each section (A, B, and C) show the changes in [Ca2+]i vs. time in a representative cell. The raster plots show [Ca2+]i vs. time for 26 representative cells from a single field. The gray scale represents the [Ca2+]i from 0 to 600 nm. Forskolin (FSK; 10 μm) treatment is indicated by the bar.
Figure 5.
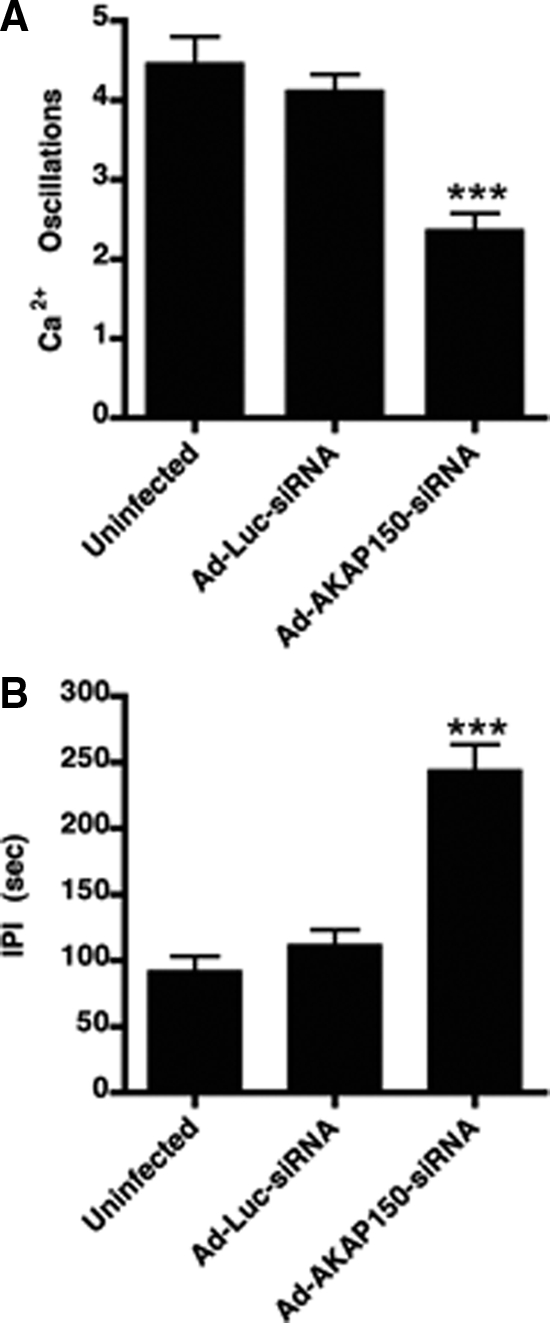
Infection with Ad-AKAP150-siRNA but not Ad-Luc-siRNA caused a decrease in the spontaneous Ca2+ oscillations. Spontaneous Ca2+ oscillations (A) and the interpulse interval (IPI; B) between the spontaneous Ca2+ oscillations in uninfected GT1-1 cells (n = 132) or GT1-1 cells infected with 5 MOI of Ad-Luc-siRNA (n = 125) or 5 MOI of Ad-AKAP150-siRNA (n = 134). Data are the mean ± sem from three separate experiments. ***, P < 0.0001 relative to uninfected and Ad-Luc-siRNA infected cells.
After recording spontaneous Ca2+ oscillations, cells were treated with forskolin to identify responsive cells. In this way any nonresponsive cells were eliminated from analysis. Forskolin treatment caused a rapid, large, pharmacological increase in cAMP levels that resulted in a dramatic increase in Ca2+ oscillations in GT1 cells as previously seen (14). Only cells showing a 20% increase in intracellular Ca2+ concentration after forskolin treatment were used for analysis of spontaneous Ca2+ oscillations.
