

Abstract
Lower respiratory tract infection by the human pneumovirus respiratory syncytial virus is a frequent cause of acute lung injury in children. Severe pneumovirus disease in humans is associated with activation of the granzyme pathway by effector lymphocytes, which may promote pathology by exaggerating pro-apoptotic caspase activity and pro-inflammatory activity. The main goal of this study was to determine whether granzymes contribute to the development of acute lung injury in pneumovirus-infected mice. Granzyme-expressing mice and granzyme A, and B-cluster single and double-gene deleted mice were inoculated with the rodent pneumovirus pneumonia virus of mice strain J3666, and were studied for markers of lung inflammation and injury. Expression of granzyme A and B is detected in effector lymphocytes in mouse lungs in response to pneumovirus infection. Mice deficient for granzyme A and the granzyme B-cluster have unchanged virus titers in the lungs, but show a significantly delayed clinical response to fatal pneumovirus infection, a feature that is associated with delayed neutrophil recruitment, diminished activation of caspase-3 and reduced lung permeability. We conclude that granzyme A and B-cluster deficiency delays the acute progression of pneumovirus disease by reducing alveolar injury.
Keywords: virus, granzyme, apoptosis, lung, lymphocyte
INTRODUCTION
The human pneumovirus, respiratory syncytial virus (RSV)3, is the leading cause of lower respiratory tract illness in young children and infants worldwide (1). In Western countries the annual incidence rate of RSV-associated hospitalization may be as high as 30 per 1000 children under 1 year of age (1,2). RSV disease in these children presents as bronchiolitis or pneumonia. However, the severe end of the RSV disease spectrum includes the development of acute lung injury and acute respiratory distress syndrome (ALI/ARDS), which is characterized by severe hypoxemia and bilateral lung infiltrates in the setting of a normal cardiac preload (3). Currently, treatment for severe RSV disease in normal healthy children is limited to supportive measures, stressing the need to further explore its pathogenesis (4).
In general, ALI/ARDS is associated with the development of diffuse neutrophilic alveolitis and loss of alveolar capillary barrier integrity as a result of enhanced lung epithelial cell death (5). Interestingly, also in humans with severe RSV disease a large number of neutrophils is recruited to the lungs (6), and caspase-dependent apoptosis of lung epithelial cells is observed (7). Similarly, in the pneumonia virus of mice (PVM) mouse model for severe RSV disease (8), pulmonary neutrophil recruitment and caspase-3 activation is associated with enhanced lung permeability and clinical signs of illness (8,9). These findings suggest that the coordinated actions of pro-inflammatory and pro-apoptotic mediators may serve to augment the pathologic responses characteristic of severe pneumovirus disease, although the nature of the mediators linking these responses are not yet fully understood.
Granzymes (Gzms) are effector molecules that may contribute to both inflammatory and apoptotic features of RSV disease. Gzms are serine proteases located in granules of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells and have traditionally been considered to be crucial to the anti-viral immune response (reviewed in (10–12)). Gzms are delivered into the cytosol of target cells by the pore-forming protein perforin, but intracellular trafficking is also known to be facilitated by adenovirus and bacterial toxins (13,14). Of the five Gzms found in humans (GzmA, B, H, K and M), the tryptase GzmA and aspartase GzmB are the most thoroughly characterized. It has been shown that GzmB induces classical (caspase-dependent) apoptosis in target cells, and in vitro micromolar concentrations of GzmA, in the presence of perforin, can induce caspase-independent DNA damage (10,15–17). However, Metkar et al. (18) reported recently that GzmA-mediated cytotoxicity at reduced, more physiologic concentrations may be relatively low (15,16). Instead, active soluble GzmA at lower concentrations induces the release of pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α from monocyte/macrophage, fibroblast and epithelial cell lines via a perforin-independent mechanism (18,19). This raises the possibility that Gzms have a broad functionality in the human immune response to viral infection in vivo, including activation of (caspase-dependent) cell death and inflammation.
Recently, our group has demonstrated that tracheal aspirates from mechanically-ventilated children with severe RSV infection contain high levels of biologically active soluble GzmA and GzmB (20). To investigate whether these Gzms may actively participate in the pathogenesis of RSV disease, specifically promoting progression to ALI/ARDS by inducing pro-inflammatory and caspase-dependent pro-apoptotic activity, we explored lung inflammatory and injury markers in acute pneumovirus infection in GzmA and/or GzmB-cluster gene deleted mice.
MATERIAL AND METHODS
Virus and mouse strains
The fully pathogenic PVM strain J3666 was originally obtained from Dr. A.J. Easton (University of Warwick, Coventry, U.K.) from virus stocks originating at the Rockefeller University. Virulence was maintained by continuous passage in mice (21). Virus titer in stock aliquots was 12 × 107 copies of PVM/ml, as determined by quantitative PCR (see below), and PVM inocula per mouse were as indicated.
