

The discovery that chiral hydrogen-bond donors effectively promote highly enantioselective reactions of cationic intermediates has opened a new direction within the field of organocatalysis.[1,2] For example, the discovery of thiourea-catalyzed enantioselective acyl-Pictet–Spengler-type cyclizations has helped bring N-acyliminium ions into the realm of viable substrates for asymmetric catalysis.[2a-d,2f,3] Harnessing N-acyliminium ions for enantioselective intermolecular addition of indoles (e.g., eq. 1) would allow direct access to functionalized indole frameworks with established biological activity,[4] and also provide useful precursors to more complex alkaloid natural product targets.[5] There have been several recent reports of intermolecular asymmetric catalytic additions of indoles to acyclic imines.[6] In most cases, the electrophilic partners have been limited to substituted benzaldehyde derivatives, although Deng and coworkers also identified successful reactions with a variety of acyclic aliphatic imines.[6c] Herein we report highly enantioselective intermolecular additions of indoles to hydroxylactam-derived cyclic N-acyliminium ions catalyzed by a new thiourea-Schiff base derivative.
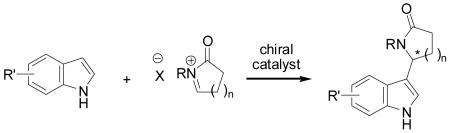 |
(1) |
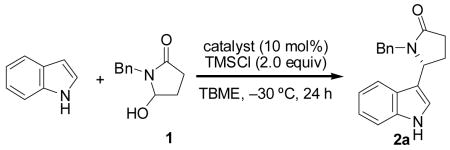
We selected as a model reaction the addition of indole to succinimide-derived hydroxylactam 1, a bench-stable, storable compound that was easily prepared and handled (eq. 2).7 A variety of thiourea and urea catalyst frameworks were evaluated as potential catalysts under conditions similar to those developed for the related intramolecular acyl-Pictet–Spengler cyclization (Table 1).2c A promising lead result was obtained in the reaction promoted by Schiff-base 4a8 in TBME at −30 °C, with adduct 2a generated in 55% ee and 30% yield (entry 2). No background reactivity was observed under these conditions in the absence of catalyst. Interestingly, pyrrole derivative 3, the optimal catalyst identified for the intramolecular acyl-Pictet–Spengler reaction,2c afforded the opposite enantiomer in low ee (entry 1).
Table 1.
Catalyst Optimization Studies
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | R1 | R2 | R3 | R4 | R5 | ee (%) |
| 1 | 3 | -- | -- | -- | -- | -- | −14 |
| 2 | 4a | Me | H | tBu | tBu | tBu | 55 |
| 3 | 4b | H | H | tBu | tBu | tBu | 35 |
| 4 | 4c | Me | H | tBu | tBu | NO2 | 13 |
| 5 | 4d | Me | H | tBu | tBu | OTIPS | 21 |
| 6 | 4e | Me | H | tBu | tBu | Cl | 40 |
| 7 | 4f | Me | H | tBu | Me | tBu | 23 |
| 8 | 4g | Me | H | tBu | SiEt3 | tBu | 65 |
| 9 | 4h | Me | (S) Me | tBu | SiEt3 | tBu | 75 |
| 10 | 4i | Me | (R) Me | tBu | SiEt3 | tBu | 80 |
| 11 | 4j | Me | (R) Me | CH2Ph | SiEt3 | tBu | 89 |
| 12 | 5 | 93 | |||||
Systematic variation of the catalyst structure around the basic framework of 4 led to substantial improvements in reaction enantioselectivity (Table 1). Diaminocyclohexane-derived catalysts were examined bearing different Schiff-base, amide, and amino acid side chain components. The primary amine resulting from hydrolysis of Schiff base 4a was found to be catalytically active, but afforded racemic product. Thus, we recognized that maintaining the integrity of the Schiff base throughout the reaction would be essential to obtaining high enantioselectivity. After extensive experimentation, it was found that catalyst 4g, bearing a triethylsilyl group at R4 and a tert-butyl group at R5, promoted formation of 2a with slightly higher enantioselectivity relative to catalyst 4a (65% vs. 55% ee).