Inhibition of spontaneous pulsatile GnRH release
Finally, we tested the effect of lowering AKAP150 expression levels on the frequency of intrinsic pulsatile GnRH release from GT1-1 cells. Uninfected GT1-1 cells or cells infected with the Ad-AKAP150-siRNA or Ad-Luc-siRNA were perifused and samples obtained every 2 min for 3 h for measurement of GnRH by RIA. A significant decrease in the number of spontaneous GnRH pulses per collection period was observed after infection with Ad-AKAP150-siRNA, whereas no difference was seen between uninfected cells and the Ad-Luc-siRNA infected cells (Table 2). The interpulse interval was significantly increased (P < 0.003) by infection with the Ad-AKAP150-siRNA (143 ± 20.25 min) vs. uninfected (32.2 ± 7.3 min) or Ad-Luc-siRNA infected cells (32.7 ± 5.2 min). Interestingly, no significant change in the pulse amplitude was observed (Fig. 6 and Table 2).
Table 2.
Summary of GnRH pulse parameters determined by cluster analysis
| WT | Ad-Luc-siRNA | Ad-AKAP150-siRNA | |
|---|---|---|---|
| Number of pulses/experiment | 3.2 ± 0.5 | 3.3 ± 0.5 | 1 ± 0.3a,b |
| (n = 7) | (n = 7) | (n = 7) | |
| Interpulse interval (min) | 32.2 ± 7.3 | 32.7 ± 5.2 | 143 ± 20.25c,d |
| (n = 22) | (n = 23) | (n = 7) | |
| Pulse amplitude (percent increase) | 184.6 ± 26.58 | 194.3 ± 37.08 | 142.5 ± 0.6e |
| (n = 22) | (n = 23) | (n = 7) |
The values are mean ± sem.
P = 0.0034 relative to wild type (WT);
P = 0.0068 relative to Ad-Luc-siRNA;
P = 0.0037 relative to WT;
P = 0.0017 relative to Ad-Luc-siRNA;
P = ns relative to WT and Ad-Luc-siRNA.
Figure 6.

Spontaneous GnRH release in uninfected GT1-1 neurons (A), neurons infected for 48 h with 5 MOI of Ad-Luc-siRNA (B), or 5 MOI of Ad-AKAP150-siRNA (C). Samples were obtained every 2 min for 180 min from perifused cells. Data are shown for three of seven experiments for each treatment. GnRH levels were measured by RIA and data were analyzed for pulsatile secretion using cluster analysis (*, pulse). IPI, Interpulse interval.
Discussion
Substantial data exist that the level of cAMP in GT1 cells regulates spontaneous cell excitability and the frequency of intrinsic GnRH pulses (11,14). GT1 GnRH neurons express several different isoforms of adenylyl cyclase including predominantly the Ca2+ insensitive forms of ACs 2, 4, and 7, the Ca2+ sensitive forms, ACs 3, 5, 6, and 9 (negatively regulated by Ca2+), and AC 1 an isoform activated by Ca2+ (9,34,35). Synthesis of cAMP in response to both intracellular Ca2+ concentrations (34) and feedback via PKA inhibition of AC 5/6 (9,36) suggest fine tuning of cAMP concentrations as a focal point for the regulation of GnRH secretion. Work by several groups has shown that small local changes in cAMP concentrations or even possibly concentration gradients of cAMP may account for the molecular specificity of cAMP signaling, emphasizing the importance of subcellular organization (for review, see Ref. 37). Increases in cAMP can initiate downstream signaling events by directly binding to target proteins, e.g. the PKA regulatory subunit, CNG channels (38) and exchange proteins directly activated by cAMP (39) or indirectly by alterations in the activity of signaling molecules after phosphorylation by activated PKA, e.g. Cav1.2 channel. The downstream signaling events mediating the effects of cAMP on cell excitability and the initiation and timing of GnRH pulses are only partially understood. Candidate components of the cAMP regulatory pathway include PKA, CNG channels, phosphodiesterases (PDEs), Cav1.2 channels and PP2B. The duration and intensity of effects of increased cAMP levels and phosphorylation of target proteins by PKA are regulated by PDE activity and protein phosphatases, respectively.
