

Abstract
Cholesterol 24S-hydroxylase (CYP46A1) is of key importance for cholesterol homeostasis in the brain. This enzyme seems to be resistant toward most regulatory factors and at present no drug effects on its activity have been described. The crystal structures of the substrate-free and substrate-bound CYP46A1 were recently determined (Mast et al., Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc. Natl. Acad. Sci. USA. 2008. 105: 9546–9551). These structural studies suggested that ligands other than sterols can bind to CYP46A1. We show here that the antifungal drug voriconazole binds to the enzyme in vitro and inhibits CYP46A1-mediated cholesterol 24-hydroxylation with a Ki of 11 nM. Mice treated with daily intraperitoneal injections of voriconazole for 5 days had high levels of voriconazole in the brain and significantly reduced brain levels of 24S-hydroxycholesterol. The levels of squalene, lathosterol, and HMG-CoA reductase mRNA were reduced in the brain of the voriconazole-treated animals as well, indicating a reduced cholesterol synthesis. Most of this effect may be due to a reduced utilization of cholesterol by CYP46A1. One of the side-effects of voriconazole is visual disturbances. Because CYP46A1 is also expressed in the neural retina, we discuss the possibility that the inhibition of CYP46A1 by voriconazole contributes to these visual disturbances.
Keywords: cholesterol biosynthesis, cholesterol elimination, brain, visual disturbances, retina, side effects
Cholesterol 24S-hydroxylase (CYP46A1) is of key importance for cholesterol homeostasis in the brain and is responsible for about two-thirds of cholesterol elimination from this organ (1, 2). Except for the enzyme induction during neonatal stage and a stimulatory effect by oxidative stress, CYP46A1 appeared to be resistant toward a broad spectrum of factors expected to regulate at transcriptional level (3). Very recently, the enzyme was crystallized in substrate-free and substrate-bound forms and found to undergo conformational changes upon substrate binding (4). This conformational flexibility of the active site prompted us to screen more than 50 different compounds for their binding and inhibition of CYP46A1. Several marketed drugs and nonpharmaceuticals were found to bind tightly to the active site and inhibit CYP46A1 activity in vitro (4). Of the eight strong CYP46A1 inhibitors identified in that study, five were imidazole-containing compounds: the H2-receptor antagonist cimetidine, the H3-receptor antagonist clobenpropit, the H3/H4 receptor antagonist thioperamide, nonpharmaceutical 4-(4-chlorophenyl)-imidazole, and the antifungal azole clotrimazole. The latter is applied only topically, therefore, in the present study we tested whether antifungal azoles that are administered systemically can inhibit CYP46A1 (Fig. 1). These drugs are known to pass the blood-brain barrier and thus have a potential to inhibit CYP46A1, which is localized almost exclusively to the brain in humans. We demonstrate here that voriconazole is an effective inhibitor of CYP46A1 in vitro and that intraperitoneal injections of the drug in mice reduce the levels of 24S- hydroxycholesterol in the brain. This finding is discussed in relation to the known side-effects of voriconazole.
Fig. 1.

Chemical structures of the CYP46A1 substrate cholesterol and four antifungal azoles tested in the present study.
MATERIALS AND METHODS
Materials
Voriconazole (vFEND) for in vitro studies was obtained from Pfizer. Aqueous solution of methanol (50%) was used to dissolve the compound. Voriconazole for the animal studies was a generous gift from the Division of Clinical Pharmacology at Karolinska University Hospital in Huddinge, Sweden. The material was dissolved in a mixture of saline containing 10% ethanol and 1% serum albumin. The final concentration of voriconazole was 100 mg/ml. Recombinant full-length human CYPs 46A1 and 27A1 and bovine CYP11A1 were expressed and purified as described (5–7).
