

Abstract
Ras proteins activate Raf and PI-3 kinases, as well as exchange factors for RalA and RalB GTPases. Many previous studies have reported that the Ral signaling cascade contributes positively to Ras-mediated oncogenesis. Here, utilizing a bioengineered tissue model of early steps in Ras-induced human squamous cell carcinoma of the skin, we found the opposite. Conversion of Ras-expressing keratinocytes from a premalignant to malignant state induced by decreasing E-cadherin function was associated with and required a knockdown of RalA to a similar degree by shRNA expression in these cells decrease in RalA expression. Moreover, direct ∼2-3 fold knockdown of RalA by shRNA expression in these cells reduced E-cadherin levels and also induced progression to a malignant phenotype. Knockdown of the Ral effector, Exo84, mimicked the effects of decreasing RalA levels in these engineered tissues. These phenomena can be explained by our finding that the stability of E-cadherin in Ras-expressing keratinocytes depends upon this RalA signaling cascade. These results imply that an important component of the early stages in squamous carcinoma progression may be a modest decrease in RalA gene expression that magnifies the effects of decreased E-cadherin expression by promoting its degradation.
Introduction
Squamous cell carcinomas (SCC) represent 90% of head and neck cancers and ∼25% of skin cancers (Dlugosz et al., 2002). A significant proportion of these cancers contain either mutations in Ras genes that lead to constitutively-activated Ras GTPases or amplified levels of Ras proteins (Dajee et al., 2003; Pierceall et al., 1991). While activated Ras is known to promote tumorigenesis, the molecular basis for its contribution is not completely understood. Ras proteins activate at least three downstream signaling cascades mediated by Raf kinases, PI-3 kinases, and Ral guanine nucleotide exchange factors (GEFs) that in turn activate RalA and RalB GTPase (Takai et al., 2001). Raf and PI-3 kinases are well-known mediators of the tumorigenic effects of oncogenic Ras. However, the role of the Ral-GEF branch of the Ras pathway is far less understood. Ral-GEFs activate two very similar Ral GTPases, RalA and RalB (Feig, 2003). When activated, Ral proteins can bind to at least four effector proteins, the RalBP-1 (Cantor et al., 1995; Jullien-Flores et al., 1995), the exocyst subunits Sec5 and Exo84 (Brymora et al., 2001; Moskalenko et al., 2002; Moskalenko et al., 2003; Polzin et al., 2002; Sugihara et al., 2002), and the transcription factor ZONAB (Frankel et al., 2005). Despite their ∼85% identity, RalA and RalB play very different roles in cells, presumably due to their distinct subcellular localization (Lim et al., 2005; Shipitsin & Feig, 2004) and differences in affinities for effectors (Fenwick et al., 2009; Shipitsin & Feig, 2004). For example, we showed that RalA, but not RalB, promotes delivery of basolateral membrane proteins to the cell surface, a function consistent with a regulator of the exocyst (Shipitsin & Feig, 2004). RalB, but not RalA, activates the atypical IkB kinase family member TBK1 through the exocyst subunit Sec5 to regulate the innate immune response (Chien et al., 2006). RalB through Sec5 and also appears to be involved in cell migration (Rosse et al., 2006) and RalA and RalB influence cytokinesis differently (Cascone et al., 2008).
Many previous studies have indicated that Ral-GEFs, RalA and RalB play positive roles in transformation of cells containing an oncogenic form of Ras (Bodemann & White, 2008). For example, expression of an activated Ral-GEF or RalA mimics (although to a significantly reduced degree) Ras in contributing to the oncogenic transformation of primary kidney cells (Hamad et al., 2002; Lim et al., 2005). RalA-GTP levels appear to be elevated in pancreatic cancer cells, and knockdown of RalA or RalB suppresses metastasis (Lim et al., 2006). In other studies, activation of RalA contributed to the growth of tumor cells in suspension (Lim et al., 2005). In addition, the tumor suppressor PP2A inhibits RalA activation by dephosphorylating its C-terminus (Sablina et al., 2007). RalB suppresses apoptosis in tumor cells through activation of the TBK1 kinase (Chien et al., 2006). Finally, RalGDS knockout mice display resistance to tumor promoter induced squamous cell carcinoma of the skin (Gonzalez-Garcia et al., 2005).
Nevertheless, there are experimental examples showing that Ral GTPases can suppress tumorigenesis. For example, expression of active RalB blocked, and inhibition of RalB expression enhanced, transformation of primary cells induced by an activated Ral-GEF (Lim et al., 2005). Active RalA also blocked migration of bladder cancer cells (Oxford et al., 2005). Finally, RalA sensitized astrocytomas to TRAIL-induced apoptosis (Panner et al., 2006).
In this report, we expanded our studies using a bioengineered 3D tissue model of Ras-induced human squamous carcinoma of the skin to reveal a tumor-suppressing role for RalA in the early steps in tumorigenesis, which functions by enhancing E-cadherin stability in keratinocytes through its effector protein, the exocyst subunit Exo84. Moreover, we found that tumor progression in this system that is driven by down-regulation of E-cadherin is associated with and requires modest down-regulation of RalA expression.
Results
Conversion of Ras-expressing keratinocytes from a premalignant to malignant state by decreasing E-cadherin expression is associated with and requires the suppression of RalA expression
In order to gain new insight into the role of Ral proteins in Ras-induced carcinoma, we turned to a well-characterized, engineered tissue model that mimics the initial stages of squamous cell carcinoma progression (Garlick, 2007). This organotypic system uses immortalized human keratinocytes (HaCaT cells) grown on top of fibroblast-populated collagen gels. Expression of oncogenic H-Ras in these cells (referred to as HaCaT-II-4, “II-4” cells) induces dysplasia but not invasion into the dermal layer, consistent with findings from transgenic mouse studies where active H-Ras was expressed specifically in keratinocytes (Bailleul et al., 1990). However, suppression of E-cadherin levels in II-4 cells induced by the expression of a dominant-negative form of E-cadherin (H-2Kd-Ecad) (Zhu & Watt, 1996) results in a transition to an invasive cell phenotype in vitro, and a switch to a high-grade malignant phenotype following transplantation of this tissue to immune-deficient mice (Margulis et al., 2005a).
We observed that decreased E-cadherin protein levels in II-4-H-2Kd-Ecad cells was associated with a modest (∼60%) decrease in the expression of both RalA and RalB proteins (Fig. 1A,B). To test whether a similar phenomenon occurs when E-cadherin levels are reduced by down-regulation of E-cadherin mRNA, a more common phenomenon in tumor progression, HaCaT-II-4 cells were stably infected with lentivirus expressing either control shRNA (“sh-Scram”) or shRNA against E-cadherin (“sh-Ecad”) (Fig. 1D). Cells that displayed reductions in E-cadherin levels comparable to those found in II-4-H-2Kd-Ecad cells also displayed comparable down-regulation of Ral proteins: ∼2-fold for RalA and ∼3-fold for RalB (Fig. 1E). Real-time PCR analysis on cDNA derived from H-2Kd-Ecad- and sh-Ecad-expressing II-4 cells showed comparable decreases in mRNA of the RALA and RALB genes, implying that E-cadherin regulates Ral proteins predominantly at the level of transcription (Fig. 1C,F).