The purpose of the current study was to better understand how cAMP signaling events are spatially and temporally organized to result in spontaneous GnRH pulses with a frequency of 24 min. In muscle and the heart, it is clear that the cAMP signaling pathways are highly organized into microdomains by the sequestration of signaling molecules. The best understood of the tethering molecules maintaining the subcellular distribution and temporal organization of signaling molecules are the AKAP proteins. We hypothesized that the AKAP79/150 (79 human/150 rodent) scaffolding protein, localized to the plasma membrane via a polybasic region (40) with a high expression in neurons (41,42) could provide a mechanism for organizing these molecules in relevant microdomains within GT1 cells. AKAP79/150 binds protein PP2B/calcineurin (28) and PKC (43) in addition to PKA. By organizing molecules that bivalently regulate signaling events, e.g. bringing both PKA and PP2B in close proximity to the Cav1.2 channel, AKAP binding provides potential feedback mechanisms timing the frequency of GnRH pulses.
AKAP150 mRNA and protein were present in GT1 cells. When RFP-labeled AKAP150 was expressed in GT1 cells, dense fluorescence was observed associated with the plasma membrane. The lowering of the levels of AKAP150 mRNA by 74% and protein by 53% by infection with the Ad-AKAP150-siRNA decreased spontaneous Ca2+ oscillations by 71% and dramatically decreased the number of spontaneous GnRH pulses (increased the interpulse frequency of GnRH pulses). No effect was observed on the amplitude of spontaneous GnRH pulses. We hypothesized that the decrease in neuron excitability and spontaneous GnRH pulsatility was a result of the disruption of the organization of signaling molecules known to be tethered to AKAP150 possibly including PKA, PKC, and PP2B.
Performing these experiments depended on the ability of expressing specific siRNAs that would sufficiently lower AKAP150 expression to alter cell function. We demonstrated that the siRNA used in the experiments dramatically lowered the level of AKAP mRNA and protein levels in transfected COS cells and in GT1 cells. No nonspecific effects were seen on nonrelated gene expression, i.e. β-actin or GnRH. A second key requirement for success of using siRNA expression in GT1 cells was the ability to direct expression to the majority of GT1 cells in the cultures. We had previously used adenovirus vectors to direct expression of PDE4D1 to GT1 cells (11). In these studies as well as the current studies, as long as the viral load was not raised above 5 MOI, little if any nonspecific effect of viral infection was observed. As a further control, the same viral vector-expressing luciferase was used to infect cells at 5 MOI. In this way we controlled for both viral infection and effects of expressing a siRNA. No effects on the frequency of Ca2+ oscillations or GnRH pulses were observed after infection with 5 MOI of Ad-Luc-siRNA relative to uninfected cells. Infection of GT1 cells with 5 MOI of an adenovirus vector expressing GFP resulted in observable green fluorescence in greater than 90% of the cells.
The decrease in cell excitability and frequency of spontaneous GnRH pulses in GT1 cells resulting from the siRNA-induced decrease in AKAP150 expression was similar to that seen by lowering cAMP levels by expression of PDE4D1 or lowering CNG channel expression using a siRNA (11,44). Also, the lack of effect on GnRH pulse amplitude of lowering AKAP150 expression was observed after the over expression of PDE4D1 and lowering CNG channel expression. These findings are consistent with the hypothesis that all three treatments involved decreased cAMP signaling at the plasma membrane resulting in decreased cell excitability. Potential mechanisms that could be involved include the integral membrane proteins CNG channels, AC 5/6, Cav1.2 channels, the inwardly rectifying potassium channel (Kir2.1/KCNJ2), β1-adrenergic receptor and PKA, PKC, and PP2B tethered to these molecules by AKAP150.
