

Abstract
Accumulating evidence shows that estrogens are protective factors in inflammatory lung diseases and are involved in the gender-related incidence of these pathologies. The aim of this study was to identify which estrogen receptor (ER), ER-α and/or ERβ, mediates hormone antiinflammatory effects in lung and how gender or aging modify this effect. Acute lung inflammation in wild type, ERα or ERβ knockout animals was induced by pleural injection of carrageenan; female mice were used and sham operated, ovariectomized, or ovariectomized and treated with 17β-estradiol (E2) before carrageenan. Our data show that ERα, and not ERβ, mediates E2-induced reduction of the inflammatory response. By real-time PCR and immunohistochemistry assays, we demonstrate ERα expression in the resident and infiltrated inflammatory cells of the lung, in which ERβ could not be detected. In these cells, E2-mediated reduction in the expression of inflammatory mediators was also due to ERα. In parallel, we observed that female mice were more prone to inflammation as compared with males, suggesting a gender-related difference in lung susceptibility to inflammatory stimuli, whereas the effect of E2 was similar in the two sexes. Interestingly, aging results in a strong increase in the inflammatory response in both sexes and in the disruption E2/ERα signaling pathway. In conclusion, our data reveal that E2 is able to regulate lung inflammation in a gender-unrelated, age-restricted manner. The specific involvement of ERα in hormone action opens new ways to identify drug targets that limit the inflammatory component of lung pathologies.
Activation of a specific intracellular receptor, estrogen receptor-αga, in inflammatory cells reduces lung inflammation in a gender-independent, age-restricted manner.
Several aspects of lung development, homeostasis, and physiopathology are regulated by estrogens. Sex differences related to lung maturation, such as alveolar type II cell activity in surfactant production or ion channel expression in the respiratory epithelium, have been extensively studied and reconciled with a direct effect of sex steroid hormones on the developing lung structures, with estrogens displaying stimulatory effects (1,2,3). Similarly, gender differences in the lung of sexually mature animals, including size and function of respiratory structures and their responsiveness to cholinergic stimulation, are controlled by estrogens (4). In line with the above-mentioned effects, interstitial and airway lung diseases were also reported to be modulated by estrogens, which either contribute or protect against disease pathogenesis, depending on the disease involved (5,6). These experimental data provide strong support to the evidence that human lung disorders are influenced by circulating levels of estrogens, which seem to affect the prevalence and severity of lung pathologies such as fibrosis, asthma, infection, and cancer (7).
Inflammation is a hallmark of lung diseases; asthma, chronic obstructive pulmonary disease, and cystic fibrosis are chronic inflammatory lung diseases each characterized by the involvement of specific molecular mediators and cellular components of inflammation (8,9). In addition, contaminant molecules that foster inflammation have been shown to exacerbate the development and severity of lung diseases. Thus, managing airway inflammation is a valuable adjunct to pulmonary therapy and an attractive field for identifying novel therapeutic targets, also considering the insensitivity of some lung disease patients to corticosteroids.
Estrogens exert their effects through the interaction with two intracellular receptors, estrogen receptor (ER)-α and ERβ. These receptors act both as potent regulators of gene transcription and as direct modulators of enzymatic complexes residing in the cytoplasm (10). Genetic manipulation of ER genes in mice allowed further understanding of the key role of ERs in lung development and physiology through distinct gene transcriptional programs (11,12).
The physiological reduction in estrogens level that occurs at menopause is associated with a general increase in the inflammatory responsiveness and exposes women to a higher risk for pathologies, such as those affecting bone and cardiovascular or central nervous systems, which are associated with inflammation (13). Our previous observations showed the influence of 17β-estradiol (E2) on inflammatory injury of the lung induced by carrageenan (CAR) injection and the involvement of ERs in protective effects of hormone; similarly, other studies addressed the positive influence of estrogens on acute lung injury models (14,15).
Despite the potency of estrogens in modulating lung inflammation and the role of the inflammatory system in lung pathologies, the specific role of each of the ERs is not yet understood. In addition, considering the negative effect that aging has on tissue integrity, possibly through an increased damage triggered by a sustained inflammatory status linked with senescence, the extent to which aging and gender modify lung responsiveness to estrogens still needs to be defined.
Therefore, we investigated the role of ERs in inflammatory lung injury and assessed the role of gender and aging in estrogens action. Our results demonstrate that activation of ERα in lung inflammatory cells might provide beneficial effects in adult, but not in aged, mice of both genders, suggesting a key role of the estrogens signaling pathway in the physiology and therapeutic opportunities of lung inflammation.
Materials and Methods
Animals and treatments
Wild-type female and male mice used throughout this study were obtained from the breeding of ERα and ERβ heterozygous mice, previously described (16). Mice have a C57B6 genetic background and were housed in the animal care facility of the Department of Pharmacological Sciences of Milan, Italy. All animal protocols were approved by the Institutional Animal Care and Use Committee at the Department of Pharmacological Sciences of the University of Milan and were in accordance with the European legislation. At 5 months of age, mice were bilaterally gonadectomized and 3 wk later injected sc with vehicle (purified corn oil) or 50 μg/kg E2 (Sigma, Milan, Italy) 2 h before the injection of CAR (Sigma). Anesthesia was induced by a sc injection of a mixture of ketamine and xylazine (78 and 6 mg/kg, respectively); higher doses (234 and 18 mg/kg) were used for lethal anesthesia. Experiments were repeated at least two times; each experimental group consisted of five to eight animals.
Materials
Unless otherwise stated, all compounds were obtained from Sigma and were of the highest commercial grade available. All stock solutions were prepared in nonpyrogenic saline.
CAR-induced pleurisy
CAR-induced pleurisy was performed as previously described (17). Mice were anesthetized and a skin incision was made at the level of the left sixth intercostals space. The underlying muscle was dissected, and saline (0.1 ml) or saline containing 2% (wt/vol) CAR (0.1 ml) was injected into the pleural cavity. The skin incision was closed with a suture and the animals were allowed to recover. After 4 h, animals were euthanized by inhalation of CO2. The chest was carefully opened and the pleural cavity rinsed with 1 ml of saline solution containing heparin (5 U/ml−1) and indomethacin (10 μg/ml). The exudate and washing solution were removed by aspiration and the total volume measured. Any exudate that was contaminated with blood was discarded. The amount of exudate was calculated by subtracting the volume injected (1 ml) from the total volume recovered. The leukocytes in the exudate were suspended in PBS and counted with an optical microscope in a Burker’s chamber after Blue Toluidine staining.