Figure 1. Loss of E-cadherin function induces down-regulation of RalA and RalB in HaCaT keratinocytes.

Western blot analysis of HaCaT, Ras-expressing HaCaT-II-4, and HaCaT-II-4 cells expressing the dominant-negative E-cadherin transgene H-2Kd-Ecad (A). Total Erk is shown as a loading control. RalA and RalB protein (B) or mRNA (C) expression were measured in the linear range by densitometry or real-time PCR, respectively. (D) Western blot analysis of HaCaT-II-4 and HaCaT-II-4 cells expressing shRNA against E-cadherin. Total Erk is shown as a loading control. RalA and RalB protein (E) or mRNA (F) expression were measured in the linear range by densitometry or real-time PCR, respectively.
To test whether other mechanisms of E-cadherin down-regulation in different cell types lead to a similar phenomenon, MSCC-1, a cell line derived from a lymph node metastasis of oral squamous cell carcinoma, was compared to an E-cadherin-deficient line, MSCC-1-Inv-1, that was derived from it (Kudo et al., 2003). Immunoblotting of lysates from these cells revealed that RalA and RalB levels were reduced ∼2-fold and 1.5 fold, respectively, in the E-cadherin-lacking MSCC-1-Inv-1 cells (Supplementary Fig. 1A,B). As before, real-time PCR demonstrated that this effect on Ral proteins could be explained by a reduction in Ral mRNA expression (Supplementary Fig. 1C). Overall, these series of experiments demonstrate that Ral levels are tied directly to E-cadherin levels in these transformed cells.
The finding that E-cadherin loss leads to the down-regulation of RalA and RalB raised the possibility that these GTPases normally suppress, rather than promote, the transition of Ras-expressing, non-invasive keratinocytes to a malignant state. It also suggested that removal of the tumor-suppressive effect of Ral proteins by a decrease in their expression by ∼50% is required for tumor progression to occur in this model system. To test these hypotheses, RalA or RalB levels in invasive II-4-H-2Kd-Ecad cells were restored back to those found in parental non-invasive II-4 cells by infection with a wild-type RalA or RalB-expressing retrovirus (Fig. 2A). While II-4 cells formed tightly packed colonies where E-cadherin was localized to cell-cell borders in 2-D cultures (Fig. 2B panels a,e), II-4 cells expressing dominant negative E-cadherin did not, and instead displayed a scattered phenotype consistent with their low E-cadherin levels (Fig. 2B panels b,f). Strikingly, II-4-H-2Kd cells in which RalA expression was restored to levels found in parental II-4 cells did form tightly packed colonies, and displayed E-cadherin at cell-cell borders (Fig. 2B panels c,g). Immunoblotting lysates from these cells showed that endogenous levels of E-cadherin are similar to those found in parental II-4 cells despite the continued expression of the dominant-negative transgene (Fig. 2A). In contrast, re-expressing RalB at levels similar to those found in control II-4 cells had no effect on cell scattering, the level of endogenous E-cadherin expression, or its subcellular localization (Fig. 2B panels d,h). We repeated these experiments in cells where E-cadherin levels were reduced (∼5-fold) by shRNA expression (Supplementary Fig. 2A) and found the same results. (Supplementary Fig. 2A and B).
Figure 2. Suppression of RalA expression is necessary for the migratory and invasive properties associated with the loss of E-cadherin function.

(A) Western blot analysis of Ras-expressing HaCaT-II-4 cells expressing empty vector or H-2Kd-Ecad transgene infected with retroviruses encoding wild-type RalA or RalB. Total Erk is shown as a loading control. (B) Phase-contrast and immunofluorescent micrographs showing colony morphology and E-cadherin membrane localization in HaCaT-II-4 (a, e), HaCaT-II-4-H-2Kd-Ecad (b, f), HaCaT-II-4-H-2Kd-Ecad-RalAwt (c, g), and HaCaT-II-4-H-2Kd-Ecad-RalBwt (d, h) cells. Bar: 100μm. (C) The indicated cell lines were incubated with 10μM cycloheximide for the indicated amount of time (n≥3). Endogenous E-cadherin protein expression was analyzed by Western blot and quantified by densitometry. (D) E-cadherin mRNA expression in the indicated cell lines was analyzed by real-time PCR. (E) Cells from the indicated lines were grown on fibroblast-populated organotypic collagen gels and tissues were stained with H&E. Bar: 100μm. Invading cells in representative images are shown with arrows and quantified from >100 microscope fields in >20 sections from multiple experiments (F). * p < 0.05 for H-2Kd-Ecad/RalAwt compared to H-2Kd-Ecad/Bleo using Mann-Whitney test.
Moreover, when cells with restored RalA expression were used to populate the epithelium of an engineered tissue (Fig. 2E panel c), their invasive properties were similar to control cells and dramatically reduced compared to II-4-H-2Kd-Ecad expressing empty vector or wild-type RalB (Fig. 2E panels b,d (insert, black arrows)). Quantification of invasion over multiple tissue sections illustrated that re-expression of wild-type RalA reduced invasion by ∼80% (Fig. 2F). Similar results were observed with sh-Ecad-expressing II-4 cells with wild-type RalA re-expressed (Supplementary Figs. 2E panel c, 2F). Thus, modest down-regulation of RalA gene expression associated with suppressed E-cadherin function is a necessary step for tumor progression by Ras-expressing keratinocytes, presumably because RalA normally functions to maintain E-cadherin levels.
How does restoring RalA to levels found in parental II-4 cells restore E-cadherin expression and function in these cells? We showed previously that RalA, but not RalB, promotes the delivery of E-cadherin from recycling endosomes to the basolateral surface of MDCK cells (Shipitsin & Feig, 2004). E-cadherin is known to be removed from the cell surface by endocytosis, where it is targeted for degradation or recycling back to the plasma membrane (Jeanes et al., 2008). Thus, we hypothesized that active RalA enhances E-cadherin levels by promoting its recycling back to the plasma membrane, and thus avoiding the degradation pathway (see model in Fig 8). It has been previously reported that H-2Kd-Ecad promotes degradation of endogeous E-cadherin (Zhu & Watt, 1996). To test the hypothesis that RalA stabilizes E-cadherin at the plasma membrane, E-cadherin turnover rate was assayed by treatment of cultures with 10μM cycloheximide (Fig. 2C). As predicted from previous studies, compared to II-4 cells (black squares), in which E-cadherin had a half-life of ∼20h, E-cadherin in II-4-H-2Kd-Ecad cells (red triangles) had a shortened half-life of ∼4h. Re-expression of wild-type RalA extended the half-life of endogenous E-cadherin to near normal levels (blue circles) while in II-4-H-2Kd-Ecad cells re-expressing wild-type RalB (green diamonds) E-cadherin turnover remained accelerated. Dominant-negative E-cadherin expression had no effect on endogenous E-cadherin mRNA expression levels, nor did the re-expression of either wild-type RalA or wild-type RalB (Fig. 2D). RalA re-expression also enhanced E-cadherin stability when E-cadherin levels were reduced by shRNA expression (Supplementary Fig. 2C, blue circles), prolonging its half-life from 24 to ∼48h. Furthermore, re-expression of either RalA or RalB did not change the expression of E-cadherin mRNA, which remained reduced by ∼80% due to the expression of sh-Ecad (Supplementary Fig. 2D). Overall, these findings support the model described in Fig. 8, where down-regulation of RalA expression associated with E-cadherin suppression potentiates E-cadherin loss by promoting its turnover in cells.