Spectral binding assay
Binding affinities of antifungal drugs were estimated as described (5, 8) using 0.3 μM purified P450. Titrations of CYP46A1 were carried out in 50 mM potassium phosphate buffer (KPi), pH 7.2, containing 100 mM NaCl and 0.02% Cymal 6. CYPs 11A1 and 27A1 were in 50 mM KPi without any additives. Antifungal drugs were added from 0.2–5 mM stocks in 50% methanol. The apparent Kd and maximal absorbance change were estimated by nonlinear least-squares fitting using the quadratic form of the single-site binding equation (9).
Determination of the in vitro IC50 and Ki
Cholesterol hydroxylation by CYP46A1 was assayed for 20 min at 37°C in 1 ml of 50 mM KPi, pH 7.2, containing 100 mM NaCl, 0.02% Cymal 6, 40 μg dilauroylglycerol-3-phosphatidylcholine, and 2U of catalase. Purified 0.4 μM CYP46A1 was reconstituted with 0.8 μM NADPH cytochrome P450 oxidoreductase, 5.4 μM cholesterol, trace amounts of [3H]cholesterol (250,000 cpm), and varying concentrations of voriconazole (from 0.000001 μM to 100 μM). The enzymatic reaction was initiated by NADPH (final 1 mM) and terminated by vortexing with 2 ml of CH2Cl2. The organic phase was isolated, evaporated, dissolved in methanol, and subjected to HPLC as described (8). To determine IC50, data were fit to the following equation by Graph-Pad Prism software:
in which I is the inhibitor concentration, the IC50 represents the inflection point and the value of 1−(A−B) is the maximum inhibition observed at an infinite inhibitor concentration. Under the assay conditions used, two types of products were formed, 24-hydroxycholesterol and 24, 25- and 24, 27-dihydroxycholesterols, which are produced upon further hydroxylation of 24- hydroxycholesterol. Dihydroxycholesterols were eluted as one peak during HPLC; therefore, we could estimate two IC50 values, one for the inhibition of cholesterol 24-hydroxylation and the other for the inhibition of 25- and 27-hydroxylations of 24S-hydroxycholesterol. In addition, we could also estimate the Ki for cholesterol 24-hydroxylation assuming a model of competitive inhibition. Determination of IC50 was carried out at cholesterol concentration equal to Km, therefore Ki = IC50/2.
Animal experiments
Seven-week-old male C57/B6 J mice from Charles River were injected intraperitoneally with voriconazole (60 mg/kg or 75 mg/kg, corresponding to 0.6 and 0.75 ml, respectively, of the drug solution). The control mice were injected with the same solutions containing no voriconazole. The mice were euthanized by cervical dislocation. Brain and liver were collected, snap- frozen in liquid nitrogen, and stored at −80°C until further analysis.
Lipid extraction
Lipids were extracted from the brain according to Folch with some modifications (10, 11). Approximately 10 mg of brain tissue was added to 1 ml of homogenization buffer (5 mM EDTA, 50 μg/ml butylated hydroxytoluene in phosphate-buffered saline, pH 7,4) in a clean glass tube and the tissue was disrupted using a polytron homogenizer. Three milliliters of chloroform-methanol (2:1, v/v) were added to the homogenate and the vials were mixed by vortexing. Samples were centrifuged at 10,000 g for 10 min. The organic phase was transferred to a new vial. The aqueous phase was reextracted as described previously three more times. The pooled organic phase was dried under a stream of argon gas, and chloroform-methanol (2:1, v/v) added to the dried samples to achieve suitable concentration.
Sterol analysis
Sterols were analyzed by isotope dilution mass-spectrometry as previously described (3, 12–14). Cholesterol was determined after saponification using [2H6] cholesterol as internal standard; [2H3] lathosterol was used as internal standard for quantification of lathosterol. 24S-Hydroxycholesterol and 27-hydroxycholesterol in the brain were measured by GC-MS (3, 12) as are squalene, lanosterol, and dehydrolanosterol (15).