Further improvement in enantioselectivity was achieved by introduction of a stereochemical element at the benzylic position of the amide, with the (R)-methyl catalyst 4i providing 2a in 80% ee (entry 10). While the majority of effective thiourea catalysts identified to date contain a tert-leucine backbone (R3 = tBu),[9] catalyst 4j derived from phenylalanine proved substantially more enantioselective than tert-leucine analog 4i (89 vs 80% ee, entry 11). Finally, constraining the chiral amide component in a cyclic framework as in catalyst 5 led to additional improvement in enantioselectivity, with generation of 2a in 93% ee (entry 12).[10-12]
 |
(2) |
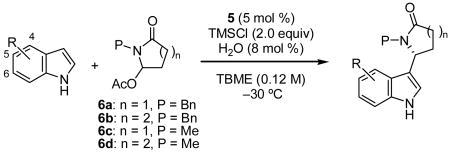
Despite the high ee's obtained with catalyst 5, the efficiency of product formation using hydroxylactam 1 was quite modest (30–40% conversion with 10 mol% catalyst). This was attributable, at least in part, to the poor solubility of 1 under the reaction conditions, so several derivatives of the hydroxylactam substrate were prepared with the aim of discovering a more suitable N-acyliminium ion precursor. Acetyl derivative 6a, prepared by acylation of 1, displayed improved solubility and underwent conversion to 2a in 93%ee and 71% isolated yield (Table 2).
Table 2.
Alkylation of Electron-Rich Indole Substrates[a]
 | ||||||
|---|---|---|---|---|---|---|
| Entry | R (indole) | lactam | Time (h) | Product | Yield [%][b] | ee [%] |
| 1 | H | 6a | 24 | 2a | 90 | 93 (99)[c] |
| 2 | H | 6c | 24 | 2b | 93 | 86 |
| 3 | 4-Me | 6a | 24 | 2c | 86 | 95 |
| 4 | 5-Me | 6a | 24 | 2d | 79 | 90 (91)[c] |
| 5 | 5-CH2CH2 | 6a | 24 | 2e | 82 | 90 |
| 6 | 6-OMe | 6a | 24 | 2f | 80 | 80 (98)[b] |
| 7 | H | 6d | 48 | 2g | 60 | 92 |
| 8 | H | 6b | 48 | 2h | 70 | 93 |
| 9 | 5-OMe | 6b | 48 | 2i | 86 | 90 |
| 10 | 5-Me | 6b | 48 | 2j | 93 | 94 |
| 11 | 5-CH2CH2 | 6b | 48 | 2k | 92 | 91 |
| 12 | 6-OMe | 6b | 48 | 2l | 76 | 88 |
Unless noted otherwise, reactions were carried out on 0.3 mmol scale.
Isolated yield after chromatographic purification.
Reaction carried out on 3 mmol scale.
Ee of product purified by trituration with Et2O in parentheses.
No reaction was observed in the absence of TMSCl or other acidic additive, and comparison of different TMS-X reagents revealed that the chloride counteranion was uniquely effective for achieving high enantioselectivity.[13-14] Use of BCl3 as an additive in place of TMSCl led to complete substrate conversion after only 8 hours, albeit with slightly diminished ee (91 vs. 93% ee).