Figure 8. Model of RalA regulation of E-cadherin function during tumorigenesis.

RalA suppresses Ras-induced tumor progression by promoting delivery and recycling of E-cadherin to the plasma membrane (solid arrows). In contrast, other Ras effectors, such as Raf, and PI-3 K enhance tumorigenesis by promoting removal of E-cadherin from the membrane and its subsequent degradation (hatched arrows).
To test this hypothesis further, we studied E-cadherin-depleted MSCC-1-Inv-1 cells, which also display reduced levels of RalA compared to their E-cadherin expressing counterparts, MSCC-1 (Supplementary Fig. 1A). Importantly, these cells display no detectable E-cadherin protein due to hypermethylation of the E-cadherin promoter (Kudo et al., 2003). Unlike in cells that express reduced levels of E-cadherin protein, re-expression of RalA in MSCC-1-Inv-1 cells back to those found in parental MSCC-1 cells did not restore E-cadherin protein expression (Supplementary Fig. 3). This finding supports the hypothesis that RalA regulates E-cadherin protein stability but not expression of its gene.
shRNA-mediated down-regulation of RalA expression by ∼60% in Ras-expressing keratinocytes reduces E-cadherin levels and induces an invasive phenotype in engineered tissues
To test whether direct suppression of RalA expression in Ras-expressing keratinocytes enhances their tumor progression in this bioengineered tissue model of SCC, RalA and RalB expression levels were directly knocked down in II-4 cells. Stable expression of shRNA against each GTPase was generated through lentiviral vector infection, and drug selected pools of II-4 cells were obtained. Knockdown efficiency was ∼60% for either RalA “sh-RalA” or RalB “sh-RalB” II-4 cells (Fig. 3A,B) when compared to control “sh-Scram,” which was comparable to the level of protein reduction found when E-cadherin levels were suppressed (see Fig. 1A). A second sequence for both RalA and RalB was also tested, with similar levels of knockdown (Supplementary Fig. 4A). When these cells were used to populate the epithelia of tissues, sh-Scram and sh-RalB showed typical dysplastic, non-invasive behavior (Fig. 3C panels a,c and Supplementary Fig. 4B panels a,c). Strikingly, both types of II-4 cells expressing either one of sh-RalA sequences (Fig. 3C panel b and Supplementary Fig. 4B panel b) showed clear invasive behavior, as significant numbers of cells (Fig. 3D), usually as clusters, breached the basement membrane to initiate invasion into the dermal layer (Fig. 3C panel b, black arrows) in amounts similar to that seen when E-cadherin function is suppressed (see Fig. 2F). This invasive phenotype was reversed by the expression of RNAi-resistant wild-type RalA but not empty vector (Supplementary Fig. 4C,D). In other systems, knockdown of RalB had the effect of reversing the phenotype of RalA knockdown (Chien & White, 2003; Oxford et al., 2005). This was not the case in the present study, as knockdown of RalB in sh-RalA cells did not abrogate their invasive phenotype (Fig. 3C panel d).
Figure 3. Suppression of RalA expression by shRNA induces invasive properties in 3D tissues.

(A) Western blot analysis of Ras-expressing HaCaT-II-4 cells expressing shRNA against RalA, RalB, or both RalA and RalB. Total Erk is shown as a loading control. (B) Quantification of RalA or RalB knockdown compared to control determined by densitometry in the linear range. (C) Scrambled (a), RalA knockdown (b), RalB knockdown (c), or RalA/RalB double-knockdown (d) HaCaT-II-4 cells were grown on collagen gels and tissues were stained with H&E. Bar: 100μm. Invading cells in representative images are shown with arrows and quantified from >100 microscope fields in >20 sections from multiple experiments (D). * p < 0.05 for sh-RalA and sh-RalA/B compared to sh-Scram using Mann-Whitney test.
To test whether RalA suppression in keratinocytes is sufficient to promote cell invasion, RalA was knocked down ∼65% in parental HaCaT keratinocytes from which Ras-expressing II-4 cells were derived (Supplementary Fig. 5A). sh-RalA HaCaT cells did not display an increase in invasion (Supplementary Fig. 5B panels a-c). Furthermore, when dominant-negative H-2Kd-Ecad was expressed in parental HaCaT cells, no decrease was observed in the expression of RalA or RalB proteins (Supplementary Fig. 5C). Thus, Ras activity, must be stimulated in order for down-regulation of RalA to promote invasion and for loss of E-cadherin to downregulate RalA and RalB.
Suppression of Exo84 expression in Ras-expressing keratinocytes mimics the phenotype of RalA knockdown
To test which RalA effector is involved, we utilized lentiviral vectors to knock down the well-characterized effectors Exo84, Sec5, and RalBP-1 (Fig. 4A) to similar degrees in Ras-expressing II-4 cells. When these cells were seeded on contracted collagen gels to generate epithelial tissues, only the cells expressing shRNA against Exo84 (“sh-Exo84”) displayed an invasive morphology (Fig. 4B panel b). The number of invasive cells (Fig. 4C) was comparable to when RalA expression was suppressed by shRNA (see Fig. 3D).
Figure 4. Suppression of the Ral effector Exo84 expression by shRNA induces invasive properties of HaCaT II-4 cells in 3D tissues.

(A) Western blot analysis of Ras-expressing HaCaT-II-4 cells expressing scrambled shRNA or shRNA against Exo84, Sec5, or RalBP-1. Total Erk is shown as a loading control. (B) Scrambled (a), Exo84 knockdown (b), Sec5 knockdown (c), or RalBP-1 knockdown (d) HaCaT-II-4 cells were used to populate tissues as described and stained with H&E. Bar: 100μm. Invading cells in representative images are shown with arrows and quantified from >100 microscope fields in >20 sections from multiple experiments (C) and two different shRNA sequences. * p < 0.01 for sh-Exo84 compared to sh-Scram using Mann-Whitney test.