Binding of cAMP to the CNG channel subunits opens the channel allowing cations that increase the depolarization drive to enter the neurons (14). The magnitude of the cAMP-regulated conductance through the CNG channels is not sufficient to trigger an action potential. Instead the increase in the resting potential presumably leads to the activation of inwardly rectified K+ channels that are responsible for triggering the action potentials in GT1 cells (45,46). By decreasing the number of CNG channels by infection with the Ad-CNG-siRNA, the cAMP levels in the neuron presumably took longer to increase the resting potential sufficiently to activate the inwardly rectified K+ channels. This resulted in fewer Ca2+ oscillations, i.e. action potentials. This decrease in spontaneous neuron excitability resulted in a decrease in the frequency of spontaneous GnRH pulses (44). The expression of PDE4D1 lowers intracellular cAMP levels and decreases cell excitability consistent with the hypothesis that the changes are mediated in part through decreases in cation conductance through CNG channels. How AKAP150 would directly affect CNG channel activity is not clear because, however, the indirect affect of decreased localized concentrations of cAMP by disruption of AKAP/AC complexes for example could account for the decrease in neuron excitabity.
Oscillatory increases in intracellular Ca2+ are generated in GT1 cells by the influx of Ca2+ through voltage-gated Ca2+ channels (4,10). The enhancement of Cav1.2 channel activity was bidirectionally regulated in cultured hippocampal neurons by PKA and PP2B tethered to the channel by AKAP150 (26). Both the activation by PKA phosphorylation and inactivation by dephosphorylation by PP2B required expression of AKAP150. Interestingly, anchored PP2B dominantly suppressed enhancement by PKA.
AKAP150 mediated feedback and potentially bidirectional regulation is the inactivation/activation of AC 5 and AC 6 activity by AKAP150-tethered PKA (19). When cAMP levels are locally increased by increased AC 5/6 activity, tethered PKA is activated resulting in phosphorylation and decreased cAMP formation. Phosphorylation of AC 5/6 by tethered PKA would be reversed by the phosphatase PP2B. Additionally, PDEs constitute another potential bidirectional feedback mechanism for regulating cAMP levels in discrete compartments. Both the Ca2+/calmodulin-sensitive PDE1B and the PKA-regulated PDE4D (specifically PDE4D3) and PDE4B subtypes are expressed in GnRH neurons (47). Phosphorylation of PDE4D3 by PKA activates the enzymes ability to hydrolyze cAMP. To date PDE subtypes have not been shown to bind to AKAP79/150; however, PDE4D3 has been shown to form a complex with Yotiao (AKAP9) (48) a membrane-associated AKAP expressed in brain (49,50); the presence of this complex in GnRH neurons has not yet been investigated. In GT1 cells treatment with the PKA inhibitor H89 increased intracellular cAMP concentrations, possibly via the removal of PDE4D3 activation, and the loss of AC 5/6 down-regulation ultimately led to an increased GnRH secretion (9). However, because H89 is known to have several nonspecific targets, these results do not conclusively prove that PKA is involved only in GnRH secretion via the regulation of negative feedback.
AKAP79/150 has also been shown to interact with the carboxy terminus of β1-adrenergic receptor (21,51), promoting receptor resensitization and recycling. The β1-adrenergic receptor is expressed in GnRH neurons and plays a role in GnRH secretion (7). Another potential target is the inwardly rectifying potassium channel Kir2.1 (KCNJ2), which is shown to have an enhanced response to elevated intracellular cAMP when complexed to AKAP79 (52).
In GT1 cells these feedback mechanisms could result in the waxing and waning of cAMP levels in the vicinity of the plasma membrane constituting a timing mechanism for cell excitability and GnRH pulsatility. The cycling of cAMP levels is a known timing mechanism for reproduction in the slime mold (53) and was proposed as a mechanism in timing the rate of contraction of the heart (54). To date direct measurements of cAMP levels in GT1 GnRH neurons have not been achieved. However, global fluctuations of cAMP concentrations in GT1 cells may not reflect the precision of localized concentrations of cAMP mediated through the assembly of macromolecular complexes involving AKAP150. Currently available cAMP sensors facilitating the measurement of cAMP concentrations in specific microdomains may be more informative (for review see Ref. 55).