Histological examination
Lung biopsies were fixed for 1 wk in buffered formaldehyde solution (10% in PBS) at room temperature, dehydrated by graded ethanol, and embedded in Paraplast (Sherwood Medical, Mahwah, NJ). Serial sections (7 μm) in sagittal plane of the whole right lobe were cut using a microtome (Microtom HM 310; Zeiss, Milan, Italy); for each animal, two sections were analyzed that were about 25 μm distant from each other, and five fields of 700 × 900 μm each were scored in each section. All animals of each experimental group were examined. Sections were deparaffinized with xylene, stained with hematoxylin and eosin, and studied using light microscopy Axiovision (Zeiss). The following morphological criteria were used for scoring lung histology under light microscopy: grade 0, normal lung; grade 1, minimal edema or inflammatory cell infiltration through alveolar or bronchiolar walls; grade 3, moderate edema and inflammatory cell infiltration without obvious damage to lung architecture; and grade 4, severe inflammatory cell infiltration with obvious damage to lung architecture. All histological studies were performed in a blinded fashion.
Myeloperoxidase (MPO) activity
MPO activity, an indicator of polymorphonucleated (PMN) cell accumulation, was determined as previously described (18). At the specified time after injection of CAR, lung tissues were obtained and weighed, each piece homogenized in a solution containing 0.5% (wt/vol) hexadecyltrimethyl-ammonium bromide dissolved in 10 mm potassium phosphate buffer (pH 7) and centrifuged for 30 min at 20,000 × g at 4 C. An aliquot of the supernatant was then allowed to react with a solution of tetramethylbenzidine (1.6 mm) and 0.1 mm hydrogen peroxide. The rate of change in absorbance was measured by spectrophotometry at 650 nm wavelength. MPO activity was defined as the quantity of enzyme degrading 1 μmol peroxide per minute−1 at 37 C and was expressed in milliunts per gram weight of wet tissue.
Measurement of cytokines
Evaluation of TNFα levels in the exudate was carried out using a colorimetric commercial ELISA kit (Calbiochem-Novabiochem Corp., Milan, Italy).
Immunohistochemistry (IHC) of intercellular adhesion molecule (ICAM)-1, TNFα, ERα, and nitrotyrosine
Tissues were processed as for histological examination. After deparaffinization, endogenous peroxidase was quenched with 0.3% (vol/vol) hydrogen peroxide in 60% (vol/vol) methanol for 30 min. The sections were permeablized with 0.1% (wt/vol) Triton X-100 in PBS for 20 min. Nonspecific adsorption was minimized by incubating the section in 2% (vol/vol) normal goat serum in PBS for 20 min. Endogenous biotin or avidin binding sites were blocked by sequential incubation for 15 min with biotin and avidin (DBA, Milan, Italy), respectively. Sections were incubated overnight with the appropriate antibody: purified hamster antimouse ICAM-1 [1:500 in PBS (wt/vol); Santa Cruz Biotechnology, Santa Cruz, CA, catalog no. SC-789]; anti-TNFα antibody [1:500 in PBS (vol/vol); Santa Cruz Biotechnology, catalog no. Sc-1350]; antinitrotyrosine rabbit polyclonal antibody [1:500 in PBS (vol/vol); Millipore, Milan, Italy, catalog no. 06-284]; anti-ERα polyclonal antibody [1:500 in PBS (vol/vol); Santa Cruz Biotechnology, catalog no. Sc-542]. Sections were washed with PBS and incubated with secondary antibody. Specific labeling was detected with a biotin-conjugated goat antirabbit IgG and avidin-biotin peroxidase complex (Santa Cruz Biotechnology; catalog no. Sc-7891). To confirm that the immunoreaction for the nitrotyrosine was specific, some sections were also incubated with the primary antibody (antinitrotyrosine) in the presence of excess nitrotyrosine (10 mm) to verify the binding specificity. To verify the binding specificity for the other antibodies, two sections were tested by IHC by omitting either the secondary or the primary antibodies. At least two sections were analyzed for each animal and each primary antibody. Densitometry analysis of IHC images was carried out using Optilab Graftek (Graftek, Voisin Le Bretonneux, France) software on a Macintosh (Milan, Italy) personal computer (CPU G3–266).
Preparation of bone marrow-derived hematopoietic cells (BMDCs) and alveolar macrophages
BMDCs
Female and male mice 4–6 months of age and 18-month-old female mice were deeply anesthetized, and BMDCs were obtained from the femurs and tibias. Briefly, legs were dissected from the animals, bones were cleaned, and marrows were flushed with Hanks’ balanced salt solution (HBSS) without calcium, magnesium, bicarbonate, or phenol red (Mediatech, Inc., Herndon, VA) using a 25-gauge needle. After gently dislodging cell aggregates with a plastic Pasteur pipette, cells were washed twice in HBSS and subjected to hypotonic lysis of red blood cells by resuspending in 1 ml of 34 mm sodium chloride, followed by the addition of 1 ml of 0.3 m sodium chloride/0.01 m sucrose and washing in Ca/Mg-free HBSS. Cells were finally placed into a 1.5-ml Eppendorf tube and centrifuged at 11,750 × g for 5 min at room temperature to elute marrow cells. Cells were immediately processed for RNA extraction.
Alveolar macrophages
Cell were harvested from both females and males mice at the age of 4–6 months and from 18-month-old female mice. Animals were deeply anesthetized and free cells were recovered from the lungs by bronchoalveolar lavage, which was performed through a tracheal incision by four sequential washes with 0.5 ml sterile physiological saline solution at room temperature injected. Cells were collected by centrifugation at 11,750 × g for 5 min and immediately processed for RNA extraction.