Knockdown of RalA or its effector Exo84 is associated with reduced E-cadherin levels in tissues
The results from Figure 2 implied that decreased RalA levels reduce E-cadherin levels. To further test this model and the notion that RalA functions through Exo84, immunofluorescent analysis of E-cadherin protein was performed on frozen sections from these engineered tissues. We observed that in tissues containing sh-RalA or sh-Exo84 expressing II-4 cells, E-cadherin was weakly expressed at cell-cell borders and was largely absent in invading cells (Fig. 5A, white arrows). By peeling the stratified epithelium away from the dermal compartment, we were able to directly assay protein and RNA expression in the epidermis. E-cadherin protein expression was reduced by ∼50% in both cases (Fig. 5B,C). Real-time PCR revealed that E-cadherin mRNA expression was not reduced, consistent with the model described in Fig. 3 showing that RalA (and Exo84) regulate E-cadherin by promoting its recycling back to the plasma membrane and thereby targeting it away from the degradation pathway.
Figure 5. Acquisition of invasive properties of II-4 cells upon RalA or Exo84 knockdown is associated with a decrease in E-cadherin protein expression.

(A) Scrambled (a), RalA knockdown (b), RalB knockdown (c), RalA/RalB double-knockdown (d), Exo84 knockdown (e), Sec5 knockdown (f), or RalBP-1 knockdown (g) HaCaT-II-4 cells were used to populate tissues and frozen sections were subjected to immunofluorescent analysis with anti-E-cadherin antibody (red). DAPI was used to counterstain the nuclei. Invading cells in representative images are shown with arrows. (B) Western blot analysis of homogenized epithelium from tissues shown in (A). Total Erk is shown as a loading control. E-cadherin expression protein (C) and mRNA (D) were measured in the linear range by densitometry or real-time PCR, respectively. * p < 0.05 for sh-RalA, shRalA/B, and sh-Exo84 compared to sh-Scram using Mann-Whitney test.
Interestingly, RalA or Exo84 knock-down II-4 cells did not display decreased E-cadherin levels when grown in standard two-dimensional cultures, nor did they display a scattered phenotype typical of cells with low E-cadherin (data not shown). Apparently, the significance of RalA and Exo84 induction of recycling E-cadherin back to the membrane in determining overall E-cadherin levels is higher in a tissue environment than in a typical cell culture environment.
RalA and Exo84 knockdown mimic E-cadherin suppression by allowing Ras-transformed keratinocytes in 3D tissues to form a highly invasive, aggressive malignant SCC after in vivo transplantation
We have previously shown that when Ras-expressing II-4 cells in fabricated 3D tissues are transplanted onto the dorsal fascia of nude mice, the transplants form hyperkeratotic surface lesions with low-grade, well-differentiated invasive keratin “pearls,” while cells with reduced E-cadherin function form large nodular and erythematous tumors with aggressively-invasive, high-grade, malignant carcinoma (Margulis et al., 2005a). When tissues comprised of RalA or Exo84 knockdown II-4 cells were transplanted to an in vivo microenvironment, they showed properties similar to transplanted tissues populated with E-cadherin-suppressed II-4 cells (Fig. 6, panels b,d), forming large nodular tumors. H&E analysis of tumor sections revealed tumor cells that had infiltrated the stroma with little evidence of morphologic differentiation (Fig. 6 panels f,h,j,l). This loss of differentiation was associated with tumor cell proliferation throughout the tumor mass as well as at the leading edge of the invasive front (Fig. 6 panels n,p). Thus, knockdown of RalA or Exo84 not only leads to suppressed levels of E-cadherin and enhanced invasive properties in these cells, but it also permits Ras-transformed cells to form aggressive carcinomas after invasion into the dermis. In contrast, transplanted tissues comprised of sh-Scram or sh-RalB cells formed small surface lesions (Fig. 6 panels a,c) harboring well-differentiated tumor islands (Fig. 6 panels e,g,i,k). Proliferating cells, identified by Ki-67 immunohistochemistry, were restricted to the basal cells at the edges of the tumor islands (Fig. 6 panels m,o). Overall, these findings imply that RalA functions through Exo84 to suppress not only Ras-induced cell invasion into the dermal compartment of engineered skin but also progression to aggressive cancer in vivo.
Figure 6. Suppression of RalA or Exo84 complements oncogenic Ras for the conversion to high-grade malignant carcinoma.

Scrambled (a, e, i, m), RalA knockdown (b, f, j, n), RalB knockdown (c, g, k, o) or Exo84 knockdown (d, h, l, p) HaCaT-II-4 cells were used to generate bioengineered tissues that were transplanted to the dorsa of nude mice. n=5 for each cohort. (a-d) Clinical appearance of surface tumor after 8 weeks. (e-l) Representative image of H&E staining from sections at 4× and 20× magnifications. (m-p) Ki-67 immunohistochemistry on serial sections of tissue. Bar: 100μm.
RalA is downregulated in head and neck squamous cell carcinoma
The experiments described here have associated the early progression of SCC with the down-regulation of RalA. We sought to determine whether down-regulation of RalA occurs in the pathogenesis of SCC. A survey of the microarray database Oncomine (Rhodes et al., 2004) revealed data comparing RalA expression in a variety of cancer types with comparable normal tissues (Fig. 7). Consistent with several previously-reported results suggesting RalA plays a positive role in tumorigenesis, RalA was up-regulated in breast, bladder, brain and prostate cancer. While data for SCC of the skin was not present in this database, RalA was significantly (p < 1×10-6) down-regulated (∼30%) in head and neck squamous cell carcinoma (HNSCC) compared to normal oral mucosa. Analysis of the differentiation status of each tumor and relative RalA expression in a contingency table (Table 1) by Fisher's exact test gave p = 0.0157 strongly associated RalA down-regulation with poorly-differentiating SCC tumors (Ginos et al., 2004). RalB was reduced to a smaller degree in HNSCC but in contrast to RaA it was also reduced in some other tumor types analyzed (Supplementary Fig. 6).
Figure 7. RalA is downregulated in head and neck squamous cell carcinoma.