The presence of controlled amounts of added water in the TMSCl-promoted reactions also had a pronounced beneficial effect on reactivity.[15] Best results were obtained using 5 mol% catalyst 5, two equivalents of TMSCl in anhydrous TBME, and 8 mol% water, and allowing the reaction to proceed for 24 h at −30 °C. Under these conditions, adduct 2 was generated reproducibly in 93% ee and 84% isolated yield. The synergistic effect of TMSCl and catalytic H2O[16] suggests that acetoxylactam 6 undergoes reaction with in situ-generated HCl to form chlorolactam 7, and that this equilibrium is driven by trapping the acetic acid byproduct with TMSCl.[17] We propose that racemic chlorolactam 7 is the actual substrate in the alkylation, and that the reaction proceeds via an SN1-type anion-binding mechanism analogous to that proposed for related thiourea-catalyzed acyl-Pictet–Spengler and oxocarbenium ion alkylation reactions (Scheme 1).[2c-e]
Scheme 1.

Proposed catalytic cycle
With a reliable protocol in hand, the scope of the reaction was investigated. Under the optimized conditions, a variety of electron-rich indoles were shown to add to both succinimide and glutarimide-derived electrophiles with high enantioselectivity (Table 2).[18] Consistent results were obtained over a ten-fold increase in the scale of the reaction (entry 1). In several cases, we found that the enantiomeric enrichment of the products could be increased simply by trituration.[19]
Encouraged by the scope of the addition with electron-rich indoles, electron-deficient indoles were examined as well. Using the optimized TMSCl conditions, these substrates provided adducts in high enantioselectivity, but in low yield. In contrast, for halogenated indole substrates, the use of 10 mol % BCl3 in the presence of 10 mol% 5 afforded useful yields and high enantioselectivity of products 2m–2u (Table 3).
Table 3.
Alkylation of Electron-Deficient Indoles
 | ||||||
|---|---|---|---|---|---|---|
| Entry | R | n | Time | Product | Yield (%)[a] | ee (%) |
| 1 | 4-F | 1 | 36 h | 2m | 57 | 96 (99)[b] |
| 2 | 4-Br | 1 | 5 d | 2n | 47 | 97 |
| 3 | 5-F | 1 | 48 h | 2o | 87 | 94 |
| 4 | 5-Br | 1 | 5 d | 2p | 80 | 93 |
| 5 | 6-F | 1 | 72 h | 2q | 75 | 96 |
| 6 | 6-Cl | 1 | 96 h | 2r | 81 | 96 |
| 7 | 4-Br | 2 | 5 d | 2s | 12 | 85 |
| 8 | 6-F | 2 | 72 h | 2t | 75 | 96 |
| 9 | 6-Cl | 2 | 5 d | 2u | 40 | 92 |
Isolated yield
Ee of product purified by trituration from Et2O in parenthesis.
In summary, we have developed a highly enantioselective addition of indoles to cyclic N-acyliminium ions using a chiral thiourea Schiff-base catalyst. The scope includes both electron-rich and electron-poor indole nucleophiles. The products are synthetically useful intermediates and are readily elaborated: for example, cleavage of the benzyl protecting group using Na/NH3 proceeds without erosion of enantiomeric excess.[19] Efforts to apply anion-binding principles to other reactions involving cationic intermediates are underway.
Experimental Section
A 10 mL, flame-dried round bottom flask is charged with indole (0.040 g, 0.343 mmol), catalyst 5 (0.012 g, 0.017 mmol) and sealed with a rubber septum. The flask is flushed with N2 and anhydrous TBME (1.6 mL) was added. The resulting yellow solution is cooled to −78 °C, and a TBME solution of acetoxy-lactam 6a (0.65 mL, 0.34 mmol, 0.53 M) was added. Next, TBME solutions of TMSCl (0.43 mL, 0.69 mmol, 1.6 M) and H2O (0.20 mL, 0.03 mmol, 0.14 M) are added sequentially and the solution is warmed to −30 °C and stirred for 24 h. Next, the heterogeneous solution is quenched by the addition of an EtOH solution of NaOEt (0.2 mL, 21 wt%), followed by immediate addition of water (1 mL). The mixture is allowed to warm to rt and is diluted with EtOAc until all solids dissolve (the amount of added EtOAc depends on particular product ∼5–10 mL). The layers are separated and the organic layer is dried (Na2SO4) and concentrated in vacuo. Purification procedures for individual products, as well as the BCl3 protocol for electron-deficient indoles, are provided as Supporting Information.