Analysis of voriconazole
Voriconazole was determined in serum and brain homogenates after the extraction with chloroform-methanol (2:1, v/v) and ethanol (10%, v/v, final), respectively. Voriconazole extract (100 μl) was then mixed with 200 μl acetonitrile, centrifuged at 10,000 g for 10 min, and 1 μl of the supernatant injected onto a Phenomenex Luna C18 3 μm column (100 × 2 mm) connected to the Agilent 1100 liquid chromatography-mass spectrometry system with an autosampler and solvent degasser. Gradient elution was with initial 35% B increasing to 100% B in 5 min (A = 1% acetonitrile with 25 mM formic acid; B = 100% acetonitrile with 25 mM formic acid). A structure analog (UK115794, Pfizer Ltd.) was used as internal standard. Ions were monitored in a positive mode at m/z 350 (voriconazole) and m/z 348 (internal standard).
Gene expression analysis
Total RNA was obtained from brain and liver tissue using Trizol (Invitrogen, Carlsbad, CA). cDNA was synthesized from 2 μg of total RNA using Capacity cDNA ReverseTranscription Kit (Applied Biosystems, Foster City, CA). Steady-state mRNA levels were estimated using Taqman probes (primer sequences available on request). All assays were run on an ABI Prism 7000 Sequense Detection System (Applied Biosystems). Estimation of a relative gene expression was performed using the comparative threshold cycle method using hypoxathine-guanine phosphoribosyl transferase as endogenous control.
Statistics
Gene expression data is expressed as mean ± SEM (16). Sterol determinations are also presented as mean ± SEM. For statistical comparisons, a two-tailed Student's t-test was performed. All in vitro assays were performed in triplicate.
Ethical aspects
All animal experiments received full approval from the animal Stockholm Experimentation Ethics Committee.
RESULTS
Binding of antifungal azoles to purified CYP46A1 and other cholesterol-metabolizing P450s
Four antifungal azoles that are used systemically were tested: voriconazole, fluconazole, ketoconazole, and itraconazole (Fig. 1 and Table 1). Of them, voriconazole had the highest affinity to CYP46A1, comparable to that of clotrimazole, with the estimated Kd value of 50 nM. Voriconazole induced a type II spectral response with a minimum at 413 nm and a maximum at 434 nm (Fig. 2). This spectral response indicates that voriconazole binds to the CYP46A1 active site and coordinates the P450 heme iron with one of its nitrogen atoms (17, 18). CYP46A1 is not the only cholesterol-metabolizing enzyme expressed in the brain. Also present in the brain are CYP27A1, which catalyzes cholesterol 27-hydroxylation and CYP11A1, which converts cholesterol to pregnenolone (19, 20). We tested voriconazole on these P450s as well. Similar to CYP46A1, voriconazole induced a type II spectral response in CYP11A1, but its binding was ∼100-fold weaker than that to CYP46A1 as assessed by the apparent Kd equal to 4.8 μM. In CYP27A1, only a very weak spectral shift was observed, indicating that either voriconazole does not bind to the enzyme at the concentrations employed (up to 100 μM) or the drug is not positioned in the P450 active site in the vicinity of the heme iron.
TABLE 1.
Binding of antifungal azoles to cholesterol-metabolizing P450s
| P450 | Antifungal Azole | Kd, nMa | ΔAmaxb | Type of a Spectral Response |
|---|---|---|---|---|
| CYP46A1 | clotrimazolec | 11 ± 2 | 0.05 ± 0.001 | type II |
| CYP46A1 | voriconazole | 50 ± 7 | 0.04 ± 0.002 | type II |
| CYP46A1 | fluconazole | 3,800 ± 100 | 0.06 ± 0.004 | type II |
| CYP46A1 | ketoconazole | 2,450 ± 50 | 0.13 ± 0.007 | type II |
| CYP46A1 | itraconazole | NDd | very weake | |
| CYP11A1 | voriconazole | 4,840 ± 80 | 0.037 ± 0.003 | type II |
| CYP27A1 | voriconazole | ND | very weak |
Calculated on the basis of the spectral binding. The results represent the average of three different titrations ± SD.