Nine- to- twelve-week-old female mice were used in all experiments. C57Bl/6 wild type mice (CD45.1+) were originally obtained from Jackson Laboratories (Bar Harbor, ME, USA) and bred in house. The GzmA−/−, GzmB−/− (generated by the group of Dr. T. Ley (22)) and GzmA−/−GzmB−/− mice on a C57Bl/6 background were originally obtained from Dr. M.M. Simon (23) and bred in house (24). GzmA+/−GzmB+/− were used to serve as a primary positive control for the GzmA−/−GzmB−/− mice: GzmA+/−GzmB+/− mice were generated by crossing GzmA−/− with GzmB−/− mice resulting in a similar mixed C57Bl/6 substrain (B6J and B6-III) background as the GzmA−/−GzmB−/− mice (22,25). Importantly, the GzmB−/− (and GzmA−/−GzmB−/−) mice lack the entire GzmB-cluster which includes GzmC, -D and -F due to knockdown by a retained phosphoglycerate kinase I gene promoter (PGK-neo) cassette (26).
Animal protocols
The animal protocols as described below were approved by the Animal Care and Experimental Research Committee of the Academic Medical Center Amsterdam, the Netherlands.
PVM diluted in Roswell Park Memorial Institute (RPMI)-medium (Invitrogen Ltd, Paisley, UK) in a total volume of 80μl was delivered via intranasal inhalation to isoflurane anesthetized mice. At day 3, 7 or 8 after inoculation the mice were euthanized with intraperitoneal pentobarbital (120mg/kg), the left lung was removed and flash-frozen in liquid nitrogen for homogenization. Bronchoalveolar lavage (BAL) was performed in the right lung by instilling four separate 0.5 ml aliquots of 0.9% NaCl containing 0.6 mM EDTA. The BAL fluid (BALF) was maintained at 4°C, and spun at 200 g for 10 min. The supernatants were stored at −80°C as individual aliquots, and the cell pellet was processed for total cell counts and differentials. For histological analysis on day 8 after PVM inoculation the right lung was fixed with 10% formalin immediately after the BAL procedure.
Measurements
Microarray and flow cytometry analysis
The microarray study as described previously (27), was probed to determine the expression profile of GzmA and GzmB transcripts in whole lung tissue in response to PVM infection. To determine intracellular expression of GzmA and GzmB in effector lymphocytes, BALF leukocytes in FACS buffer (0.5% BSA, 0.02% potassium-EDTA in PBS) were probed with FITC-labeled anti-CD8, PerCP-Cy5-labeled anti-NK1.1 and APC-labeled anti-CD3 (all anti-mouse from eBioscience, San Diego, CA, USA). Next, cells were fixed and permeabilized with fixation/permeabilization solution (BD Biosciences, Franklin Lakes, NJ) and stained with PE-labeled mouse anti-mouse GzmA mAb (Santa Cruz Biotechnology Inc., Heidelberg, Germany) or with PE-labeled rat anti-mouse GzmB mAb (eBioscience) in 10% normal rat serum to block non-specific binding. The FACSCalibur flow cytometer (BD Biosciences) and FlowJo software (Tree Star Inc, Ashland, OR) were used for analysis.
Lung virus titers
Lungs were homogenized in Trizol reagent to extract RNA, according to the manufacturer's description (Invitrogen Ltd., Paisley, UK). RNA was resuspended in DEPC-treated water. After DNaseI treatment of 2 μg RNA (Applied Biosystems, Foster City, CA) cDNA synthesis was performed using a random hexamer cDNA synthesis kit (Applied Biosystems). Copies of the PVM sh gene (GenBank: AY573815) were detected in quantitative real-time PCR reactions containing 1 μl cDNA, Taqman PCR Master Mix (Applied Biosystems), 77 nM TAMRA probe (5'-6FAM-CGCTGATAATGGCCTGCAGCA TAMRA-3'), 200 nM primers (5'-GCCTGCATCAACACAGTGTGT-3' and 5'-GCCTGATGTGGCAGTGCTT-3'). The gapdh housekeeping gene was detected in cDNA samples using rodent gapdh primers (100nM) and VIC-probe (200nM) (Ambion, Applied Biosystems). Standard curves with known concentrations of the full-length sh-gene and gapdh decatemplate (Applied Biosystems) were used for quantification. Results are expressed as copies of PVM-sh per 109 copies of gapdh (28).
Lung inflammatory markers
Total BALF leukocyte counts were performed on an aliquot of the BALF cells using a Bürker bright line counting chamber. Cytospins were stained with Romanovsky (Diff-Quick) and differential WBC counts were obtained by counting 200 leukocytes using a standard light microscope. Lung cytokines in BALF were measured by multiplex fluorescent bead assay for TNFα, IFNγ, IL-6, IL-1β, KC and MIP-2 (Luminex, R&D systems, Minneapolis, MN).