RNA preparation and real-time PCR
Total RNA was purified from BMDCs or liver and lung using RNeasy minikit (QIAGEN, Milan, Italy) according to the manufacturer’s instructions including a step with deoxyribonuclease incubation. Liver was homogenized using a rotor-stator homogenizer (Ultra-Turrax T10; IKA-WERKE GMBH & Co., Staufen, Germany), whereas lung was homogenized using potter (Wheaton, Millville, NJ). One microgram RNA was used for cDNA preparation using the Moloney murine leukemia virus reverse transcriptase (Promega, Milan, Italy) as previously described (19). Control reactions without addition of the enzyme were performed for each sample. Five microliters of a 1:25 dilution of the cDNA were amplified using TaqMan technology. PCR was carried out in triplicate on 96-well plate using TaqMan 2× universal PCR master mix No AmpErase UNG (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol using 7900HT fast real-time PCR system (Applied Biosystems) with the following thermal profile: 2 min at 50 C; 10 min at 95 C; 40 cycles (15 sec at 95 C, 1 min at 60 C). Data were analyzed using the sequence detection system software version 2.3 (Applied Biosystems) and the 2-ΔΔCt method (20). Gene expression assays (Applied Biosystems) were used for Esr1 (Mm00433149_m1) and Esr2 (Mm01281854_m1) gene amplifications and for the reference gene 36B4 (forward primer, 5′-GGCGACCTGGAAGTCCAACT-3′; reverse primer, 5′-CCATCAGCACCACGGCCTTC-3′; probe, ATCTGCTGCAT-CTGCTTGGAGCCCA, 5′-fluor label, 6-VIC).
Statistical evaluation
All values are expressed as mean ± sem of n observations. The results were analyzed by one-way ANOVA followed by a Bonferroni post hoc test for multiple comparisons. P < 0.05 was considered significant.
Results
Endogenous estrogen and lung inflammation
CAR-induced pleurisy is a well-accepted animal model of acute lung inflammation, which consists of a cellular exudate formation mainly made of PMN cells that invade lung parenchyma. To determine the role of endogenous estrogens, ovaries were removed to eliminate their endogenous production. Sham-operated or ovariectomized (ovx) animals were injected with CAR in the pleural cavity. As shown in Fig. 1A, histological inspection of the lungs showed the presence of numerous round-shaped cells in close proximity of blood vessels, in addition to edema and epithelial lesions in both sham and ovx animals. A histological score was used to grade this inflammatory reaction, which is shown in Fig. 1B. In parallel, PMN cells recovered from the pleural exudate are shown in Fig. 1C. No macroscopic differences were noted in the lungs of the two groups, whereas an increase in the number of PMN cells was observed in ovx mice, suggesting that ovary removal increases the inflammatory response in the lungs. Inflammation was absent in CAR-untreated sham and ovx mice, with a histological score of 0 and about 0.5 × 10−6 PMN cells per milliliter in both groups (data not shown).
Figure 1.

Estrogen and lung inflammation in female mice. A, Histology of lung sections stained with hematoxylin-eosin obtained from CAR-treated sham, ovx, or ovx females mice treated with E2 (ovx+E2). Representative samples are shown with magnifications on the right. B, As measure of tissue morphology, a histological score was assigned to each animal through the assessment of histological parameters during tissue observation at the microscope, as described in Materials and Methods. Sham, Open boxes; ovx, black boxes; ovx+E2, dashed boxes. C, Pleural exudate was recovered and analyzed for infiltrated PMN cell number; bar legend as in B). Bars represent the average ± sem of all the animals (n = 8), each analyzed in triplicate. *, P < 0.05; ***, P < 0.001.
To link more specifically the effect of ovx with the lack of endogenous estrogens, a group of ovx animals received an injection of 50 μg/kg E2 2 h before CAR injection (17). E2-treated animals showed both an improvement in the histological score (Fig. 1, A and B) and a lower number of infiltrated cells when compared with ovx mice; interestingly, these parameters were even lower than those observed in the sham group (Fig. 1C). CAR-untreated ovx+E2 mice had no inflammatory signs (data not shown).
Altogether these results suggest that deprivation of endogenous estrogens increases the inflammatory response in the mouse lung and that E2 is an effective pharmacological agent to reduce lung inflammation. These results are in agreement with our previous observation on the effect of E2 on CAR-induced pleurisy in female rats (17).
ERs and lung inflammation
Because we established that E2 is involved in lung inflammation, we asked which ER mediated this effect. Pleurisy was induced with CAR injection in ER-knockout animals (ER-KO) which were either sham-operated, ovx, or ovx injected with E2 before pleurisy induction. Representative histological sections of ER-KO mice are shown in Fig. 2, A and B. Ovx per se did not alter the histological score, PMN cell infiltration, and activity (as measured by MPO levels) in ER-KO mice (Fig. 2, C–E). Interestingly, the effect of E2 on lung architecture and inflammatory parameters was absent in ERα-KO mice. In fact, the histological structure as well as PMN infiltration and MPO levels were not reduced by E2 administration in ERα-KO mice (Fig. 2, C–E). On the other hand, inflammation was still regulated by E2 in ERβ-KO mice similarly to wild-type (wt) animals. It is important to underline that CAR-untreated ER-KO mice did not reveal any difference in tissue morphology or cellular components as compared with wild-type animals (data not shown). These data are in contrast to the observation by Patrone et al. (12), in which female ERβ-KO mice showed impaired lung morphology. However, animals in that study were 1 yr old; thus, it is possible that females ERβ-KO mice show alterations in lung physiology in relation with aging.
Figure 2.

Role of ER isoforms in lung inflammation. A and B, Histology of lung sections stained with hematoxyline-eosin from ERα- and ERβ-KO mice injected with CAR in the pleural space. Representative samples are shown. C, Histological score. D and E, Pleural exudate was recovered and analyzed (D) for infiltrated PMN cell number and (E) for MPO. Bars represent the average ± sem of all the animals (wt, n = 8; ERα- and ERβ-KO, sham and ovx n = 8; ovx+E2 n = 5), each analyzed in triplicate. *, P < 0.05; ***, P < 0.001.
Altogether these results show that the antiinflammatory effect of E2 in this experimental model of lung inflammation is mediated by ERα, not ERβ.
ER expression in lung and inflammatory cells
The involvement of ERα in regulating lung responsiveness was unexpected due to the fact that it has been shown that ERβ is the most abundant ER in lung (21). We thus reasoned that the effect of E2 could be mediated by a small population of ERα-positive cells, residing and/or infiltrating in lung. We thus analyzed ERα expression by IHC on lung tissue using an ERα-specific antibody. This assay revealed the expression of ERα in the nucleus of endothelial cells along lung vessels (Fig. 3A). After CAR injection, ERα labeling is observed in the nuclei of infiltrated leukocytes (Fig. 3B) and endothelial cells, suggesting that the intracellular localization of the receptor is not modified by CAR ± E2. These IHC data thus demonstrate that ERα protein is expressed by endothelial cells and myeloid cells that infiltrate lung parenchyma during inflammation.