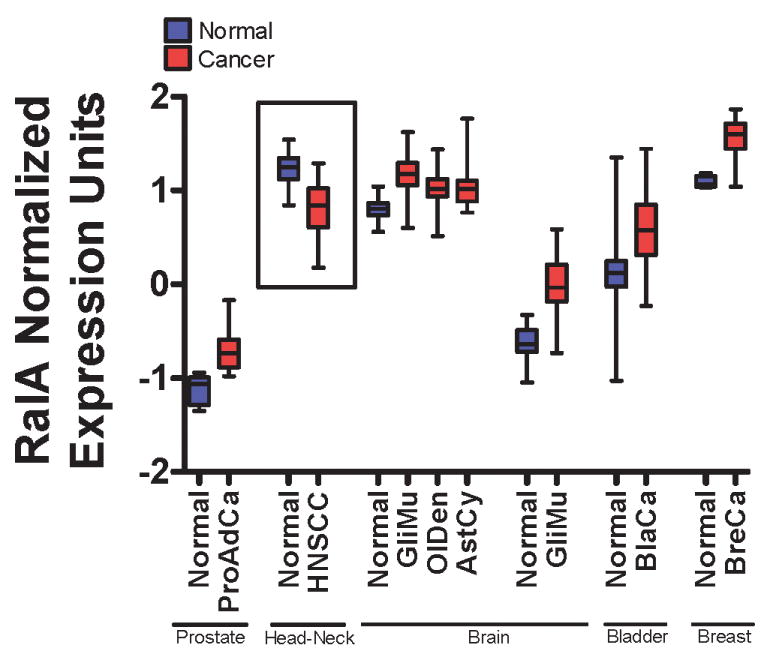
Survey of Oncomine microarray database for RalA expression in normal and diseased tissues. Data is represented as a box-and-whiskers plot with the box representing the 1st and 3rd quartiles and the whiskers denoting the data range. Center bar denotes median. Significance cutoff is p < 1×10-4 for each cancer type compared to normal. ProAdCa: Prostate adenocarcinoma (normal, n=8; cancer n=27). HNSCC: Head and neck squamous cell carcinoma (normal, n=13; cancer n=41). GliMu: Glioblastoma multiforme (normal, n=23; cancer n=77; normal, n=7; cancer n=25;). OlDen: Oligodendroglioma (normal, n=23; cancer n=50). AstCy: Astrocytoma (normal, n=23; cancer n=26). BlaCa: Bladder carcinoma (normal, n=48; cancer n=109). BreCa: Breast Carcinoma (normal, n=7; cancer n=41).
Table 1. Contingency table of RalA expression and carcinoma differentiation diagnosis.
Microarray data was sorted by differentiation status (poor vs. better than poor) and sub-classified based on RalA expression relative to the median for the entire sample set. p = 0.157 by Fisher's exact test.
| Tumor Differentiation | RalA Expression | Total | |
|---|---|---|---|
| Above Median | Below Median | ||
| Poor | 1 | 7 | 8 |
| Mod/Poor, Mod Well | 21 | 12 | 33 |
| Total | 22 | 19 | 41 |
Discussion
This study reveals that RalA suppresses early steps in Ras-induced tumor progression in an engineered human tissue model of squamous cell carcinoma of stratified squamous epithelia by promoting the stability of a well-characterized suppressor of tumor cell invasion, the cell-cell adhesion receptor E-cadherin. RalA performs this function through its effector Exo84, a component of the exocyst complex. Adding to the importance of this finding is the related discovery that RalA gene expression is tied to E-cadherin levels in squamous carcinoma cells. Therefore, an important component of early carcinoma progression driven by oncogenic events that reduce E-cadherin gene expression may be the concomitant decrease of RalA levels. This magnifies abrogation of E-cadherin expression and function by suppressing its recycling back to the plasma membrane through Exo84, thereby enhancing its degradation (see model in Fig. 8).
These conclusions are based on our observation that preventing the decrease in RalA gene expression, which accompanies E-cadherin down-regulation in Ras-expressing keratinocytes, elevates E-cadherin levels by decreasing its turnover rate. This, in turn, blocks the invasive properties of these cells in engineered tissues thus preventing the initiation of SCC. In addition, direct down-regulation of RalA or Exo84 through appropriate shRNA expression decreases E-cadherin levels. This converts Ras-expressing keratinocytes from pre-malignancy to malignancy, which enables them to invade into the dermis and generate high-grade invasive carcinomas after these engineered human tissues are transplanted onto the dorsal fascia of nude mice. Finally, the potential translational impact of these findings is further supported by screening a human cancer gene expression database, which revealed that RalA expression is specifically reduced in head and neck SCCs and strongly associated RalA down-regulation with poorly-differentiating tumors.
In this system, RalA signaling opposes other Ras effector pathways, since Raf and PI-3 kinase signaling have been shown to enhance progression of SCC (Pons & Quintanilla, 2006). One explanation for this antagonism appears to be their opposing effects on E-cadherin. Both Erk and PI-3K have been shown to drive E-cadherin from the membrane resulting in its degradation (Potempa & Ridley, 1998), while RalA does the opposite. How RalA suppresses E-cadherin degradation is not entirely clear. However, we have shown previously that active RalA functions in a way consistent with positive regulation of the exocyst, as it enhances the rate of delivery of vesicles carrying E-cadherin from the perinuclear recycling endosome specifically to the basolateral surface of MDCK epithelial cells (Shipitsin & Feig, 2004). Enhancing the recycling of proteins back to the plasma membrane through RalA effects on the exocyst could stabilize E-cadherin by targeting vesicles carrying it away from the degradation pathway (see Fig. 8). Our finding that RalA stabilizes E-cadherin in keratinocytes through the exocyst component Exo84 supports this model.
While suppression of RalA expression causes down-regulation of E-cadherin and tumor progression in oncogenic Ras-expressing HaCaT-II-4 keratinocytes, it does not do so in parental HaCaT cells. This implies that RalA tumor-suppressing activity becomes increasingly important as forces suppressing E-cadherin expression become significant during tumor progression. Because RalA displays its tumor-suppressing function at the level of protein stabilization, the recycling function of RalA may no longer be significant in tumor cells that have little or no E-cadherin. This idea is supported by our finding that while RalA levels are reduced in highly invasive oral squamous carcinoma cells that lack any detectable E-cadherin protein, E-cadherin levels were not elevated when RalA expression was restored.
Our experiments supporting the idea that Exo84 is the mediator of the tumor-suppressing function of RalA has interesting implications since this represents the first demonstration that a component of the exocyst complex can function to suppress tumor progression. Second, the exocyst complex has been implicated in many cell functions including delivery of membrane proteins specifically to the basolateral membrane (Hsu et al., 2004), secretion of proteins out of the cell (Sugita, 2008), cell migration (Rosse et al., 2006) and cytokinesis (Chen et al., 2006). There is growing evidence that distinct subcomplexes of the eight known exocyst subunits mediate these distinct processes (Moskalenko et al., 2003). Here we show that although it is known that both Sec5 and Exo84 exocyst subunits are downstream effectors of active Ral proteins, knockdown of Exo84, but not Sec5, phenocopies RalA knockdown. This finding implies that RalA signals through an exocyst subcomplex that includes Exo84, but not Sec5, to regulate E-cadherin membrane delivery and tumor progression. Interestingly, RalB was previously shown to signal through Sec5, but not Exo84 to activate the TBK1 kinase and suppress apoptosis to support tumor progression (Chien et al., 2006).