No changes in the magnitude of spontaneous GnRH pulses was consistently observed in GT1 cells after decreased AKAP150 and CNG channel expression and increased PDE4D1 expression. Clearly large global pharmacologically induced increases in cAMP levels induced by forskolin or 8-bromoadenosine-cAMP caused dramatic increases in GnRH release from GT1 cells (56). The physiological relevance of pharmacological induced changes have frequently been questioned, but the emergence of data on the importance of the organization of signaling complexes in discrete domains further challenges the relevance of these findings. Spontaneous GnRH pulses are likely to be regulated by highly organized signaling complexes within discrete intracellular domains, a hypothesis consistent with the findings of this paper. The magnitude of pulses may be restricted by the participation of only small populations of signaling molecules organized within these signaling complexes.
We conclude that AKAP150 plays a significant role in organizing cAMP signaling complexes involved in regulating GT1 cell excitability and the frequency of intrinsic pulsatile GnRH secretion. Additional experiments addressing the dynamics of bidirectional regulation of cAMP signaling molecules are necessary to draw further conclusions. These results also raise numerous questions on the organization of additional signaling pathways involved. Understanding the spatial and temporal organization of signaling complexes will vastly increase our understanding of the regulation of intrinsic pulsatile secretion.
Acknowledgments
We acknowledge Lili Zhang for the analysis of the Ca2+ data and development of the JPULSAR software, a program modified from PULSAR (32), to detect peaks in [Ca2+]i oscillations.
Footnotes
This work was supported by National Institutes of Health Grant HD041996.
Disclosure Summary: Q.C., R.I.W., and B.E.B. have nothing to declare.
First Published Online November 3, 2009
Abbreviations: AC, Adenylyl cyclase; AKAP, A-kinase anchoring protein; [Ca2+]i, intracellular Ca2+ concentration; Cav, voltage sensitive Ca2+ channel; CNG, cyclic nucleotide gated channel; GFP, green fluorescent protein; luc, luciferase; MOI, multiplicity of infection; PDE, phosphodiesterase; PKA, protein kinase A; PKC, protein kinase C; PP2B, calcium/calmodulin-dependent phosphatase; RFP, red fluorescent protein; siRNA, small interfering RNA.
References
- Duittoz AH, Batailler M 2000 Pulsatile GnRH secretion from primary cultures of sheep olfactory placode explants. J Reprod Fertil 120:391–396 [PubMed] [Google Scholar]
- Funabashi T, Daikoku S, Shinohara K, Kimura F 2000 Pulsatile gonadotropin-releasing hormone (GnRH) secretion is an inherent function of GnRH neurons, as revealed by the culture of medial olfactory placode obtained from embryonic rats. Neuroendocrinology 71:138–144 [DOI] [PubMed] [Google Scholar]
- Terasawa E, Keen KL, Mogi K, Claude P 1999 Pulsatile release of luteinizing hormone-releasing hormone (LHRH) in cultured LHRH neurons derived from the embryonic olfactory placode of the rhesus monkey. Endocrinology 140:1432–1441 [DOI] [PubMed] [Google Scholar]