Normalized to nmol of P450.
Clotrimazole is not used systemically. This data is from reference (4).
ND, not determined.
The spectral response ΔA413-434 was < 0.002 absorbance units when 0.3 μM P450 was titrated with up to a 100 μM drug.
Fig. 2.
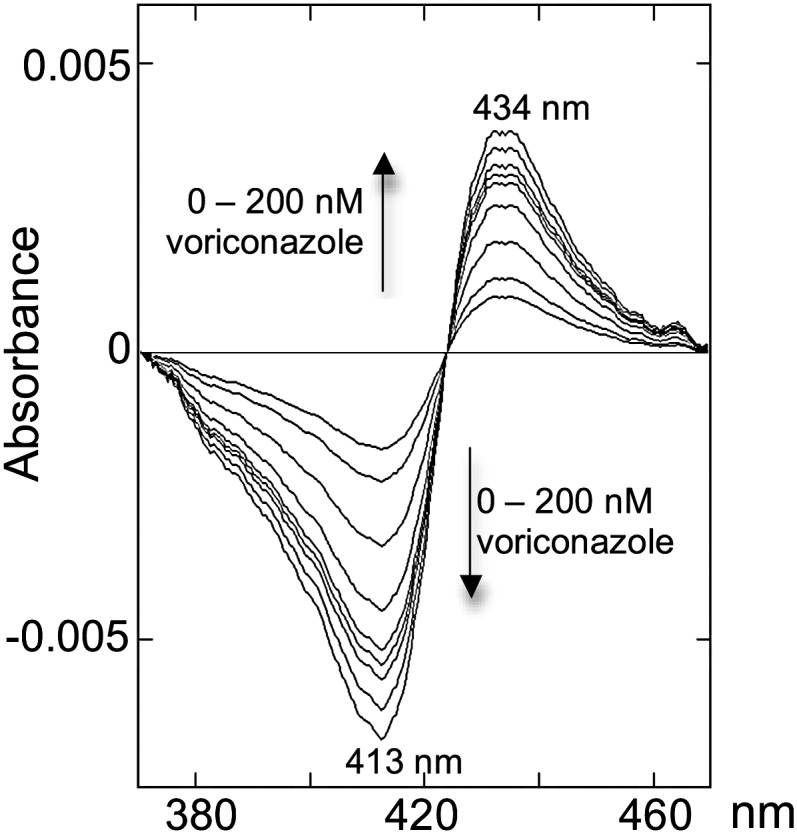
Spectral response in human recombinant CYP46A1 induced by voriconazole binding.
Inhibition of cholesterol 24S-hydroxylase in vitro
Of the three cholesterol-metabolizing P450s, CYP46A1 demonstrated the tightest binding of voriconazole. Therefore, we next tested whether voriconazole inhibits the enzyme activity in vitro. CYP46A1 converts cholesterol to 24S-hydroxycholesterol and then can further metabolize 24S-hydroxycholesterol to 24, 25-, and 24,27-dihydroxycholesterols (5). The IC50 for cholesterol 24-hydroxylation was 22 nM and that for 25- and 27-hydroxylations of 24S-hydroxycholesterol was 47 nM (Fig. 3). Because cholesterol concentration in the inhibition assay was equal to the Km, the Ki for cholesterol 24-hydroxylation was estimated to be 11 nM.
Fig. 3.

Inhibition of CYP46A1 activity by voriconazole in vitro. IC50 plots for the inhibition of cholesterol 24-hydroxylation and of 25- and 27-hydroxylations of 24S-hydroxycholesterol are shown in closed squares and open circles, respectively. Voriconazole concentration is plotted on a log scale.