Why does RalA play a suppressing role in tumor progression in this tissue model system of human squamous carcinoma, while it has been found to play a tumor-promoting role in other cancer models? One possibility is that RalA plays different roles in different tumor cell types or different stages of tumor progression. For example, a suppressing role for RalA may be important in squamous carcinomas. This explanation is supported by our analysis of the Oncomine human cancer gene expression database, which shows that RalA expression is reduced in oral SCCs, and another report that found RalA is reduced in lung SCC (Smith et al., 2007). In contrast, our analysis of Oncomine revealed that RalA levels are elevated in other cancer types including bladder, prostate and brain cancer. Others have also found elevated RalA levels in liver and pancreatic cancer (Smith et al., 2007). Also, the assay system we have used in this study follows Ral function in early stages of tumor development, whereas in many other reports RalA expression was manipulated in already established tumor cell lines such as pancreatic cancer cells.
A related issue is that our studies using engineered human tissues reveal that RalA is particularly important in suppressing tumor progression through its positive effects on E-cadherin function particularly when E-cadherin levels are limiting. Others have found tumor promoting activity for RalA in soft agar growth assays or in tumor forming assays after injection into mice, where E-cadherin levels may not be as important (Lim et al., 2005; Lim et al., 2006). Finally, we have observed a tumor-promoting consequence of a modest ∼2-3 fold decrease in RalA expression, a drop that occurs naturally when E-cadherin levels fall during tumor progression. Previous studies have shown tumor inhibition in response more complete RalA ablation. The molecular basis for these differences could be that RalA uses different effector proteins under these different conditions.
However, these explanations do not account for the results from a previous study showing that Ras-induced squamous carcinoma is reduced in knockout mice lacking an activator of Ral GTPases, RalGDS (Gonzalez-Garcia et al., 2005). However, we recently found that RalGDS has a Ral-independent scaffold function that promotes Akt activation by growth factors (Hao et al., 2008). This raises the possibility that the failure to generate carcinomas in mice depleted of RalGDS was due to an absence of Akt-mediated survival signaling, rather than Ras-mediated Ral activation. In addition, the knockout mice lacked RalGDS in all cells, not solely in keratinocytes, and it is not clear which specific cell populations are responsible for inhibiting tumor progression in these mice.
Finally, one of the most surprising conclusions from this study is that a modest (∼60%) reduction in RalA gene expression induced by E-cadherin suppression is a necessary step for early tumor progression of Ras-expressing keratinocytes in this model system. Reduction of RalA magnifies the effect of tumorigenic forces that suppress E-cadherin expression by enhancing its degradation. Interestingly, the expression of the RalA effector involved in this process, Exo84, is not regulated by E-cadherin, implying that RalA is the key sensor for E-cadherin levels in this cascade. Since RalA is regulated at the level of gene expression, an interesting possibility is that RalA expression can be reversed later in tumor progression, such as during metastasis, where RalA has been shown to play a positive role (Lim et al., 2006).
Materials and Methods
Three-dimensional cell culture
Three-dimensional tissues were prepared as previously described (Carlson et al., 2008). Briefly, human foreskin fibroblasts were mixed with Bovine Type I collagen (2.5×104 cells/ml) and allowed to contract for seven days in deep-well polycarbonate tissue culture inserts (Organogenesis, Canton, MA). 5×105 keratinocytes were seeded on contracted collagen gels and grown submerged for three days in low-calcium epidermal growth medium. Cultures were then maintained for two days in normal-calcium epidermal growth medium and subsequently fed only from the bottom for seven days.
Protein Turnover Assays
Subconfluent monolayer cultures were treated with 10μm cycloheximide (Sigma) or vehicle for 0-24h. Plates were washed and lysed as described above before being subjected to immunoblot analysis.
Quantification of invasion
For each tissue generated, invasive cells were counted and averaged from approximately 150 microscope fields (≥5 serial sections from ≥2 different depths). Averages shown represent ≥3 independent experiments ±S.D. P values, where shown, are calculated by the Mann-Whitney test.
Data Analysis
Data represented in graphs and analyzed by Mann-Whitney, Student's t, and Fisher's exact tests were performed using GraphPad Prism 4.0 for Windows.
Tissue transplantation
All animal experiments were performed according to a protocol approved by the Tufts University Institutional Animal Care and Use Committee as described (Greenberg et al., 2005). Briefly, a 1.3cm dorsal skin section was removed from 5 nude mice per cell line. Three-dimensional tissues were grown as described above and transplanted onto fascia at the site of skin excision. Bandages were removed after 2 weeks and mice were sacrificed eight weeks after transplantation. Skin tumors were excised, trimmed, and processed as described (Margulis et al., 2005b).
Supplementary Material
(A) Western blot analysis of E-cadherin positive MSCC-1 and E-cadherin negative MSCC-1-Inv-1 cells. Total Erk is shown as a loading control. RalA and RalB protein (B) or mRNA (C) expression were measured in the linear range by densitometry or real-time PCR, respectively.
(A) Western blot analysis of Ras-expressing HaCaT-II-4 cells expressing empty vector or shRNA against E-cadherin infected with retroviruses encoding wild-type RalA or RalB. Total Erk is shown as a loading control. (B) Phase-contrast or immunofluorescent micrographs showing colony morphology and E-cadherin membrane localization in HaCaT-II-4 (a, e), HaCaT-II-4-sh-Ecad (b, f), HaCaT-II-4-sh-Ecad-RalAwt (c, g), and HaCaT-II-4-sh-Ecad-RalBwt (d, h) cells. Bar: 100μm. (C) The indicated cell lines were incubated with 10μM cycloheximide for the indicated amount of time (n≥3). E-cadherin protein expression was analyzed by Western blot and quantified by densitometry. (D) E-cadherin mRNA expression in the indicated cell lines was analyzed by real-time PCR. * p < 0.05 for sh-Ecad lines compared to control using student's t-test. (E) Cells from the indicated lines were grown on fibroblast-populated collagen gels and tissues were stained with H&E. Bar: 100μm. Invading cells in representative images are shown with arrows and quantified from >100 microscope fields in >20 sections from multiple experiments (F). * p < 0.05 for sh-Ecad/RalAwt compared to sh-Ecad/Bleo using Mann-Whitney test.
Western blot analysis of MSCC-1 or MSCC-1-Inv-1 cells expressing control or wild-type RalA or RalB retroviruses. Total Erk is shown as a loading control.
(A) Western blot analysis of Ras-expressing HaCaT-II-4 cells expressing an alternative sequence to RalA or RalB. Total Erk is shown as a loading control. (B) Scrambled (a), RalA knockdown (b), or RalB knockdown (c) HaCaT-II-4 cells were grown on contracted organotypic collagen gels and FFPE tissues were stained with H&E. Bar: 100μm. Invading cells in representative images are shown with arrows. (C) Western blot analysis of HaCaT-II-4 cells expressing Scrambled or RalA shRNA and either empty vector or RNAi-resistant wild-type RalA. (D) II-4 cells knocked down in RalA (a, b) and overexpressing RNAi-resistant wild-type RalA (c) were grown on fibroblast-populated collagen gels and tissues were stained with H&E. Bar: 100μm. Invading cells in representative images are shown with arrows.