- Krsmanović LZ, Stojilković SS, Merelli F, Dufour SM, Virmani MA, Catt KJ 1992 Calcium signaling and episodic secretion of gonadotropin-releasing hormone in hypothalamic neurons. Proc Natl Acad Sci USA 89:8462–8466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez de la Escalera G, Choi AL, Weiner RI 1992 Generation and synchronization of gonadotropin-releasing hormone (GnRH) pulses: intrinsic properties of the GT1-1 GnRH neuronal cell line. Proc Natl Acad Sci USA 89:1852–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetsel WC, Valença MM, Merchenthaler I, Liposits Z, López FJ, Weiner RI, Mellon PL, Negro-Vilar A 1992 Intrinsic pulsatile secretory activity of immortalized luteinizing hormone-releasing hormone-secreting neurons. Proc Natl Acad Sci USA 89:4149–4153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez de la Escalera G, Choi AL, Weiner RI 1992 β1-Adrenergic regulation of the GT1 gonadotropin-releasing hormone (GnRH) neuronal cell lines: stimulation of GnRH release via receptors positively coupled to adenylate cyclase. Endocrinology 131:1397–1402 [DOI] [PubMed] [Google Scholar]
- Uemura T, Nishimura J, Yamaguchi H, Hiruma H, Kimura F, Minaguchi H 1997 Effects of noradrenaline on GnRH-secreting immortalized hypothalamic (GT1-7) neurons. Endocr J 44:73–78 [DOI] [PubMed] [Google Scholar]
- Vitalis EA, Costantin JL, Tsai PS, Sakakibara H, Paruthiyil S, Iiri T, Martini JF, Taga M, Choi AL, Charles AC, Weiner RI 2000 Role of the cAMP signaling pathway in the regulation of GnRH secretion in GT1 cells. Proc Natl Acad Sci USA 97:1861–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles AC, Hales TG 1995 Mechanisms of spontaneous calcium oscillations and action potentials in immortalized hypothalamic (GT1-7) neurons. J Neurophysiol 73:56–64 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Beltran-Parrazal L, Butler P, Conti M, Charles AC, Weiner RI 2003 Lowering cyclic adenosine-3′,5′-monophosphate (cAMP) levels by expression of a cAMP-specific phosphodiesterase decreases intrinsic pulsatile gonadotropin-releasing hormone secretion from GT1 cells. Mol Endocrinol 17:1982–1990 [DOI] [PubMed] [Google Scholar]
- Paruthiyil S, El Majdoubi M, Conti M, Weiner RI 2002 Phosphodiesterase expression targeted to gonadotropin-releasing hormone neurons inhibits luteinizing hormone pulses in transgenic rats. Proc Natl Acad Sci USA 99:17191–17196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Majdoubi M, Weiner RI 2002 Localization of olfactory cyclic nucleotide-gated channels in rat gonadotropin-releasing hormone neurons. Endocrinology 143:2441–2444 [DOI] [PubMed] [Google Scholar]
- Charles A, Weiner R, Costantin J 2001 cAMP modulates the excitability of immortalized hypothalamic (GT1) neurons via a cyclic nucleotide gated channel. Mol Endocrinol 15:997–1009 [DOI] [PubMed] [Google Scholar]
- Beene DL, Scott JD 2007 A-kinase anchoring proteins take shape. Curr Opin Cell Biol 19:192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua ML, Smith KE, Gorski JA, Horne EA, Gibson ES, Gomez LL 2006 Regulation of neuronal PKA signaling through AKAP targeting dynamics. Eur J Cell Biol 85:627–633 [DOI] [PubMed] [Google Scholar]
- Dodge-Kafka KL, Bauman A, Kapiloff MS 2008 A-kinase anchoring proteins as the basis for cAMP signaling. Handb Exp Pharmacol 186:3–14 [DOI] [PubMed] [Google Scholar]
- Smith FD, Langeberg LK, Scott JD 2006 The where’s and when’s of kinase anchoring. Trends Biochem Sci 31:316–323 [DOI] [PubMed] [Google Scholar]
- Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DM, Dessauer CW, Scott JD 2006 Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell 23:925–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser ID, Cong M, Kim J, Rollins EN, Daaka Y, Lefkowitz RJ, Scott JD 2000 Assembly of an A kinase-anchoring protein-β(2)-adrenergic receptor complex facilitates receptor phosphorylation and signaling. Curr Biol 10:409–412 [DOI] [PubMed] [Google Scholar]
- Gardner LA, Tavalin SJ, Goehring AS, Scott JD, Bahouth SW 2006 AKAP79-mediated targeting of the cyclic AMP-dependent protein kinase to the β1-adrenergic receptor promotes recycling and functional resensitization of the receptor. J Biol Chem 281:33537– 33553 [DOI] [PubMed] [Google Scholar]