Inhibition of cholesterol 24S-hydroxylase in vivo
In a pilot study, we exposed mice to different doses of voriconazole, euthanized experimental and control animals 6 h after the injection, and measured the levels of 24S-hydroxycholesterol in the brain. The treatment appeared to reduce the levels of 24S-hydroxycholesterol. The levels were slightly lower after the injection with 60 mg/kg body weight than after injection with 75 mg/kg body weight (results not shown). In a subsequent experiment, we treated five mice with voriconazole, 60 mg/kg body weight, and euthanized the animals and their controls 2 h later. The levels of 24S-hydroxycholesterol were reduced by 20% but this effect was not statistically significant (P > 0.05, Student's t-test). There was no significant effect on levels of lathosterol or total cholesterol in the brain (results not shown).
Then, we treated six mice with voriconazole, 60 mg/kg body weight, once a day for 5 days and observed a significant reduction (by 37%) in the levels of 24S-hydroxycholesterol (P = 0.03) (Fig. 4A). There was no effect on the levels of cholesterol (Fig. 4B) or 27-hydroxycholesterol (Fig. 4C) (P > 0.05). However, the level of lathosterol in the brain was significantly reduced (Fig. 4D). Lathosterol serves as a marker for cholesterol biosynthesis (21). Therefore, reduced levels of this sterol in voriconazole-treated mice indicate a reduction in cholesterol synthesis (P < 0.05).
Fig. 4.

Effect of 5 daily intraperitoneal injections (60 mg/kg body weight) with voriconazole on levels of 24S-hydroxycholesterol (A), cholesterol (B), 27-hydroxycholesterol (C), and lathosterol (D) in the mouse brain . Error bars indicate SEM.
Because most of the 24S-hydroxycholesterol present in the circulation originates from the brain, a reduction of brain synthesis should lead to a reduction of plasma 24S-hydroxycholesterol, unless voriconazole has an effect on the metabolism in the liver. A pool of plasma from the voriconazole-treated mice had a concentration of 24S- hydroxycholesterol of 13.2 ng/ml. A pool of plasma from the control mice had a concentration of 24S-hydroxycholesterol of 18.8 ng/ml. Thus, voriconazole appeared to suppress the plasma levels by about 30%. It should be pointed out that the plasma level of 27-hydroxycholesterol was not significantly affected by the voriconazole treatment. This oxysterol was at 46 ng/ml and 52 ng/ml in the voriconazole-treated mice and in the controls, respectively.
Effects of voriconazole on the levels of cholesterol precursors upstream of lathosterol
The reduced levels of lathosterol in the voriconazole-treated mice suggest a reduced cholesterol synthesis. Part of this reduction could be due to the inhibition of the cytochrome P450 enzyme CYP51, which is responsible for demethylation of lanosterol, an upstream precursor of lathosterol. In accordance with this, we found that the brain levels of lanosterol increased from 6.1 ± 2.0 ng/mg tissue in the controls to 27 ± 10 ng/mg tissue in the voriconazole-treated mice (P < 0.001). The levels of dihydrolanosterol were 0.14 ± 0.05 ng/mg tissue and 1.9 ± 0.7 ng/mg tissue, respectively. The levels of a lanosterol precursor squalene were 2.2 ± 0.8 ng/mg tissue in the controls and 1.7 ± 0.6 ng/mg tissue in the voriconazole-treated mice (P = 0.05). The latter is consistent with a reduction of cholesterol synthesis, also, at a step prior to lanosterol demethylation.
Effects of voriconazole on mRNA levels of genes involved in cholesterol homeostasis
Voriconazole had no significant effect on expression of CYP46A1 mRNA (Fig. 5A) or HMG-CoA synthase (Fig. 5C). However, there was a significant suppressive effect on the HMG-CoA reductase mRNA levels (Fig. 5B). The latter is consistent with the inhibitory effect of voriconazole on the brain levels of squalene and lathosterol.