Lung caspase-activation
Caspase-3 immunohistochemistry on 5-μm-thick paraffin embedded lung sections was performed using rabbit anti-cleaved (active) caspase-3 mAb (Cell Signaling, Danvers, MA) followed by secondary labelling with anti-rabbit IgG HRP polymer (Immunologic, Duiven, The Netherlands). HRP activity was detected with Peroxidase Substrate Kit (Vector®NovaRED, Vector Laboratories, Burlingame, CA). Quantification of the overall lung caspase-3 activity in lung homogenates corrected for lung weight was performed using the caspase-3/CPP32 Fluorometric Assay kit from Biovision, Mountain View (CA) as described previously (29).
Lung permeability
Leakage of the high molecular weight serum protein α-macroglobulin or IgM into the lungs was measured by immunoassay for mouse α-macroglobulin (Life Diagnostics, West Chester, PA) or mouse IgM (Bethyl Laboratories, West Chester, PA) performed on BALF.
Lung histology
A blinded histology analysis for evidence of lung injury was performed on haematoxylin and eosin stained lung sections. A pathological grade for each lung section was determined according to the following criteria: 1 = presence of areas with cellular alveolitis involving 25% or less of the lung parenchyma; 2 = lesions involving 25–50% of the lung; 3 = lesions involving 50% or more of the lung. An additional point was given when there were alveolar wall alterations such as thickening or capillary congestion outside the aforementioned inflammatory areas.
Clinical response
Total body weight and a clinical scoring system previously validated in multiple PVM mouse studies was used: score 1 = healthy, no signs of illness, 2 = subtle ruffled fur, 3 = evident ruffled fur with hunched posture, 4 = evident lethargy with abnormal breathing pattern, 5 = moribund, 6 = death; modified from Cook et al. (30)). The endpoint for sacrifice used in this study was a score of 5 and/or loss > 20% of starting body weight.
Statistical analysis
To detect differences among groups we performed either unpaired t-test (for comparison of two groups) or one-way ANOVA (for comparison of multiple groups) followed by LSD post hoc analysis on log10 transformed data. A p value of < 0.05 was considered statistical significant.
RESULTS
Expression of GzmA and GzmB in effector lymphocytes in lungs of PVM-infected mice
Severe pneumovirus (RSV) disease in children is associated with increased local levels of biologically active GzmA and GzmB, and cellular expression of GzmB predominantly in BALF CTLs (20). To determine whether infection with the murine pneumovirus PVM induces GzmA and GzmB expression in mice we analyzed the temporal gene expression profiles of transcripts encoding GzmA and GzmB from a microarray study performed on total lung RNA from C57Bl/6 mice inoculated with a non-lethal dose of PVM (27). Transcripts encoding both GzmA and GzmB were detected in lung tissue from PVM infected mice (Figure 1A). Levels of both the GzmA and GzmB transcript reached a peak at day 7 and remained above baseline levels until day 14 after inoculation. Immunoreactive GzmA was detected predominantly in CD3−/NK1.1+ (NK) cells, and GzmB in CD8+/CD3+ (CTL) cells isolated from BALF at day 7 of infection (Figure 1B).
Figure 1.

PVM infection induces expression of GzmA and GzmB. A, gene expression profiles of transcripts encoding GzmA and GzmB in total lung RNA of C57Bl/6 mice (RNA from 5 mice pooled per time point) at time zero and days 1–7, 10, 14, 21 and 28 after inoculation with PVM J3666 (30 pfu). Peak of GzmA and GzmB expression at day 7 (green highlight). B, histogram plots of intracellular GzmA and GzmB expression in BALF CD8+CD3+ cells (CTLs, blue plot), NK1.1+CD3− (NK cells, green plot) and CD8−CD3+ (CD8− T lymphocytes, red plot) of PVM-infected mice at day 7 (BALF leukocytes from 3 mice pooled).
PVM infection in GzmA and GzmB-cluster gene deleted mice
Based on the Gzm gene expression profile, we hypothesized that if the Gzm response contributes either positively or negatively to severe pneumovirus disease, this would become apparent at or beyond peak expression at day 7. To address this question we investigated lung virus titers, caspase-3 activation and cytokine release in wild type, GzmA or GzmB single-gene deleted and GzmAGzmB double-gene deleted mice on day 7 and day 8 after inoculation with PVM (Figure 2).
Figure 2.

Lung response to PVM infection in GzmA and GzmB-cluster gene deleted mice. A, virus titer in the lung, expressed as number of PVM-sh copies per 109 gapdh copies; B, lung homogenate caspase-3 activity (arbitrary fluorescence units); C, concentration of α-macroglobulin in BALF; D, BALF total neutrophil (PMN) counts; E, concentration of IL-6 (pg/ml) in BALF; and F, concentration of TNFα (pg/ml) in BALF, in the wild type (B6 wt), GzmA−/−, GzmB−/− and GzmA−/−GzmB−/− mice on day 3 (n=3 per group), 7 (n=6 per group) and 8 (n=4–6 per group) after PVM inoculation (6 × 103 copies). * p<0.05. Data are shown as bar graphs depicting mean and standard error.