Supplementary Material
Footnotes
This research was supported by the NIH NIGMS (PO1 GM-69721) and by NIH NRSA grant to E. A. Peterson.
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Dr. Emily A. Peterson, Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford St., Cambridge, MA 02138
Prof. Dr. Eric N. Jacobsen, Email: jacobsen@chemistry.harvard.edu, Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford St., Cambridge, MA 02138, Fax: (+1) 617-496-1880.
References
- 1.For recent general reviews of chiral H-bond catalysts, see:; a) Taylor MS, Jacobsen EN. Angew Chem Int Ed. 2006;45:1520–1543. doi: 10.1002/anie.200503132. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2006;118:1550–1573. [Google Scholar]; b) Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]
- 2.a) Taylor MS, Jacobsen EN. J Am Chem Soc. 2004;126:10558–10559. doi: 10.1021/ja046259p. [DOI] [PubMed] [Google Scholar]; b) Taylor MS, Tokunaga N, Jacobsen EN. Angew Chem Int Ed. 2005;44:6700–6704. doi: 10.1002/anie.200502277. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2005;117:6858–6862. [Google Scholar]; c) Raheem IT, Thiara PS, Peterson EA, Jacobsen EN. J Am Chem Soc. 2007;129:13404–13405. doi: 10.1021/ja076179w. [DOI] [PubMed] [Google Scholar]; d) Raheem IT, Thiara PS, Jacobsen EN. Org Lett. 2008;10:1577–1580. doi: 10.1021/ol800256j. [DOI] [PubMed] [Google Scholar]; e) Reisman SE, Doyle AG, Jacobsen EN. J Am Chem Soc. 2008;130:7198–7199. doi: 10.1021/ja801514m. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Pan PC, List B. Chem Asian J. 2008;3:430–437. doi: 10.1002/asia.200700327. [DOI] [PubMed] [Google Scholar]; g) Zhang Z, Schreiner PR. Chem Soc Rev. 2009;38 doi: 10.1039/b801793j. For a review of anion-binding thiourea organocatalysis, see: in press. [DOI] [PubMed] [Google Scholar]
- 3.For seminal applications of N-acyliminium ions in Al-catalyzed Reissert reactions, see:; a) Takamura M, Funabashi K, Kanai M, Shibasaki M. J Am Chem Soc. 2000;122:6327–6328. doi: 10.1021/ja010654n. [DOI] [PubMed] [Google Scholar]; b) Takamura M, Funabashi K, Kanai M, Shibasaki M. J Am Chem Soc. 2001;123:6801–6808. doi: 10.1021/ja010654n. [DOI] [PubMed] [Google Scholar]; c) Funabashi K, Ratni H, Kanai M, Shibasaki M. J Am Chem Soc. 2001;123:10784–10785. doi: 10.1021/ja016935c. [DOI] [PubMed] [Google Scholar]; d) Ichikawa E, Suzuki M, Yabu K, Albert M, Kanai M, Shibasaki M. J Am Chem Soc. 2004;126:11808–11809. doi: 10.1021/ja045966f. [DOI] [PubMed] [Google Scholar]
- 4.Baldwini IR, Bamborough P, Christopher JA, Kerns JK, Longstaff T, Miller DD. PCT Int Appl. 2005 WO 2005067923. [Google Scholar]
- 5.a) Atta-ur-Rahman B. Indole Alkaloids. Harwood Academic; Chchester, UK: 1998. [Google Scholar]; b) Amat M, Hadida S, Llor N, Sathyanarayana S, Bosch J. J Org Chem. 1996;61:3878–3882. doi: 10.1021/jo952251+. [DOI] [PubMed] [Google Scholar]; c) Shimizu S, Ohori K, Arai T, Sasai H, Shibasaki M. J Org Chem. 1998;63:7547–7551. doi: 10.1021/jo981069g. [DOI] [PubMed] [Google Scholar]