- Malbon CC, Tao J, Wang HY 2004 AKAPs (A-kinase anchoring proteins) and molecules that compose their G-protein-coupled receptor signalling complexes. Biochem J 379:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman AL, Goehring AS, Scott JD 2004 Orchestration of synaptic plasticity through AKAP signaling complexes. Neuropharmacology 46:299–310 [DOI] [PubMed] [Google Scholar]
- Higashida H, Hoshi N, Zhang JS, Yokoyama S, Hashii M, Jin D, Noda M, Robbins J 2005 Protein kinase C bound with A-kinase anchoring protein is involved in muscarinic receptor-activated modulation of M-type KCNQ potassium channels. Neurosci Res 51:231–234 [DOI] [PubMed] [Google Scholar]
- Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW 2007 Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry 46:1635–1646 [DOI] [PubMed] [Google Scholar]
- Oliveria SF, Dell'Acqua ML, Sather WA 2007 AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 55:261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, Scott JD 1995 Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science 267:108–111 [DOI] [PubMed] [Google Scholar]
- Dell'Acqua ML, Dodge KL, Tavalin SJ, Scott JD 2002 Mapping the protein phosphatase-2B anchoring site on AKAP79. Binding and inhibition of phosphatase activity are mediated by residues 315–360. J Biol Chem 277:48796–48802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, Scott JD 2008 Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci USA 105:12557–12562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Paruthiyil S, Butler P, Weiner RI 2004 Role of cAMP signaling in the mediation of dopamine-induced stimulation of GnRH secretion via D1 dopamine receptors in GT1-7 cells. Neuroendocrinology 80:2–10 [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY 1985 A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260:3440–3450 [PubMed] [Google Scholar]
- Merriam GR, Wachter KW 1982 Algorithms for the study of episodic hormone secretion. Am J Physiol 243:E310–E318 [DOI] [PubMed] [Google Scholar]
- Ostroveanu A, Van der Zee EA, Dolga AM, Luiten PG, Eisel UL, Nijholt IM 2007 A-kinase anchoring protein 150 in the mouse brain is concentrated in areas involved in learning and memory. Brain Res 1145:97–107 [DOI] [PubMed] [Google Scholar]
- Krsmanovic LZ, Mores N, Navarro CE, Tomić M, Catt KJ 2001 Regulation of ca(2+)-sensitive adenylyl cyclase in gonadotropin-releasing hormone neurons. Mol Endocrinol 15:429–440 [DOI] [PubMed] [Google Scholar]
- Martin C, Jacobi JS, Nava G, Jeziorski MC, Clapp C, Martínez de la Escalera G 2007 GABA inhibition of cyclic AMP production in immortalized GnRH neurons is mediated by calcineurin-dependent dephosphorylation of adenylyl cyclase 9. Neuroendocrinology 85:257–266 [DOI] [PubMed] [Google Scholar]
- Iwami G, Kawabe J, Ebina T, Cannon PJ, Homcy CJ, Ishikawa Y 1995 Regulation of adenylyl cyclase by protein kinase A. J Biol Chem 270:12481–12484 [DOI] [PubMed] [Google Scholar]
- Cooper DM, Crossthwaite AJ 2006 Higher-order organization and regulation of adenylyl cyclases. Trends Pharmacol Sci 27:426–431 [DOI] [PubMed] [Google Scholar]
- Biel M, Zong X, Distler M, Bosse E, Klugbauer N, Murakami M, Flockerzi V, Hofmann F 1994 Another member of the cyclic nucleotide-gated channel family, expressed in testis, kidney, and heart. Proc Natl Acad Sci USA 91:3505–3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL 2003 Epac: a new cAMP target and new avenues in cAMP research. Nat Rev Mol Cell Biol 4:733–738 [DOI] [PubMed] [Google Scholar]