Surprisingly, whole lung virus titers from PVM-infected wild type mice were indistinguishable from those measured in lungs from PVM-infected GzmA−/−, GzmB−/− and GzmA−/−GzmB−/− mice, both on day 7 and 8 after inoculation as determined by quantitative RT-PCR (Figure 2A). However, caspase-3 activity in lung homogenates of the GzmA−/−GzmB−/− mice was significantly reduced compared to the wild type and to the GzmA−/− mice on day 7 and 8 after PVM inoculation (Figure 2B). Previously we have identified that reduced whole lung caspase-3 activity is associated with decreased leakage of the high molecular weight serum protein α-macroglobulin into the alveolar spaces in severe PVM disease in mice (9). In line with this, GzmB−/− and GzmA−/−GzmB−/− mice had reduced concentrations of α-macroglobulin in BALF, as compared to wild type and GzmA−/− mice, although this reached statistical significance only for the GzmB−/− mice (Figure 2C).
PVM infection in mice promotes robust neutrophil influx into the lungs (8). Neutrophil recruitment, as determined by BALF total neutrophil counts, was not different among the wild type, GzmA−/−, and GzmB−/− mice; in contrast neutrophil recruitment was delayed in the GzmA and the GzmB-cluster double-gene deleted mice (Figure 2D). However, this delay was not associated with the release of the pro-inflammatory cytokines IL-1β (not detected in any of the mice), IL-6 (Figure 2E) and TNFα (Figure 2F) in the lungs.
PVM infection in GzmA−/−GzmB−/− and GzmA+/−GzmB+/− mice
The findings thus far suggest that Gzms may alter the response to PVM, including caspase-mediated apoptosis and lung permeability, but do not affect viral clearance or the release of the pro-inflammatory cytokines IL-1β, IL-6 and TNFα. We found a more pronounced phenotype in the double-gene deleted mice consistent with previous findings (31). As such, we proceeded with a focus on the GzmA−/−GzmB−/− mice, and to account for the differences in the genetic background of the substrains that may have affected the results above, we compared the lung responses to PVM infection of GzmA−/−GzmB−/− mice to GzmA+/−GzmB+/− mice (22,25).
Viral titers and caspase activation
Consistent with our findings above, whole lung virus titers remained indistinguishable (Figure 3A), while there was a marked reduction in caspase-3 activity in lung homogenates in the GzmA−/−GzmB−/− mice as compared to the GzmA+/−GzmB+/− mice (Figure 3B). To investigate the lung tissue distribution of active caspase-3 in the mice, we performed immunohistochemical localization for cleaved caspase-3 in lung tissue sections of day 8 after PVM inoculation. The lung tissues of GzmA+/−GzmB+/− mice showed widespread cleaved caspase-3 staining in alveolar wall cells and among infiltrating leukocytes (Figure 3C). Interestingly, although PVM replication is detected primarily in bronchial epithelial cells (27), cleaved caspase-3 was not evident in these cells.
Figure 3.

PVM clearance and apoptosis in GzmA and GzmB-cluster gene deleted mice. A, virus titer in the lung, expressed as number of PVM-sh copies per 109 gapdh copies; B, lung homogenate caspase-3 activity (arbitrary fluorescence units), in the GzmA+/−GzmB+/− mice and GzmA−/−GzmB−/− mice on day 7 (n=6 per group) and 8 (n=6 per group) after PVM inoculation (6 × 103 copies). * p<0.05. Data are shown as box plots depicting the median, interquartile range and full range. C, representative images of cleaved caspase-3 immunohistochemistry in lung tissues of the GzmA+/−GzmB+/− mice and GzmA−/−GzmB−/− mice on day 8 after PVM inoculation. Note the relative positive staining in alveolar wall cells (arrows) as compared to bronchial epithelial cells in the GzmA+/−GzmB+/− mice.
Lung permeability
As compared to the GzmA+/−GzmB+/− mice, the GzmA−/−GzmB−/− mice displayed diminished concentrations of α-macroglobulin and IgM in BALF, both markers for lung permeability (9) (Figure 4A and B).
Figure 4.
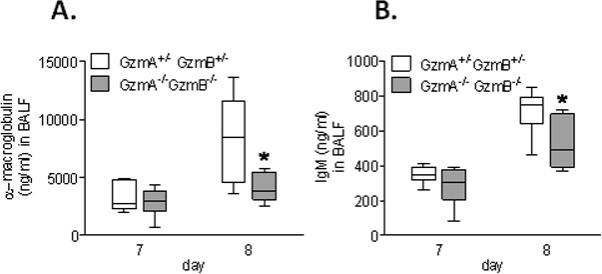
Lung permeability response to PVM infection in GzmA and GzmB-cluster gene deleted mice. Concentration of α-macroglobulin (A) and IgM (B) in BALF of the GzmA+/−GzmB+/− mice and GzmA−/−GzmB−/− mice on day 7 (n=6 per group) and 8 (n=6 per group) after PVM inoculation (6 × 103 copies). * p<0.05. Data are shown as box plots depicting the median, interquartile range and full range.