Figure 3.

ERα expression in lung and inflammatory cells. Representative immunohistochemistry images of lung from vehicle (A) and CAR-treated sham (B) or untreated (C) ERα-KO female mice analyzed with an ERα-specific antibody. ERα-positive cells are visible in endothelial cells of the vessel wall (arrow in A) and in round-shaped cells in proximity of vessels (arrow in B). D, Expression levels of the ERα gene was evaluated by real-time PCR on the mRNA extracted from BMDCs, alveolar macrophages (alv. MØ), and lung tissue and represented in relation with ERα mRNA from liver. Each data point represent the average ± sem of samples from three female mice analyzed in triplicate.
To confirm and extend these evidences to other lung inflammatory cells, which are eliminated by washings during IHC, we analyzed ERα expression at the mRNA levels in BMDCs and alveolar macrophages, which are the blood-derived and resident innate immunity cells of the lung, respectively. Real-time PCR analyses revealed that ERα is expressed in alveolar macrophages and BMDCs freshly isolated from female mice; expression in these cells is represented in comparison with that of liver, a classic ERα target tissue (Fig. 3D). Considerable amounts of ERα mRNA are also found in lung, which sustains previous evidence (21) and our present data on ERα expression in lung endothelial cells (Fig. 3A). CAR injection does not significantly change ERα mRNA levels in lung tissue (data not shown), suggesting that transcription of ERα gene is not modulated by pleural inflammation, at least in the experimental conditions used in this study, and that the infiltration of ERα-positive cells after CAR treatment (Fig. 3B) does not significantly increase overall ERα mRNA levels. Consistently with the dispensable role of ERβ reported in Figure 2, mRNA levels of this receptor were undetectable by real-time PCR in alveolar macrophages or BMDCs, whereas its expression in lung did not change after CAR injection (data not shown).
Altogether our data show that the effect of E2 on lung inflammation can be ascribed to ER-α expressed in inflammatory cells of the lung, residing in the air compartment or recruited from the bloodstream, and in vascular endothelial cells.
ERs and expression of lung inflammatory mediators
With the aim to demonstrate that ERα is active in lung inflammatory cells and mediates the observed antiinflammatory effect of E2, we evaluated the expression levels of inflammatory mediators induced by CAR injection in control or E2-treated wt and ER-KO mice. The short time frame of our experimental design and the use of IHC allowed us to directly link and visualize E2 activity within specific cell types. Lung tissue sections from intact wt, ERα-KO, and ERβ-KO mice injected with CAR were analyzed by IHC using antibodies against ICAM-1, a protein involved in leukocyte-endothelial adhesion; a specific staining along the vascular endothelium in sections from wt (data not shown), ERα-KO, and ERβ-KO mice was detected after CAR injection in both sham and ovx female mice (Fig. 4, A and B); through a semiquantitative analysis of the IHC images, we observed that the ERα-KO genotype is generally associated with a higher ICAM-1 expression in all experimental groups as compared with wt, whereas ERβ-KO mice had a similar profile as wt (Fig. 4D).
Figure 4.

ICAM-1 immunolabeling in lung of ER-KO animals. Lung tissue from ERα- (A) or ERβ-KO (B) mice, sham, ovx, or ovx+E2, were analyzed by IHC after CAR injection with specific antibody against ICAM-1. C, IHC of lung sections from ERα- and ERβ-KO untreated mice. Representative immunhistochemistry images are shown. Densitometry evaluation (D) of ICAM-1 immunolabeling in wt, ERα-KO, and ERβ-KO mice. Bar legend as in Fig. 1B. Data are expressed as means of percent of total tissue area ± sem. * vs. sham, ^ vs. ovx; ° vs. corresponding treatment in wt. **, °°, P < 0.01; ***, °°°, P < 0.001.
E2 treatment is highly potent in reducing ICAM-1 labeling in both wt and ERβ-KO, whereas it is ineffective in ERα-KO. Similar results were obtained when we assayed the expression of another adhesion molecule, P-selectin (supplemental Fig. S1, published as supplemental data on The Endocrine Society’s Journals Online web site at http://endo.endojournals.org). These results suggest that indeed endothelial cells of the lung vessels are endogenous targets for the antiinflammatory activity of E2 through the selective involvement of ERα. However, because ovariectomy results in increased ICAM-1 levels in ERα-KO mice, a role for ERβ in the lung inflammatory response cannot be excluded, at least in this genetically modified animal model.
We next evaluated the expression of TNFα, an inflammatory cytokine that is immediately synthesized by inflammatory cells in response to stimuli. The IHC analysis revealed the expression of TNFα in endothelial cells of the vessel wall and in infiltrated leukocytes after CAR injection (supplemental Fig. S2). Quantification of IHC labeling images showed that the ERα-KO genotype is associated with higher levels of TNFα expression in all three experimental groups as compared with wt, whereas the absence of ERβ does not alter E2 responsiveness of lung cells (Fig. 5A). Again these data show that ERα is involved in E2 action in lung inflammation, whereas ERβ seems to be involved in lung responsiveness after hormone deprivation in the absence of ERα.
Figure 5.