Fig. 5.

Relative levels of mRNA expression in mouse brain after 5 daily intraperitoneal injections (60 mg/kg body weight) with voriconazole. A: CYP46A1; B: HMG-CoA Reductase; C: HMG-CoA Synthase. Error bars indicate SEM.
Brain levels of voriconazole
The level of voriconazole in the brain of the mice after five daily intraperitoneal injections (60 mg/kg body weight) was 43 ± 8 μg/g wet weight corresponding to 123 μM. Kinetics of brain levels of voriconazole after a single injection (60 mg/kg) is shown in Fig. 6.
Fig. 6.

Kinetics of brain levels of voriconazole after a single intraperitoneal injection (60 mg/kg body weight). The data is from one mouse at each time point.
DISCUSSION
In the present study, we tested four antifungal azoles that are used systemically and three cholesterol-metabolizing P450s that are known to be expressed in the brain. First, we established that voriconazole binds with high affinity to CYP46A1 in vitro and efficiently inhibits CYP46A1-catalyzed cholesterol hydroxylations in the reconstituted system. Then, in accordance with this finding and the fact that voriconazole can cross the blood-brain barrier, we detected a statistically significant decrease in 24S- hydroxycholesterol levels in the brain in mice injected intraperitoneally with voriconazole for 5 days. We also observed a reduction in the levels of 24S-hydroxycholesterol in the circulation.
There are two different explanations for the reduced levels of 24S-hydroxycholesterol in the brain of the treated animals. The first explanation is a direct inhibition of the enzyme by voriconazole. The very high levels of voriconazole measured in the brain are in accord with this. The second possibility is related to the fact that voriconazole is an inhibitor of cholesterol synthesis. It has been shown that voriconazole exerts its antifungal effect through inhibition of cytochrome P450 14-α sterol demethylase, CYP51, which is responsible for the demethylation of lanosterol in the ergosterol biosynthesis pathway (22). Therefore, a reduced synthesis of the brain cholesterol may lead to a reduced substrate availability for CYP46A1 and reduced production of 24S-hydroxycholesterol. These two alternative mechanisms whereby voriconazole may inhibit the production of 24S-hydroxycholesterol are shown in Fig. 7.
Fig. 7.

Interactions between voriconazole and synthesis of cholesterol and 24S-hydroxycholesterol in the brain.
The reduced levels of cholesterol precursors squalene and lathosterol as well as HMG-CoA reductase mRNA indicate that brain cholesterol synthesis was reduced in the voriconazole-treated mice. The fact that the treatment had no significant effect on the size of the pool of cholesterol in the brain does not favor the hypothesis that reduced substrate availability is of importance for the reduced production of 24S-hydroxycholesterol. A primary effect of voriconazole on CYP46A1 can be expected to lead to a reduced metabolism of brain cholesterol and, thus, a reduced need for de novo synthesis. If the reduced rate of lanosterol demethylation had been the most important effect, a compensatory increase of HMG-CoA reductase mRNA would have been expected. In this connection it is of interest that a complete gene knock-out of CYP46A1 in mice is associated with a reduction of cholesterol synthesis by about 40% (2).
The expression of the HMG-CoA reductase gene is regulated by sterol-regulatory element binding protein (SREBP)-2, and we had expected a significant effect on expression of another SREBP-2 regulated gene, HMG-CoA synthase. Our failure to demonstrate this may be related to the interindividual variations that may be too great to detect a relatively small difference. Addition of 24S-hydroxycholesterol to cultures of primary neuronal cells from embryonic rat pups was also shown to significantly decrease the protein level of HMG-CoA synthase (23). Levels of cholesterol are low in such embryonic cells and the cultures were carried out in cholesterol-deficient medium. Therefore, the rate of cholesterol synthesis should be high. These conditions are markedly different from those of adult neuronal cells in vivo, and the levels of transcriptional factors may be very different in embryonic cells.