Lung inflammation
As compared to the GzmA+/−GzmB+/− mice, the GzmA−/−GzmB−/− mice had decreased total neutrophil counts in BALF on day 7 after PVM inoculation (Figure 5A). However, this apparent delay in neutrophil influx was not associated with the release of cytokines IL-1β (not detected in any of the mice), IL-6 (Figure 5B), TNFα (Figure 5C), MIP-2 (Figure 5E) or KC (Figure 5F). IFNγ is among the critical regulators of the neutrophil response in PVM infection (32), and was the only cytokine that correlated with the delay in neutrophil recruitment in the GzmA−/−GzmB−/− mice (Figure 5D).
Figure 5.
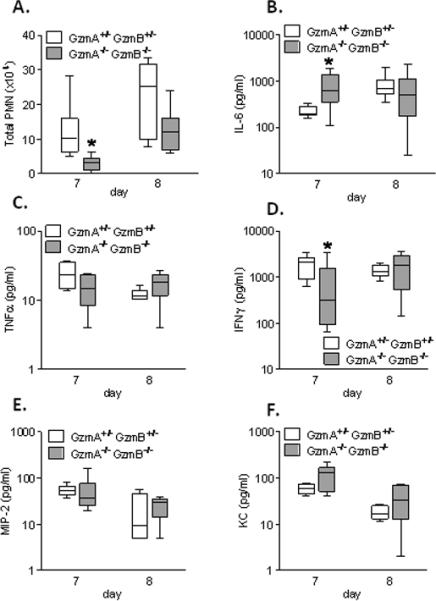
Lung inflammatory response to PVM infection in GzmA and GzmB-cluster gene deleted mice. Total neutrophil (PMN) counts (A), and concentrations of IL-6 (B), TNFα (C), IFNγ (D), MIP-2 (E) and KC (F) in BALF of the GzmA+/−GzmB+/− mice and GzmA−/−GzmB−/− mice on day 7 (n=6 per group) and 8 (n=6 per group) after PVM inoculation (6 × 103 copies). * p<0.05. Data are shown as box plots depicting the median, interquartile range and full range.
Lung histology
On day 8 after inoculation with PVM, the lung tissues from all mice showed extensive peri-bronchial and peri-vascular cellular infiltrates, as well as areas with acute alveolitis and alveolar wall alterations such as thickening and (mild hemorrhagic) capillary congestion, similar to findings described previously (8). However, cellular alveolitis and capillary congestion was more diffuse in the GzmA+/−GzmB+/− mice, when compared to GzmA−/−GzmB−/− mice (Figure 6): mean (± SE) histopathology scores were 3.2 (0.3) in the GzmA−/+GzmB−/+ mice, and 2.2 (0.2) in the GzmA−/−GzmB−/− mice (p<0.05).
Figure 6.

Lung tissue response to PVM infection in GzmA and GzmB-cluster gene deleted mice. Representative images of haematoxylin and eosin stained lung tissue from the GzmA+/−GzmB+/− mice and GzmA−/−GzmB−/− mice on day 8 after PVM inoculation (6 × 103 copies). Magnification 100x.
Clinical response
Finally, to address whether these Gzm-mediated alterations of the lung response to PVM affect the clinical phenotype, we followed clinical scores and body weight up to day 12 after inoculation with a lethal inoculum of PVM. The GzmA−/−GzmB−/− mice showed a statistically significant delay in reaching the maximum clinical response to PVM (Figure 7A). All the GzmA+/−GzmB+/− mice reached the defined endpoint (clinical score of 5 and/or body weight loss > 20%) at day 10 after PVM inoculation, whereas at this point this was reached by 67% of the GzmA−/−GzmB−/− mice (p<0.05). A delayed response to PVM infection among the GzmA−/−GzmB−/− mice was also observed when weight loss alone as a sole, purely objective measure of the clinical response was examined (Figure 7B).
Figure 7.

Clinical response to PVM infection in GzmA and GzmB-cluster gene deleted mice. A, Kaplan-Meier curves showing the percentages of GzmA+/−GzmB+/− (black dots, n=9) and GzmA−/−GzmB−/− (white dots, n=9) mice reaching the endpoint of a clinical score of 5 (moribund) and/or weight loss > 20% after PVM inoculation (1 × 104 copies). p < 0.05 by log-rank (Mantel-Cox) test. B, percentage of baseline (day 0) weight of the GzmA+/−GzmB+/− (black dots) and GzmA−/−GzmB−/− (white dots) mice after PVM inoculation (1 × 104 copies). * p < 0.05. Bars show the mean.