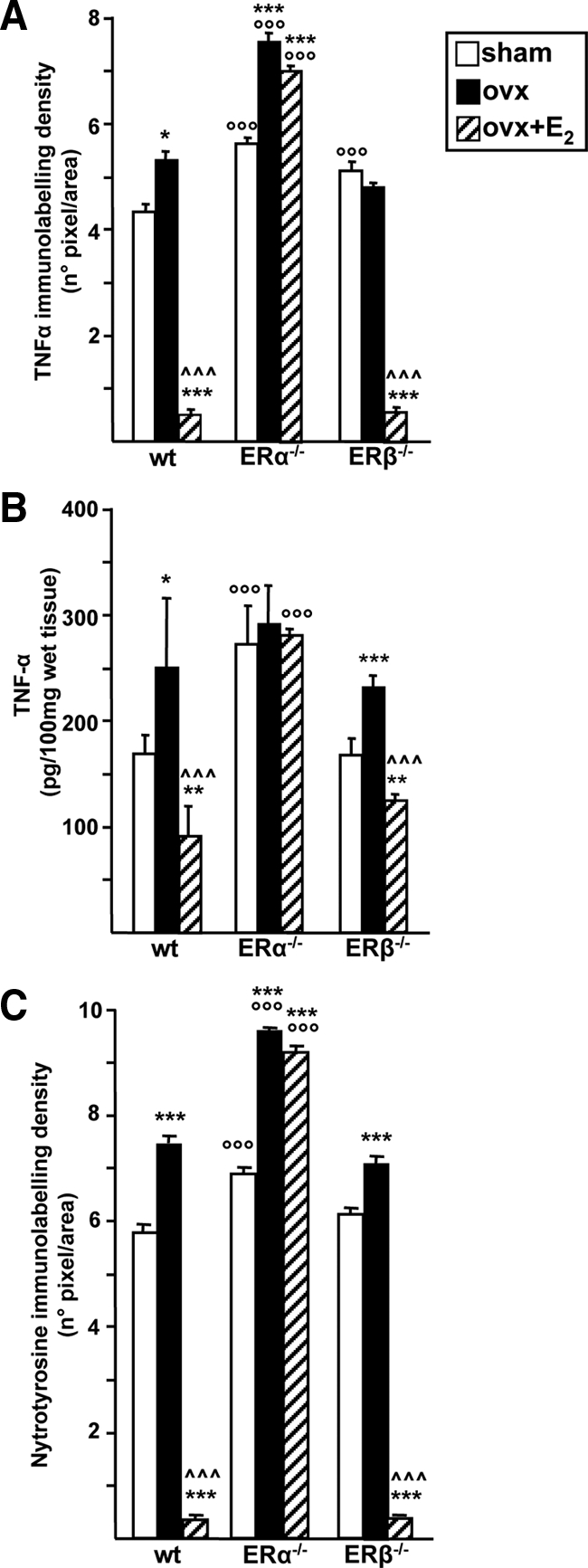
TNF-α and nytrotyrosine immunolabeling in ER-KO animals. Lung tissue from wt, ERα-KO, or ERβ-KO mice, sham operated, ovx, or ovx+E2 (bar legend as in Fig. 1B) were analyzed after CAR injection with specific antibodies against TNF-α (A) or nytrotyrosine residues (C); densitometry of IHC images were evaluated and data are expressed as means of percent of total tissue area ± sem (B) TNF-α levels were analyzed by immunoenzymatic assay in the pleural exudate. Bars represent the average ± sem of all the animals, each analyzed in triplicate. * vs. sham; ^ vs. ovx; ° vs. corresponding treatment in wt. *, P < 0.05; **, P < 0.01; ***, °°°, ^^^, P < 0.001.
To further confirm that reduced mRNA levels corresponded to decreased levels of TNFα protein, this cytokine was analyzed by immunoenzymatic assay; Fig. 5B shows that TNFα protein levels are regulated by E2 through ERα, in that both hormone deprivation and replacement induce changes only when ERα is expressed. The slight differences observed in TNFα levels between IHC and ELISA from the same experimental group (i.e. compare ovx ERα-KO in Fig. 5, A and B) are not understood yet; however, they could be reconciled with the fact that only part of TNFα synthesized by lung cells and visualized by IHC reaches the pleural exudate; in addition, it is not known whether and how alterations in the ER genotype may influence cytokine secretion, stability, and reabsorption in pleural exudate. Accordingly with the TNFα data, IL-1β expression was also down-regulated by E2 through the specific involvement of ERα (supplemental Fig. S3).
Finally, we analyzed the levels of nytrosylated proteins, a parameter that in addition to being a reliable sign of ongoing inflammation, allows evaluation of the possibility that E2 acts as a scavenger molecule, without the mediation of intracellular receptor proteins. If this hypothesis were true, a reduction in protein nitrosylation in all knockout animals would occur after E2 administration. On the contrary, E2 failed to modulate protein nytrosylation in ERα-KO mice, as shown in Fig. 5C. Again, this inflammatory parameter is also modulated by E2 signaling specifically in inflammatory cells of the lung (supplemental Fig. S4).
Altogether these data show that resident and infiltrated cells of the lung are targets for the E2 antiinflammatory activity through the specific intervention of ERα. In addition, genetic ablation of ERα results in a higher expression of inflammatory markers in the lung without altering tissue susceptibility to hormone deprivation.
Lung inflammation and gender
It is known that several parameters in lung biology and pathology are sexually dimorphic. To determine whether E2 could act as a protective, antiinflammatory molecule also in males, we analyzed CAR-induced pleurisy in male mice that underwent gonadectomy (gdx) to remove the endogenous source of estrogenic precursors. In parallel, a group of gdx animals received E2 administration before pleurisy induction. As shown in Fig. 6, A and B, lower values of inflammatory parameters were observed in sham males as compared with females; this is shown by both a lower histological score and a reduced number of infiltrated cells in the lungs of 5-month-old male mice. gdx per se does not alter inflammatory parameters. Interestingly, E2 administration reduces lung inflammation, resulting in a lower grade of inflammation which is even smaller than intact animals. Consistently, ERα mRNA levels in alveolar macrophages and BMDCs from males are similar to those found in females of the same age (data not shown).
Figure 6.

Effect of gender and aging on E2 signaling in lung inflammation. Lung inflammation was induced by CAR injection in female and male mice at the age of 5 (bar legend as in Fig. 1B) or 18 months (either intact or treated with E2), as indicated. Histological score (A) or infiltrated PMN cells numbers (B) are shown; bars represent the average ± sem of all the animals (n = 8), each analyzed in triplicate. * vs. sham; ^ vs. 5-month sham; *, P < 0.05; **, P < 0.01; ***, ^^^, P < 0.001. C, Expression levels of the ERα gene was evaluated by real-time PCR on the mRNA extracted from BMDCs, alveolar macrophages (alv. MØ), and lung tissue from 5-month-old (black bars) and 18-month-old (open bars) female mice. Each data point represents the average ± sem of samples from three mice analyzed in triplicate. D, Representative images of immunohistochemistry assays performed using a specific ERα antibody in lung sections from 18-month-old female mice injected with CAR (left panel) or with E2 before CAR (right panel). A cytoplasmic localization of ERα is observed in PMN cells, independently of E2 administration.