Treatment with voriconazole represents a less drastic experimental model for inhibition of CYP46A1 than the knock-out model. Thus, voriconazole may be used for detailed studies on the relation between CYP46A1 activity and cholesterol homeostasis. In such studies, it is necessary to define the degree of inhibition of the enzyme by measurement of the product 24S-hydroxycholesterol in the brain.
One of the most important side effects of voriconazole is visual disturbances. Because CYP46A1 is expressed in the neural retina as well (24, 25), the possibility may be considered that the voriconazole-induced visual disturbances are related to an interaction between the drug and retinal CYP46A1. If this is the case, visual disturbances would be expected in CYP46A1 gene knock-out mice. Consistent with this contention, abnormalities in the ERG signals have recently been demonstrated in such mice (D. Russell, personal communication). Further work on a possible relation between vision and cholesterol 24S-hydroxylase is in progress.
Footnotes
This work was supported by grants from the Swedish Science Council and Brain Power (to I.B.), from the National Institutes of Health (GM062882 and AG024336 to I.A.P.), and by the Jules and Doris Stein Professorship from Research to Prevent Blindness Foundation (to I.A.P.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Björkhem I., Lutjohann D., Breuer O., Sakinis A., Wennmalm A. 1997. Importance of a novel oxidative mechanism for elimination of brain cholesterol. Turnover of cholesterol and 24(S)-hydroxycholesterol in rat brain as measured with 18O2 techniques in vivo and in vitro. J. Biol. Chem. 272: 30178–30184. [DOI] [PubMed] [Google Scholar]
- 2.Lund E. G., Xie C., Kotti T., Turley S. D., Dietschy J. M., Russell D. W. 2003. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 278: 22980–22988. [DOI] [PubMed] [Google Scholar]
- 3.Ohyama Y., Meaney S., Heverin M., Ekstrom L., Brafman A., Shafir M., Andersson U., Olin M., Eggertsen G., Diczfalusy U., et al. 2006. Studies on the transcriptional regulation of cholesterol 24- hydroxylase (CYP46A1): marked insensitivity toward different regulatory axes. J. Biol. Chem. 281: 3810–3820. [DOI] [PubMed] [Google Scholar]
- 4.Mast N., White M. A., Björkhem I., Johnson E. F., Stout C. D., Pikuleva I. A. 2008. Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc. Natl. Acad. Sci. USA. 105: 9546–9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mast N., Norcross R., Andersson U., Shou M., Nakayama K., Björkhem I., Pikuleva I. A. 2003. Broad substrate specificity of human cytochrome P450 46A1 which initiates cholesterol degradation in the brain. Biochemistry. 42: 14284–14292. [DOI] [PubMed] [Google Scholar]
- 6.Murtazina D. A., Andersson U., Hahn I. S., Björkhem I., Ansari G. A., Pikuleva I. A. 2004. Phospholipids modify substrate binding and enzyme activity of human cytochrome P450 27A1. J. Lipid Res. 45: 2345–2353. [DOI] [PubMed] [Google Scholar]
- 7.Pikuleva I. A., Puchkaev A., Björkhem I. 2001. Putative helix F contributes to regioselectivity of hydroxylation in mitochondrial cytochrome P450 27A1. Biochemistry. 40: 7621–7629. [DOI] [PubMed] [Google Scholar]
- 8.Mast N., Andersson U., Nakayama K., Björkhem I., Pikuleva I. A. 2004. Expression of human cytochrome P450 46A1 in Escherichia coli: effects of N- and C-terminal modifications. Arch. Biochem. Biophys. 428: 99–108. [DOI] [PubMed] [Google Scholar]