- Tao J, Shumay E, McLaughlin S, Wang HY, Malbon CC 2006 Regulation of AKAP-membrane interactions by calcium. J Biol Chem 281:23932–23944 [DOI] [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser ID, Cone RD, Scott JD 1992 Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP 79. J Biol Chem 267:16816–16823 [PubMed] [Google Scholar]
- Sarkar D, Erlichman J, Rubin CS 1984 Identification of a calmodulin-binding protein that co-purifies with the regulatory subunit of brain protein kinase II. J Biol Chem 259:9840–9846 [PubMed] [Google Scholar]
- Faux MC, Rollins EN, Edwards AS, Langeberg LK, Newton AC, Scott JD 1999 Mechanism of A-kinase-anchoring protein 79 (AKAP79) and protein kinase C interaction. Biochem J 343(Pt 2):443–452 [PMC free article] [PubMed] [Google Scholar]
- Blackman BE, Yoshida H, Paruthiyil S, Weiner RI 2007 Frequency of intrinsic pulsatile gonadotropin-releasing hormone secretion is regulated by the expression of cyclic nucleotide-gated channels in GT1 cells. Endocrinology 148:3299–3306 [DOI] [PubMed] [Google Scholar]
- Bosma MM 1993 Ion channel properties and episodic activity in isolated immortalized gonadotropin-releasing hormone (GnRH) neurons. J Membr Biol 136:85–96 [DOI] [PubMed] [Google Scholar]
- Costantin JL, Charles AC 2001 Modulation of Ca(2+) signaling by K(+) channels in a hypothalamic neuronal cell line (GT1-1). J Neurophysiol 85:295–304 [DOI] [PubMed] [Google Scholar]
- Sakakibara H, Conti M, Weiner RI 1998 Role of phosphodiesterases in the regulation of gonadotropin-releasing hormone secretion in GT1 cells. Neuroendocrinology 68:365–373 [DOI] [PubMed] [Google Scholar]
- Terrenoire C, Houslay MD, Baillie GS, Kass RS 2009 The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J Biol Chem 284:9140–9146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JW, Wyszynski M, Madhavan R, Sealock R, Kim JU, Sheng M 1998 Yotiao, a novel protein of neuromuscular junction and brain that interacts with specific splice variants of NMDA receptor subunit NR1. J Neurosci 18:2017–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piggott LA, Bauman AL, Scott JD, Dessauer CW 2008 The A-kinase anchoring protein Yotiao binds and regulates adenylyl cyclase in brain. Proc Natl Acad Sci USA 105:13835–13840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner LA, Naren AP, Bahouth SW 2007 Assembly of an SAP97-AKAP79-cAMP-dependent protein kinase scaffold at the type 1 PSD-95/DLG/ZO1 motif of the human β(1)-adrenergic receptor generates a receptosome involved in receptor recycling and networking. J Biol Chem 282:5085–5099 [DOI] [PubMed] [Google Scholar]
- Dart C, Leyland ML 2001 Targeting of an A kinase-anchoring protein, AKAP79, to an inwardly rectifying potassium channel, Kir2.1. J Biol Chem 276:20499–20505 [DOI] [PubMed] [Google Scholar]
- Halloy J, Lauzeral J, Goldbeter A 1998 Modeling oscillations and waves of cAMP in Dictyostelium discoideum cells. Biophys Chem 72:9–19 [DOI] [PubMed] [Google Scholar]
- Sculptoreanu A, Rotman E, Takahashi M, Scheuer T, Catterall WA 1993 Voltage-dependent potentiation of the activity of cardiac L-type calcium channel α1 subunits due to phosphorylation by cAMP-dependent protein kinase. Proc Natl Acad Sci USA 90:10135–10139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrera M, Dodoni G, Monterisi S, Pertegato V, Zamparo I, Zaccolo M 2008 A toolkit for real-time detection of cAMP: insights into compartmentalized signaling. Handb Exp Pharmacol 186:285–298 [DOI] [PubMed] [Google Scholar]
- Martínez de la Escalera G, Choi AL, Weiner RI 1995 Signaling pathways involved in GnRH secretion in GT1 cells. Neuroendocrinology 61:310–317 [DOI] [PubMed] [Google Scholar]