Thus, these data reveal a gender difference in the lung physiopathology because females show a more robust inflammatory response. In addition, there results suggest that the E2 signaling pathway is conserved in male mice and leads to a reduction in the lung inflammatory response.
Role of aging in lung inflammation and E2 action
Increasing evidence shows that aging is associated with a higher level of inflammatory response, in that increased levels of inflammatory mediators, such as TNFα, are found in the blood of old subjects. Thus, we analyzed lung inflammation after CAR injection in 18-month-old mice and evaluated the effect of E2 on the inflammatory response; we chose not to gonadectomize this group of old mice because we could not exclude that the endocrine ablation in old mice would affect lung homeostasis and response to CAR injection. As shown in Fig. 6, A and B, both male and female mice at 18 months of age show a robust inflammatory reaction, as shown by a dramatic worsening of the histological score and massive infiltration of PMN cells after CAR injection; however, E2 administration does not cause inflammation in either gender. We thus analyzed ERα expression levels in lung, alveolar macrophages, and BMDCs; Fig. 6C shows that there is no variation in old as compared with young animals. Interestingly, the IHC assay for ERα revealed that this receptor is exclusively in the cytoplasm of PMN cells, both in the absence or presence of E2 (see Fig. 6D), in contrast to the nuclear immunoreactivity for this receptor in infiltrated inflammatory cells of young animals in the absence or presence of CAR ± E2 (as shown in Fig. 3B). Intracellular localization of ERα in endothelial cells was very similar to that reported in Fig. 3A for young mice (data not shown). Thus, aging is associated with the nuclear exclusion of ERα specifically in inflammatory cells; this might help to explain the inefficacy of E2 treatment in aged mice. Altogether, these data show that the dramatic changes in the inflammatory response that occur in the lung of aged animals are gender independent and associated with the disruption of E2/ERα signaling pathway.
Discussion
This work provides novel findings related to estrogens action in lung: 1) ERα is the molecular mediator of E2 inhibitory activity on CAR-induced lung inflammation; 2) circulating myeloid precursors, resident macrophages and endothelial cells are direct targets of this activity; 3) E2 activity is conserved among females and males; and 4) aging is associated with a high inflammatory reaction that is gender independent and E2 unresponsive.
We believe that these observations give a deeper understanding of estrogens action in lung physiopathology. In fact, although previous studies have shown that E2 reduces inflammation in lung, the molecular and cellular details of this action were mostly unknown. Whereas both ERs are required for pulmonary alveolar formation (22), lung cells were shown to express higher levels of ERβ than ERα mRNAs (21), and the ERβ protein was detected by IHC in pulmonary airway structures, whereas ERα protein expression could not be detected (12). Despite the key role of ERβ in lung homeostasis, we here observed that ERα was required for the activity of E2 on the inflammatory response of the lung. Thus, we reasoned that the target cells for E2 could reside in either the air or the blood compartments of the lung. Our real-time PCR and IHC studies indeed confirmed our hypothesis and showed ERα, and not ERβ, expression in alveolar macrophages and endothelial cells of lung vessels, which to our knowledge is the first observation reporting ER expression in these cell types as well as in BMDCs, as previously shown (23). Thus, we here define that ERα is a key player in the inflammatory response of lung and that its activity proceeds through the control of resident and circulating inflammatory cells.
Several lines of evidence are now converging on the key role played by ERα in lung disease; in fact, a previous study by Carey et al. (24) reported that ERα deletion also affects the protective role played by E2 in allergic airway hyperresponsiveness. Interestingly, the lung inflammatory reaction associated with this mouse model was not influenced by ERα. Although the reasons for the different results between the study by Carey et al. and the present work are not known, some major differences in the two experimental models might provide the explanation: 1) allergens do not directly act on lung tissue to induce inflammation, which occurs in response to molecules, such as IL-5, secreted by the spleen, and 2) different cellular populations, namely eosinophils, are involved in allergic inflammation of the lung, whereas neutrophils and monocytes are recruited by local inflammatory signals. It is thus possible that E2-activated ERα controls inflammation induced by direct stimulators of lung innate immunity, whereas it is not involved in pulmonary inflammation driven by allergens. Consistent with our hypothesis, E2 has been shown to selectively require ERα to control the inflammatory response in other tissues and cells (25,26,27,28). Accordingly, it has also been shown that ERα mediates the effect of estrogens on alveolar loss and regeneration in adult female mice (29), whereas ERβ appears to mediate the protective effects of estrogens in lung injury induced by trauma-hemorrhage (30). Importantly, ERβ agonists failed to have an impact on lung inflammatory response (31), which confirm ER specificity avoiding the use of animal models endowed with genetic as well as endocrine diversity. Our present work thus leads to consolidate the hypothesis that ERα ablation highly impacts on the protective effect of endogenous estrogens against both inflammatory and, as demonstrated by others, allergic pathologies of the lung.
The identification of a specific receptor isoform mediating the beneficial effects of E2 leads to interesting pharmacologic and pharmacogenetic consequences. The clinical availability of ERα selective ligands that act as potent antiinflammatory drugs might represent a therapeutic opportunity of particular relevance because patients with inflammatory obstructive disorders of the lung often do not benefit from classic antiinflammatory agents, such as glucocorticoids (32,33). Although ERα modulation is associated with toxic effects on reproductive tissues, novel hints from basic research allow the exploitation of the beneficial effects of selective ER modulators, which act as agonists in selected tissue, including lung, and antagonists in reproductive organs, or propose the use of labile estrogenic compounds that are degraded before reaching distant tissues (34). The observation that ERα is present in inflammatory cells of the lung also implies the existence of intracellular partners of receptor action that might prove to be valuable drug target candidates for the therapeutic management of lung inflammation. Mechanistically the E2/ERα molecular pathway that leads to a reduction in inflammatory gene expression has been previously investigated and reconciled with both nuclear and cytoplasmic activities; as a transcription factor, E2-activated ERα interferes with the availability of transcriptional activators and reduces inflammatory gene expression; in addition, the cytoplasmic activity of ERα modulates the intracellular traffic of nuclear factor-κB, thus preventing its activity on inflammatory gene promoters (35). Future molecular investigations are thus needed to identify the hormonal partners that allow the modulation of the inflammatory response. Finally, gene polymorphisms that alter ERα expression or activity in lung might predispose individuals carrying those genetic alterations to inflammatory and allergic lung pathologies. Interestingly, the protective activity of the exogenous administration of E2 against lung inflammation is conserved also in male mice, suggesting that our therapeutic and genetic considerations might apply independently of gender.