- 6.a) Johannsen M. Chem Comm. 1999:2233–2234. [Google Scholar]; b) Jia YX, Xie JH, Duan HF, Wang LX, Zhou QL. Org Lett. 2006;8:1621–1624. doi: 10.1021/ol0602001. [DOI] [PubMed] [Google Scholar]; c) Wang YQ, Song J, Hong R, Deng LH. J Am Chem Soc. 2006;128:8156–8157. doi: 10.1021/ja062700v. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rowland GB, Rowland EB, Liang Y, Perman JA, Antilla JC. Org Lett. 2007;9:2609–2611. doi: 10.1021/ol0703579. [DOI] [PubMed] [Google Scholar]; e) Jia YX, Zhong J, Zhu SF, Zhang CM, Zhou QL. Angew Chem Int Ed. 2007;46:5565–5567. doi: 10.1002/anie.200701067. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2008;119:5661–5663. [Google Scholar]; f) Kang Q, Zhao ZA, You SL. J Am Chem Soc. 2008;129:1484–1485. doi: 10.1021/ja067417a. [DOI] [PubMed] [Google Scholar]; g) Terada M, Yokoyama S, Sorimachi K, Uraguchi D. Adv Synth Catal. 2007;349:1863–1867. [Google Scholar]; Kang Q, Zheng XJ, You SL. Chem Eur J. 2008;14:3539–3542. doi: 10.1002/chem.200800263. [DOI] [PubMed] [Google Scholar]
- 7.Hubert JC, Wunberg JBPA, Speckamp WN. Tetrahedron. 1975;31:1437–1441. [Google Scholar]
- 8.Wenzel AG, Jacobsen EN. J Am Chem Soc. 2002;124:12964–12965. doi: 10.1021/ja028353g. [DOI] [PubMed] [Google Scholar]
- 9.For important exceptions, see:; a) Fang YQ, Jacobsen EN. J Am Chem Soc. 2008;130:5660–5661. doi: 10.1021/ja801344w. [DOI] [PubMed] [Google Scholar]; b) Klausen RS, Jacobsen EN. Org Lett. 2009;11:887–890. doi: 10.1021/ol802887h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For an efficient asymmetric synthesis of 2-phenylpyrrolidines, see:; Campos KR, Klapars A, Waldman JH, Dormer PG, Chen C. J Am Chem Soc. 2006;128:3538–3539. doi: 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]
- 11.The diastereomer of catalyst 5, prepared from (R)-2-phenylpyrrolidinone, D-phenylalanine, and (S),(S)-diaminocyclohexane provided product 2 in −79% ee.
- 12.2-Arylpyrrolidine-derived thiourea catalysts were also applied successfully to enantioselective alkylations of oxocarbenium ions. See ref. 2e.
- 13.Adduct 2 was obtained in 26% ee using TMSBr with catalyst 4a, and as a racemate using TMSI.
- 14.It has been established previously that thioureas interact much more strongly with chloride than bromide or iodide:; a) Kondo S, Nagamine M, Tano Y. Tetraheron Lett. 2003;44:8801–8804. [Google Scholar]; b) Kondo S, Sato M. Tetrahedron. 2006;62:4844–4850. [Google Scholar]
- 15.Very poor reactivity was observed under strictly anhydrous conditions. Elevated levels of added water (>20 mol%) led to diminished enantioselectivity.
- 16.Reactions carried out with HCl alone proceeded with comparable enantioselectivity but were substantially slower.
- 17.Conversion of 6a to chlorolactam 7 was established and monitored by 1H NMR. Spectra (1H and 13C NMR) of 7 and of related compounds are provided in the Supporting Information.
- 18.The absolute configuration of addition products was determined by single crystal X-Ray analysis of addition product 2s (Table 3), with all other products assigned by analogy.
- 19.See Supporting Information for complete details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.