DISCUSSION
The primary goal of this study was to determine whether Gzms promote pathophysiologic responses, including acute lung injury, in pneumovirus-infected mice. In this work, we detect both GzmA (predominantly in NK cells) and GzmB (predominantly in CTLs) in association with severe respiratory sequelae induced by the mouse pneumovirus pathogen, PVM. We found that mice deficient for both GzmA and the GzmB-cluster (GzmA−/−GzmB−/−) have a delayed clinical response to fatal PVM infection, although Gzm expression has no impact on virus replication or clearance per se. Instead, we determine that the absence of Gzms results in delayed neutrophil recruitment, diminished activation of caspase-3 and reduced lung permeability.
The Gzm system exploited by effector lymphocytes plays a major role in host anti-viral immunity by activating cell death programs in infected target cells (10–12). Providing the first in vivo evidence for Gzm-mediated viral elimination, Mullbacher et al. (31) showed that the survival and clearance of the mouse poxvirus, ectromelia, in mice lacking both GzmA and the GzmB-cluster was markedly depressed as compared to wild type mice. Systematic challenge of Gzm-deficient mice to different virus families has not been undertaken, but replication of cytomegalovirus and herpes simplex virus have also found to be controlled by Gzm activity (33,34). At the same time, there is much evidence that effector lymphocyte-mediated immunopathogenic mechanisms form the basis of enhanced disease in certain virus infections, and this includes the development of severe human pneumovirus (RSV) disease (35,36). For example, both CTLs and NK cells are recruited to the lungs during primary RSV infection in mice (36–38). Depletion of CTLs in mice reduces clinical signs of RSV disease, despite delaying viral clearance (36), while intravenous transfer of high dose CTLs into CTL-depleted mice causes severe hemorrhage and neutrophilic inflammation (39). Frey et al. (40) studied the PVM mouse model, which, unlike RSV infection in mice, reproduces several features of severe RSV disease in humans, including robust viral replication, pro-inflammatory alterations in the airways and alveoli, alveolar injury and importantly, overt clinical signs of illness (8). They found similar results, namely, that T-cell deficient mice have a delayed clearance of PVM, but reduced pulmonary inflammatory infiltrates and weight loss (40). These findings suggested that effector lymphocytes may contribute to the immunopathophysiology associated with human RSV infection, but whether the Gzm system is involved herein remained unclear from those studies.
Recently, we found high levels of active GzmA and GzmB in tracheal aspirates from children with severe RSV disease (20). Surprisingly, in the present study, virus titers in lung tissue were not altered by Gzm-targeting, despite a major reduction in lung caspase-3 activation in GzmA and GzmB-cluster deficient mice. Similarly, Frey et al. (40) found that mice deficient for perforin, which facilitates granzyme-trafficking into virus-infected target cells, did not alter PVM lung titers. These findings suggest that alternative pathways may play more prominent roles in PVM clearance. Certain viruses, such as adenovirus and parainfluenza virus type 3, are known to have evolved strategies that specifically inhibit GzmB (41,42), and poxvirus can block caspase signaling (11,43). Interestingly, in our study we found little to no caspase-3 activation in bronchial epithelial cells, clearly the most important site for PVM replication (27). However it is currently unknown whether PVM directly induces anti-apoptotic pathways in infected bronchial epithelial cells.
Similar to previous findings (9), PVM infection was associated with increased caspase-3 activation in alveolar wall cells, and this was markedly reduced in Gzm-deficient mice. As such, this finding may explain at least in part the observed decreased lung permeability in the Gzm-deficient mice. Accumulating evidence implicates caspase-dependent apoptosis as an important mechanism leading to loss of alveolar capillary barrier integrity and ultimately enhanced lung permeability (44). Patients who die with ALI/ARDS have increased caspase-3 staining in alveolar wall cells (45), and numerous experimental studies modelling both indirect and direct ALI/ARDS have shown enhanced apoptosis of lung epithelial cells is associated with leakage of serum proteins into the lungs and histopathological alterations, which can be attenuated by inhibiting pro-apoptotic pathways (44,46,47). Our findings suggest the Gzm pro-apoptotic pathway can contribute to the development of alveolar injury in severe pneumovirus disease. Interestingly, both GzmA and GzmB mRNA expression is augmented in patients with septic ARDS and in mice intratracheally instilled with LPS (47,48), suggesting the Gzm pathway may also be of relevance to bacterial-induced ALI/ARDS .