Our results indicate that alveolar macrophages and BMDCs express ERα and lead to postulate novel physiological activities for estrogens in lung homeostasis as well as in regenerative processes. It has been suggested that activation of macrophages that reside in the alveolar and pleural cavities are the main regulators of lung inflammatory response (36). Thus, the involvement of resident macrophages in E2 action suggests that estrogenic drugs could be used topically to reduce the acute inflammatory response associated with pulmonary disorders. On the other hand, the presence of ERα in BMDCs and the demonstration of E2 responsiveness in myeloid progenitor cells (37) imply that E2 signaling has the potential to participate in lung repair and regeneration because stem cell-related therapies are widely viewed as promising treatment options for patients suffering from various types of pulmonary diseases (38).
We here also show that inflammatory cells in the lung of aged animals are dramatically more reactive to inflammatory agents and that this increased reactivity is both gender independent and E2 unresponsive. This suggests that aging is associated with an alteration in signal transduction pathways and overall reactivity of lung inflammatory cells (39). This conclusion is also further sustained by our observation that ERα is mainly localized in the cytoplasm of inflammatory cells even after E2 administration, whereas its expression remains unchanged, in aged as compared with young animals. Thus, aging appears to be associated with a loss of ERα nuclear activity, its cytoplasmic role still waiting to be understood in both normal and age-related pathological conditions. Future studies will shed more light on these aspects.
In conclusion, our data reveal that exogenous E2 is able to regulate lung reactivity in a gender-unrelated, age-restricted manner and through the involvement of a specific intracellular pathway. These observation suggest the existence of E2/ERα targets in lung inflammatory cells that foster beneficial effects on lung inflammation. The identification of the molecular players underlying this physiological advantage will lead to deepen our understanding of lung physiopathology and to improve the therapeutic strategies for lung inflammatory disorders.
Supplementary Material
Acknowledgments
We thank Clara Meda and Monica Rebecchi for their technical skill; Paolo Sparaciari, Giuseppina Monteleone, and Joanna Illner for their experimental support; and Pierre Chambon for helpful discussion.
Footnotes
This work was supported by the European Commission (Estrogen and Women Aging, EWA LSHM-CT-2005-518245) and National Institutes of Health (Menopause: decreased response to increasing inflammation, MADRI RO1-AG027713-01) (to A.M.) and the Italian Ministry of Research (Programma di Ricerca Scientifica di Rilevante Interesse Nazionale, PRIN 20006065483_003) (to E.V.).
Disclosure Summary: The authors have nothing to disclose.
First Published Online December 1, 2009
Abbreviations: BMDC, Bone marrow-derived hematopoietic cell; CAR, carrageenan; E2, 17β-estradiol; ER, estrogen receptor; ER-KO, ER-knockout; gdx, gonadectomy; HBSS, Hanks’ balanced salt solution; ICAM, intercellular adhesion molecule; IHC, immunohistochemistry; MPO, myeloperoxidase; ovx, ovariectomized; PMN, polymorphonucleated; wt, wild type.
References
- Sweezey N, Tchepichev S, Gagnon S, Fertuck K, O'Brodovich H 1998 Female gender hormones regulate mRNA levels and function of the rat lung epithelial Na channel. Am J Physiol 274:C379–C386 [DOI] [PubMed] [Google Scholar]
- Torday JS, Nielsen HC 1987 The sex difference in fetal lung surfactant production. Exp Lung Res 12:1–19 [DOI] [PubMed] [Google Scholar]
- Carey MA, Card JW, Voltz JW, Germolec DR, Korach KS, Zeldin DC 2007 The impact of sex and sex hormones on lung physiology and disease: lessons from animal studies. Am J Physiol Lung Cell Mol Physiol 293:L272–L278 [DOI] [PubMed] [Google Scholar]
- Massaro GD, Mortola JP, Massaro D 1996 Estrogen modulates the dimensions of the lung’s gas-exchange surface area and alveoli in female rats. Am J Physiol 270:L110–L114 [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Santagati S, Sautebin L, Mazzon E, Calabrò G, Serraino I, Caputi AP, Maggi A 2000 17β-Estradiol antiinflammatory activity in CAR-induced pleurisy. Endocrinology 141:1455–1463 [DOI] [PubMed] [Google Scholar]
- Ligeiro de Oliveira AP, Oliveira-Filho RM, da Silva ZL, Borelli P, Tavares de Lima W 2004 Regulation of allergic lung inflammation in rats: interaction between estradiol and corticosterone. Neuroimmunomodulation 11:20–27 [DOI] [PubMed] [Google Scholar]
- Carey MA, Card JW, Voltz JW, Arbes Jr SJ, Germolec DR, Korach KS, Zeldin DC 2007 It’s all about sex: gender, lung development and lung disease. Trends Endocrinol Metab 18:308–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greally P, Hussein MJ, Cook AJ, Sampson AP, Piper PJ, Price JF 1993 Sputum tumour necrosis factor-α and leukotriene concentrations in cystic fibrosis. Arch Dis Child 68:389–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, Berger M 1995 Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med 152:2111–2118 [DOI] [PubMed] [Google Scholar]
- Cheskis BJ, Greger JG, Nagpal S, Freedman LP 2007 Signaling by estrogens. J Cell Physiol 213:610–617 [DOI] [PubMed] [Google Scholar]
- Massaro D, Massaro GD 2006 Estrogen receptor regulation of pulmonary alveolar dimensions: alveolar sexual dimorphism in mice. Am J Physiol Lung Cell Mol Physiol 290:L866–L870 [DOI] [PubMed] [Google Scholar]
- Patrone C, Cassel TN, Pettersson K, Piao YS, Cheng G, Ciana P, Maggi A, Warner M, Gustafsson JA, Nord M 2003 Regulation of postnatal lung development and homeostasis by estrogen receptor β. Mol Cell Biol 23:8542–8552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegeto E, Ciana P, Maggi A 2002 Estrogen and inflammation: hormone generous action spreads to the brain. Mol Psychiatry 7:236–238 [DOI] [PubMed] [Google Scholar]