(A) Western blot analysis of parental HaCaT cells expressing scrambled shRNA or shRNA against RalA or RalB. Total Erk is shown as a loading control. (B) Scrambled (a), RalA knockdown (b), or RalB knockdown (c) parental HaCaT cells were grown fibroblast-populated collagen gels and tissues were stained with H&E. Bar: 100μm. Experiment was performed in duplicate with both sets of RalA or RalB shRNA sequences. RalA expression is not suppressed when E-cadherin is lost in cells lacking oncogenic Ras. (C) Western blot analysis of parental HaCaT cells expressing empty vector or the dominant-negative E-cadherin transgene H-2Kd-Ecad.
Survey of Oncomine microarray database for RalB expression in normal and diseased tissues described in Figure 7. Data is represented as a box-and-whiskers plot with the box representing the 1st and 3rd quartiles and the whiskers denoting the data range. Center bar denotes median. Significance cutoff is p < 1×10-4 for each cancer type compared to normal unless noted.
Acknowledgments
We thank M. W. Carlson, Y. Szwec-Levin, S. Ezell, T. DesRochers and S. Dong for excellent technical assistance. This work was supported by grants to LAF from NIGMS and to JAG from NIDCR. We thank. N. Fusenig for HaCaT cells and their derivatives and F. Watt for the H-2Kd-Ecad vectors.
References
- Bailleul B, Surani MA, White S, Barton SC, Brown K, Blessing M, Jorcano J, Balmain A. Cell. 1990;62:697–708. doi: 10.1016/0092-8674(90)90115-u. [DOI] [PubMed] [Google Scholar]
- Bodemann BO, White MA. Nat Rev Cancer. 2008;8:133–40. doi: 10.1038/nrc2296. [DOI] [PubMed] [Google Scholar]
- Brymora A, Valova VA, Larsen MR, Roufogalis BD, Robinson PJ. J Biol Chem. 2001;276:29792–7. doi: 10.1074/jbc.C100320200. [DOI] [PubMed] [Google Scholar]
- Cantor S, Urano T, Feig LA. Mol Cell Biol. 1995;15:4578–4584. doi: 10.1128/mcb.15.8.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson MW, Alt-Holland A, Egles C, Garlick JA. Curr Protoc Cell Biol. 2008;Chapter 19(Unit 19 9) doi: 10.1002/0471143030.cb1909s41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascone I, Selimoglu R, Ozdemir C, Del Nery E, Yeaman C, White M, Camonis J. Embo J. 2008;27:2375–87. doi: 10.1038/emboj.2008.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XW, Inoue M, Hsu SC, Saltiel AR. J Biol Chem. 2006;281:38609–16. doi: 10.1074/jbc.M512847200. [DOI] [PubMed] [Google Scholar]
- Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, Balakireva MG, Romeo Y, Kopelovich L, Gale M, Jr, Yeaman C, Camonis JH, Zhao Y, White MA. Cell. 2006;127:157–70. doi: 10.1016/j.cell.2006.08.034. [DOI] [PubMed] [Google Scholar]
- Chien Y, White MA. EMBO Rep. 2003;4:800–6. doi: 10.1038/sj.embor.embor899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, Marinkovich MP, Tao S, Lin Q, Kubo Y, Khavari PA. Nature. 2003;421:639–43. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- Dlugosz A, Merlino G, Yuspa SH. J Investig Dermatol Symp Proc. 2002;7:17–26. doi: 10.1046/j.1523-1747.2002.19631.x. [DOI] [PubMed] [Google Scholar]
- Feig LA. Trends Cell Biol. 2003;13:419–425. doi: 10.1016/s0962-8924(03)00152-1. [DOI] [PubMed] [Google Scholar]
- Fenwick R, Prasannan S, Campbell L, Nietlispach D, Evetts K, Camonis J, Mott H, Owen D. Biochemistry. 2009 doi: 10.1021/bi802129d. [DOI] [PubMed] [Google Scholar]
- Frankel P, Aronheim A, Kavanagh E, Balda MS, Matter K, Bunney TD, Marshall CJ. Embo J. 2005;24:54–62. doi: 10.1038/sj.emboj.7600497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlick JA. Adv Biochem Eng Biotechnol. 2007;103:207–39. doi: 10.1007/b137206. [DOI] [PubMed] [Google Scholar]
- Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, Ondrey FG, Adams GL, Gaffney PM. Cancer Res. 2004;64:55–63. doi: 10.1158/0008-5472.can-03-2144. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia A, Pritchard CA, Paterson HF, Mavria G, Stamp G, Marshall CJ. Cancer Cell. 2005;7:219–26. doi: 10.1016/j.ccr.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Greenberg S, Margulis A, Garlick JA. Methods Mol Biol. 2005;289:425–30. doi: 10.1385/1-59259-830-7:425. [DOI] [PubMed] [Google Scholar]
- Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, Der CJ, Counter CM. Genes Dev. 2002;16:2045–57. doi: 10.1101/gad.993902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Wong R, Feig LA. Mol Cell Biol. 2008;28:2851–9. doi: 10.1128/MCB.01917-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SC, TerBush D, Abraham M, Guo W. Int Rev Cytol. 2004;233:243–65. doi: 10.1016/S0074-7696(04)33006-8. [DOI] [PubMed] [Google Scholar]
- Jeanes A, Gottardi CJ, Yap AS. Oncogene. 2008;27:6920–9. doi: 10.1038/onc.2008.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullien-Flores V, Dorseuil O, Romero F, Letourneur F, Saragosti S, Berger R, Tavitian A, Gacon G, Camonis JH. J Biol Chem. 1995;270:22473–22477. doi: 10.1074/jbc.270.38.22473. [DOI] [PubMed] [Google Scholar]
- Kudo Y, Kitajjma S, Sato S, Miyauchi M, Ogawa I, Takata T. Oral Oncol. 2003;39:515–20. doi: 10.1016/s1368-8375(03)00015-0. [DOI] [PubMed] [Google Scholar]
- Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD, Der CJ, Counter CM. Cancer Cell. 2005;7:533–45. doi: 10.1016/j.ccr.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Lim KH, O'Hayer K, Adam SJ, Kendall SD, Campbell PM, Der CJ, Counter CM. Curr Biol. 2006;16:2385–94. doi: 10.1016/j.cub.2006.10.023. [DOI] [PubMed] [Google Scholar]
- Margulis A, Zhang W, Alt-Holland A, Crawford HC, Fusenig NE, Garlick JA. Cancer Res. 2005a;65:1783–91. doi: 10.1158/0008-5472.CAN-04-3399. [DOI] [PubMed] [Google Scholar]
- Margulis A, Zhang W, Garlick JA. Methods Mol Biol. 2005b;289:61–70. doi: 10.1385/1-59259-830-7:061. [DOI] [PubMed] [Google Scholar]
- Moskalenko S, Henry DO, Rosse C, Mirey G, Camonis JH, White MA. Nat Cell Biol. 2002;4:66–72. doi: 10.1038/ncb728. [DOI] [PubMed] [Google Scholar]
- Moskalenko S, Tong C, Rosse C, Mirey G, Formstecher E, Daviet L, Camonis J, White MA. J Biol Chem. 2003;278:51743–8. doi: 10.1074/jbc.M308702200. [DOI] [PubMed] [Google Scholar]
- Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Cancer Res. 2008;68:3645–54. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- Oxford G, Owens CR, Titus BJ, Foreman TL, Herlevsen MC, Smith SC, Theodorescu D. Cancer Res. 2005;65:7111–20. doi: 10.1158/0008-5472.CAN-04-1957. [DOI] [PubMed] [Google Scholar]
- Panner A, Nakamura JL, Parsa AT, Rodriguez-Viciana P, Berger MS, Stokoe D, Pieper RO. Mol Cell Biol. 2006;26:7345–57. doi: 10.1128/MCB.00126-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierceall WE, Goldberg LH, Tainsky MA, Mukhopadhyay T, Ananthaswamy HN. Mol Carcinog. 1991;4:196–202. doi: 10.1002/mc.2940040306. [DOI] [PubMed] [Google Scholar]
- Polzin A, Shipitsin M, Goi T, Feig LA, Turner TJ. Mol Cell Biol. 2002;22:1714–22. doi: 10.1128/MCB.22.6.1714-1722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons M, Quintanilla M. Clin Transl Oncol. 2006;8:466–74. doi: 10.1007/s12094-006-0046-4. [DOI] [PubMed] [Google Scholar]
- Potempa S, Ridley AJ. Mol Biol Cell. 1998;9:2185–200. doi: 10.1091/mbc.9.8.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosse C, Hatzoglou A, Parrini MC, White MA, Chavrier P, Camonis J. Mol Cell Biol. 2006;26:727–34. doi: 10.1128/MCB.26.2.727-734.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablina AA, Chen W, Arroyo JD, Corral L, Hector M, Bulmer SE, DeCaprio JA, Hahn WC. Cell. 2007;129:969–82. doi: 10.1016/j.cell.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipitsin M, Feig LA. Mol Cell Biol. 2004;24:5746–56. doi: 10.1128/MCB.24.13.5746-5756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal SS, Yadav S, Singhal J, Zajac E, Awasthi YC, Awasthi S. Biochem Pharmacol. 2005;70:481–8. doi: 10.1016/j.bcp.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Smith SC, Oxford G, Baras AS, Owens C, Havaleshko D, Brautigan DL, Safo MK, Theodorescu D. Clin Cancer Res. 2007;13:3803–13. doi: 10.1158/1078-0432.CCR-06-2419. [DOI] [PubMed] [Google Scholar]
- Sugihara K, Asano S, Tanaka K, Iwamatsu A, Okawa K, Ohta Y. Nat Cell Biol. 2002;4:73–8. doi: 10.1038/ncb720. [DOI] [PubMed] [Google Scholar]
- Sugita S. Acta Physiol (Oxf) 2008;192:185–93. doi: 10.1111/j.1748-1716.2007.01803.x. [DOI] [PubMed] [Google Scholar]
- Takai Y, Sasaki T, Matozaki T. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- Zhu AJ, Watt FM. J Cell Sci. 1996;109(Pt 13):3013–23. doi: 10.1242/jcs.109.13.3013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Western blot analysis of E-cadherin positive MSCC-1 and E-cadherin negative MSCC-1-Inv-1 cells. Total Erk is shown as a loading control. RalA and RalB protein (B) or mRNA (C) expression were measured in the linear range by densitometry or real-time PCR, respectively.
(A) Western blot analysis of Ras-expressing HaCaT-II-4 cells expressing empty vector or shRNA against E-cadherin infected with retroviruses encoding wild-type RalA or RalB. Total Erk is shown as a loading control. (B) Phase-contrast or immunofluorescent micrographs showing colony morphology and E-cadherin membrane localization in HaCaT-II-4 (a, e), HaCaT-II-4-sh-Ecad (b, f), HaCaT-II-4-sh-Ecad-RalAwt (c, g), and HaCaT-II-4-sh-Ecad-RalBwt (d, h) cells. Bar: 100μm. (C) The indicated cell lines were incubated with 10μM cycloheximide for the indicated amount of time (n≥3). E-cadherin protein expression was analyzed by Western blot and quantified by densitometry. (D) E-cadherin mRNA expression in the indicated cell lines was analyzed by real-time PCR. * p < 0.05 for sh-Ecad lines compared to control using student's t-test. (E) Cells from the indicated lines were grown on fibroblast-populated collagen gels and tissues were stained with H&E. Bar: 100μm. Invading cells in representative images are shown with arrows and quantified from >100 microscope fields in >20 sections from multiple experiments (F). * p < 0.05 for sh-Ecad/RalAwt compared to sh-Ecad/Bleo using Mann-Whitney test.
Western blot analysis of MSCC-1 or MSCC-1-Inv-1 cells expressing control or wild-type RalA or RalB retroviruses. Total Erk is shown as a loading control.
(A) Western blot analysis of Ras-expressing HaCaT-II-4 cells expressing an alternative sequence to RalA or RalB. Total Erk is shown as a loading control. (B) Scrambled (a), RalA knockdown (b), or RalB knockdown (c) HaCaT-II-4 cells were grown on contracted organotypic collagen gels and FFPE tissues were stained with H&E. Bar: 100μm. Invading cells in representative images are shown with arrows. (C) Western blot analysis of HaCaT-II-4 cells expressing Scrambled or RalA shRNA and either empty vector or RNAi-resistant wild-type RalA. (D) II-4 cells knocked down in RalA (a, b) and overexpressing RNAi-resistant wild-type RalA (c) were grown on fibroblast-populated collagen gels and tissues were stained with H&E. Bar: 100μm. Invading cells in representative images are shown with arrows.
(A) Western blot analysis of parental HaCaT cells expressing scrambled shRNA or shRNA against RalA or RalB. Total Erk is shown as a loading control. (B) Scrambled (a), RalA knockdown (b), or RalB knockdown (c) parental HaCaT cells were grown fibroblast-populated collagen gels and tissues were stained with H&E. Bar: 100μm. Experiment was performed in duplicate with both sets of RalA or RalB shRNA sequences. RalA expression is not suppressed when E-cadherin is lost in cells lacking oncogenic Ras. (C) Western blot analysis of parental HaCaT cells expressing empty vector or the dominant-negative E-cadherin transgene H-2Kd-Ecad.
Survey of Oncomine microarray database for RalB expression in normal and diseased tissues described in Figure 7. Data is represented as a box-and-whiskers plot with the box representing the 1st and 3rd quartiles and the whiskers denoting the data range. Center bar denotes median. Significance cutoff is p < 1×10-4 for each cancer type compared to normal unless noted.