In addition to pro-apoptotic functions of Gzms, recent renewed attention has been given to their potential pro-inflammatory effects, specifically of GzmA (18,19). Metkar et al. (18) found that active human GzmA induces the release of IL-1β, IL-6 and TNF-α from monocytes, which can be attenuated by the IL-1β convertase (caspase-1) inhibitor WEHD-FMK, and similarly, that mouse macrophages produce IL-1β in response to treatment with active mouse GzmA. Interestingly, GzmA and GzmB-cluster deficient mice showed a delayed lung neutrophil response to PVM infection, but this was not associated with the release of IL-1β, IL-6, TNF-α, KC and MIP-2. The fact that we did not detect IL-1β in the BALF of any of the mice may suggest that the above mentioned mechanism of GzmA-mediated pro-inflammatory activity through caspase-1 may not be directly relevant in the PVM natural infection model. IFNγ, potentially released from activated NK cells, has been identified as a critical regulator of neutrophil influx into the lungs in severe PVM disease in mice (32), and followed the same pattern as the total neutrophil counts in the GzmA and GzmB-cluster deficient mice. However, it remains to be elucidated whether mouse Gzm may directly mediate the release of IFNγ, as for example, human GzmA does not induce IFNγ specifically in monocytes (18). In addition, one needs to recognize that a number of different events, including adhesion, migration and clearance, affect the number of neutrophils in the lungs. In this light, the potent proteolytic degradation of extracellular matrix proteins by both GzmA and GzmB is of particular interest, because Gzm-mediated remodelling of the extracellular environment has been shown to have important consequences for cellular migration and survival (49).
As stated above, in the most current paradigm, GzmA elicits predominantly pro-inflammatory activity, while GzmB mediates caspase-dependent apoptosis, based mostly on observations in vitro (15,16). However, deciphering the specific functions of the individual Gzm family members in vivo has proven to be difficult and not straightforward (11,16,18,31). Interestingly, studies in mice have shown that the combined deficiency for GzmA and the GzmB-cluster can result in a more pronounced or paradoxically diminished phenotype, as compared to single-gene (GzmA or the GzmB-cluster) deficiency (18,31), suggesting complex and synergistic interaction between Gzms. The interpretation of experiments involving Gzm targeted mice is further complicated by the fact that the GzmB-gene deleted mice lack the entire GzmB-cluster which includes GzmC, -D and -F (26). In addition, the differences in substrain background between wild type, single-gene and double-gene Gzm deleted mice may affect host responses to pathogens. Finally, in vivo, through indirect mechanisms involving inflammatory cells and extra-cellular matrix remodeling, Gzms may influence several processes, including cell death pathways (49), to which they are not directly attributed to: e.g. while GzmA does not directly cause caspase activation, in our study, GzmA single-gene deleted mice showed some reduction in lung caspase-3 activity. Taking into account these several issues, we have primarily focused on the effects of combined deficiency for GzmA and the GzmB-cluster, but this obviously limits our interpretation of the functions of the individual Gzm family members in acute pneumovirus infection.
Finally, it is important to recognize that mouse and human Gzm differ in structure, enzyme activity and inhibitory regulation (50). For example, human GzmB is more cytotoxic than mouse GzmB in vitro (50). In addition, unlike mouse GzmB, the human form requires Bid cleavage for full caspase-3 activation, and is controlled by the inhibitor PI-9 (50). Furthermore, the evolution of the Gzm system, potentially driven by host specific pathogens, may compromise some of the meaning of any study of Gzm responses to human virus pathogens inoculated in mice. As such, the use of the natural mouse pneumovirus pathogen, PVM, in this work may represent an important advantage, as it permits us to explore the roles and functions of Gzms in response to a physiologically and evolutionarily relevant challenge. On the other hand, although PVM and RSV are both pneumoviruses, they are not the same virus, and we need to stress there is no one animal model that displays all features of human RSV disease.
In conclusion, pneumovirus infection in mice induces expression of GzmA and GzmB by effector lymphocytes. The combined deficiency of GzmA and the GzmB-cluster results in a delayed progression of pneumovirus disease by reducing alveolar injury in mice, without altering viral clearance. We speculate that targeting of the Gzm system in humans may be beneficial in diminishing RSV-ALI/ARDS immunopathogenesis.
ACKNOWLEDGEMENTS
The authors thank P.J. van de Berg for expert advice on FACS analysis, and J. de Jong for assistance with animal breeding.
Gants/funding: supported in part by Ars Donandi, Amsterdam. H.F. Rosenberg supported by National Institute of Allergy and Infectious Diseases Division of Intramural Research (Z01-AI000943).