- Card JW, Carey MA, Bradbury JA, DeGraff LM, Morgan DL, Moorman MP, Flake GP, Zeldin DC 2006 Gender differences in murine airway responsiveness and lipopolysaccharide-induced inflammation. J Immunol 177:621–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speyer CL, Rancilio NJ, McClintock SD, Crawford JD, Gao H, Sarma JV, Ward PA 2005 Regulatory effects of estrogen on acute lung inflammation in mice. Am J Physiol Cell Physiol 288:C881–C890 [DOI] [PubMed] [Google Scholar]
- Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M 2000 Development. 127:4277–4291 [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Mazzon E, Sautebin L, Serraino I, Dugo L, Calabró G, Caputi AP, Maggi A 2001 The protective role of endogenous estrogens in carrageenan-induced lung injury in the rat. Mol Med 7:478–487 [PMC free article] [PubMed] [Google Scholar]
- Mullane KM, Kraemer R, Smith B 1985 Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J Pharmacol Methods 14:157–167 [DOI] [PubMed] [Google Scholar]
- Vegeto E, Bonincontro C, Pollio G, Sala A, Viappiani S, Nardi F, Brusadelli A, Viviani B, Ciana P, Maggi A 2001 Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J Neurosci 21:1809–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD 2001 Analysis of relative gene expression data using real-time quantitative PCR and the 2[-ΔΔC(T)] method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS 1997 Tissue distribution and quantitative analysis of estrogen receptor-α (ERα) and estrogen receptor-β (ERβ) messenger ribonucleic acid in the wild-type and ERα-knockout mouse. Endocrinology 138:4613–4621 [DOI] [PubMed] [Google Scholar]
- Massaro D, Massaro GD 2004 Estrogen regulates pulmonary alveolar formation, loss, and regeneration in mice. Am J Physiol Lung Cell Mol Physiol 287:L1154–L1159 [DOI] [PubMed] [Google Scholar]
- Igarashi H, Kouro T, Yokota T, Comp PC, Kincade PW 2001 Age and stage dependency of estrogen receptor expression by lymphocyte precursors. Proc Natl Acad Sci USA 98:15131–15136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey MA, Card JW, Bradbury JA, Moorman MP, Haykal-Coates N, Gavett SH, Graves JP, Walker VR, Flake GP, Voltz JW, Zhu D, Jacobs ER, Dakhama A, Larsen GL, Loader JE, Gelfand EW, Germolec DR, Korach KS, Zeldin DC 2007 Spontaneous airway hyperresponsiveness in estrogen receptor-α-deficient mice. Am J Respir Crit Care Med 175:126–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM 2001 Estrogen receptor α, not β, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci USA 98:1952–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegeto E, Belcredito S, Etteri S, Ghisletti S, Brusadelli A, Meda C, Krust A, Dupont S, Ciana P, Chambon P, Maggi A 2003 Estrogen receptor-α mediates the brain antiinflammatory activity of estradiol. Proc Natl Acad Sci USA 100:9614–9619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanczyk M, Yellayi S, Zamora A, Subramanian S, Tovey M, Vandenbark AA, Offner H, Zachary JF, Fillmore PD, Blankenhorn EP, Gustafsson JA, Teuscher C 2004 Estrogen receptor-1 (Esr1) and -2 (Esr2) regulate the severity of clinical experimental allergic encephalomyelitis in male mice. Am J Pathol 164:1915–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF 2001 Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-α but not estrogen receptor-β. Circulation 103:423–428 [DOI] [PubMed] [Google Scholar]
- Massaro D, Clerch LB, Massaro GD 2007 Estrogen receptor-α regulates pulmonary alveolar loss and regeneration in female mice: morphometric and gene expression studies. Am J Physiol Lung Cell Mol Physiol 293:L222–L228 [DOI] [PubMed] [Google Scholar]
- Yu HP, Hsieh YC, Suzuki T, Shimizu T, Choudhry MA, Schwacha MG, Chaudry IH 2006 Salutary effects of estrogen receptor-β agonist on lung injury after trauma-hemorrhage. Am J Physiol Lung Cell Mol Physiol 290:L1004–L1009 [DOI] [PubMed] [Google Scholar]
- Catley MC, Birrell MA, Hardaker EL, de Alba J, Farrow S, Haj-Yahia S, Belvisi MG 2008 Estrogen receptor β: expression profile and possible anti-inflammatory role in disease. J Pharmacol Exp Ther 326:83–88 [DOI] [PubMed] [Google Scholar]
- Adcock IM, Ford PA, Bhavsar P, Ahmad T, Chung KF 2008 Steroid resistance in asthma: mechanisms and treatment options. Curr Allergy Asthma Rep 8:171–178 [DOI] [PubMed] [Google Scholar]
- Falk JA, Minai OA, Mosenifar Z 2008 Inhaled and systemic corticosteroids in chronic obstructive pulmonary disease. Proc Am Thorac Soc 5:506–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labaree DC, Reynolds TY, Hochberg RB 2001 Estradiol-16α-carboxylic acid esters as locally active estrogens. J Medicinal Chem 44:1802–1814 [DOI] [PubMed] [Google Scholar]
- Ghisletti S, Meda C, Maggi A, Vegeto E 2005 17β-Estradiol inhibits inflammatory gene expression by controlling NF-κB intracellular localization. Mol Cell Biol 25:2957–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cailhier JF, Sawatzky DA, Kipari T, Houlberg K, Walbaum D, Watson S, Lang RA, Clay S, Kluth D, Savill J, Hughes J 2006 Resident pleural macrophages are key orchestrators of neutrophil recruitment in pleural inflammation. Am J Respir Crit Care Med 173:540–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paharkova-Vatchkova V, Maldonado R, Kovats S 2004 Estrogen preferentially promotes the differentiation of CD11c+ CD11b(intermediate) dendritic cells from bone marrow precursors. J Immunol 172:1426–1436 [DOI] [PubMed] [Google Scholar]
- Gharaee-Kermani M, Gyetko MR, Hu B, Phan SH 2007 New insights into the pathogenesis and treatment of idiopathic pulmonary fibrosis: a potential role for stem cells in the lung parenchyma and implications for therapy. Pharm Res 24:819–841 [DOI] [PubMed] [Google Scholar]
- Corsini E, Di Paola R, Viviani B, Genovese T, Mazzon E, Lucchi L, Marinovich M, Galli CL, Cuzzocrea S 2005 Increased carrageenan-induced acute lung inflammation in old rats. Immunology 115:253–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.