- 9.Copeland R. A. 2000. Protein-ligand binding equilibria. Enzymes. Copeland R. A., editor A John Wiley & Sons, Inc, New York: 76–108. [Google Scholar]
- 10.Folch J., Lees M., Sloane Stanley G. H. 1957. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226: 497–509. [PubMed] [Google Scholar]
- 11.Folch J., Ascoli I., Lees M., Meath J. A., Le B. N. 1951. Preparation of lipide extracts from brain tissue. J. Biol. Chem. 191: 833–841. [PubMed] [Google Scholar]
- 12.Dzeletovic S., Breuer O., Lund E., Diczfalusy U. 1995. Determination of cholesterol oxidation products in human plasma by isotope dilution-mass spectrometry. Anal. Biochem. 225: 73–80. [DOI] [PubMed] [Google Scholar]
- 13.Heverin M., Bogdanovic N., Lutjohann D., Bayer T., Pikuleva I., Bretillon L., Diczfalusy U., Winblad B., Björkhem I. 2004. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer's disease. J. Lipid Res. 45: 186–193. [DOI] [PubMed] [Google Scholar]
- 14.Björkhem I., Lutjohann D., Diczfalusy U., Stahle L., Ahlborg G., Wahren J. 1998. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 39: 1594–1600. [PubMed] [Google Scholar]
- 15.Tamboli I. Y., Prager K., Thelen K. M., Dewachter I., Pietrzik C. U., St George-Hyslop P., Sisodia S. S., De Srooper B., Heneka M. T., Filippov M. A., et al. 2008. Loss of gamma-secretase function impairs endocytosis of lipoprotein particles and membrane cholesterol homeostasis. J. Neurosci. 28: 12097–12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livak K. J., Schmittgen T. D. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 17.Dawson J. H., Andersson L. A., Sono M. 1982. Spectroscopic investigations of ferric cytochrome P-450-CAM ligand complexes. Identification of the ligand trans to cysteinate in the native enzyme. J. Biol. Chem. 257: 3606–3617. [PubMed] [Google Scholar]
- 18.Poulos T. L., Howard A. J. 1987. Crystal structures of metyrapone- and phenylimidazole-inhibited complexes of cytochrome P-450cam. Biochemistry. 26: 8165–8174. [DOI] [PubMed] [Google Scholar]
- 19.Brown J., 3rd, Theisler C., Silberman S., Magnuson D., Gottardi-Littell N., Lee J. M., Yager D., Crowley J., Sambamurti K., Rahman M. M., et al. 2004. Differential expression of cholesterol hydroxylases in Alzheimer's disease. J. Biol. Chem. 279: 34674–34681. [DOI] [PubMed] [Google Scholar]
- 20.Stoffel-Wagner B. 2001. Neurosteroid metabolism in the human brain. Eur. J. Endocrinol. 145: 669–679. [DOI] [PubMed] [Google Scholar]
- 21.Lund E., Sisfontes L., Reihner E., Bjorkhem I. 1989. Determination of serum levels of unesterified lathosterol by isotope dilution-mass spectrometry. Scand. J. Clin. Lab. Invest. 49: 165–171. [DOI] [PubMed] [Google Scholar]
- 22.Jeu L., Piacenti F. J., Lyakhovetskiy A. G., Fung H. B. 2003. Voriconazole. Clin. Ther. 25: 1321–1381. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y., Muneton S., Sjovall J., Jovanovic J. N., Griffiths W. J. 2008. The effect of 24S-hydroxycholesterol on cholesterol homeostasis in neurons: quantitative changes to the cortical neuron proteome. J. Proteome Res. 7: 1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bretillon L., Diczfalusy U., Björkhem I., Maire M. A., Martine L., Joffre C., Acar N., Bron A., Creuzot-Garcher C. 2007. Cholesterol-24S-hydroxylase (CYP46A1) is specifically expressed in neurons of the neural retina. Curr. Eye Res. 32: 361–366. [DOI] [PubMed] [Google Scholar]
- 25.Ramirez D. M., Andersson S., Russell D. W. 2008. Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J. Comp. Neurol. 507: 1676–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]