Footnotes
Abbreviations used in this paper: RSV, respiratory syncytial virus; ALI/ARDS, acute lung injury/acute respiratory distress syndrome; PVM, pneumonia virus of mice; Gzm, granzyme; BAL(F), bronchoalveolar lavage (fluid)
REFERENCES
- 1.Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, Grijalva CG, Zhu Y, Szilagyi P. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 2009;360:588–598. doi: 10.1056/NEJMoa0804877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shay DK, Holman RC, Newman RD, Liu LL, Stout JW, Anderson LJ. Bronchiolitis-associated hospitalizations among US children, 1980–1996. JAMA. 1999;282:1440–1446. doi: 10.1001/jama.282.15.1440. [DOI] [PubMed] [Google Scholar]
- 3.Dahlem P, van Aalderen WM, Hamaker ME, Dijkgraaf MG, Bos AP. Incidence and short-term outcome of acute lung injury in mechanically ventilated children. Eur. Respir. J. 2003;22:980–985. doi: 10.1183/09031936.03.00003303. [DOI] [PubMed] [Google Scholar]
- 4.Nokes JD, Cane PA. New strategies for control of respiratory syncytial virus infection. Curr. Opin. Infect. Dis. 2008;21:639–643. doi: 10.1097/QCO.0b013e3283184245. [DOI] [PubMed] [Google Scholar]
- 5.Ware LB, Matthay MA. The acute respiratory distress syndrome. N. Engl. J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 6.Wang SZ, Forsyth KD. The interaction of neutrophils with respiratory epithelial cells in viral infection. Respirology. 2000;5:1–10. doi: 10.1046/j.1440-1843.2000.00219.x. [DOI] [PubMed] [Google Scholar]
- 7.Welliver TP, Reed JL, Welliver RC., Sr. Respiratory syncytial virus and influenza virus infections: observations from tissues of fatal infant cases. Pediatr. Infect. Dis. J. 2008;27:S92–S96. doi: 10.1097/INF.0b013e318168b706. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg HF, Domachowske JB. Pneumonia virus of mice: severe respiratory infection in a natural host. Immunol. Lett. 2008;118:6–12. doi: 10.1016/j.imlet.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bem RA, van Woensel JB, Bos AP, Koski A, Farnand AW, Domachowske JB, Rosenberg HF, Martin TR, Matute-Bello G. Mechanical ventilation enhances lung inflammation and caspase activity in a model of mouse pneumovirus infection. Am. J Physiol Lung Cell Mol. Physiol. 2009;296:L46–L56. doi: 10.1152/ajplung.00467.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat. Rev. Immunol. 2003;3:361–370. doi: 10.1038/nri1083. [DOI] [PubMed] [Google Scholar]
- 11.Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 2002;20:323–370. doi: 10.1146/annurev.immunol.20.100201.131730. [DOI] [PubMed] [Google Scholar]
- 12.Bots M, Medema JP. Granzymes at a glance. J Cell Sci. 2006;119:5011–5014. doi: 10.1242/jcs.03239. [DOI] [PubMed] [Google Scholar]
- 13.Browne KA, Blink E, Sutton VR, Froelich CJ, Jans DA, Trapani JA. Cytosolic delivery of granzyme B by bacterial toxins: evidence that endosomal disruption, in addition to transmembrane pore formation, is an important function of perforin. Mol. Cell Biol. 1999;19:8604–8615. doi: 10.1128/mcb.19.12.8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Froelich CJ, Orth K, Turbov J, Seth P, Gottlieb R, Babior B, Shah GM, Bleackley RC, Dixit VM, Hanna W. New paradigm for lymphocyte granule-mediated cytotoxicity. Target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J. Biol. Chem. 1996;271:29073–29079. doi: 10.1074/jbc.271.46.29073. [DOI] [PubMed] [Google Scholar]
- 15.Froelich CJ, Pardo J, Simon MM. Granule-associated serine proteases: granzymes might not just be killer proteases. Trends Immunol. 2009;30:117–123. doi: 10.1016/j.it.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Trapani JA, Bird PI. A renaissance in understanding the multiple and diverse functions of granzymes? Immunity. 2008;29:665–667. doi: 10.1016/j.immuni.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Martinvalet D, Dykxhoorn DM, Ferrini R, Lieberman J. Granzyme A cleaves a mitochondrial complex I protein to initiate caspase-independent cell death. Cell. 2008;133:681–692. doi: 10.1016/j.cell.2008.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metkar SS, Menaa C, Pardo J, Wang B, Wallich R, Freudenberg M, Kim S, Raja SM, Shi L, Simon MM, Froelich CJ. Human and mouse granzyme A induce a proinflammatory cytokine response. Immunity. 2008;29:720–733. doi: 10.1016/j.immuni.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 19.Sower LE, Klimpel GR, Hanna W, Froelich CJ. Extracellular activities of human granzymes. I. Granzyme A induces IL6 and IL8 production in fibroblast and epithelial cell lines. Cell Immunol. 1996;171:159–163. doi: 10.1006/cimm.1996.0187. [DOI] [PubMed] [Google Scholar]
- 20.Bem RA, Bos AP, Bots M, Wolbink AM, van Ham SM, Medema JP, Lutter R, van Woensel JB. Activation of the granzyme pathway in children with severe respiratory syncytial virus infection. Pediatr Res. 2008;63:650–655. doi: 10.1203/PDR.0b013e31816fdc32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Domachowske JB, Bonville CA, Gao JL, Murphy PM, Easton AJ, Rosenberg HF. The chemokine macrophage-inflammatory protein-1 alpha and its receptor CCR1 control pulmonary inflammation and antiviral host defense in paramyxovirus infection. J Immunol. 2000;165:2677–2682. doi: 10.4049/jimmunol.165.5.2677. [DOI] [PubMed] [Google Scholar]
- 22.Heusel JW, Wesselschmidt RL, Shresta S, Russell JH, Ley TJ. Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell. 1994;76:977–987. doi: 10.1016/0092-8674(94)90376-x. [DOI] [PubMed] [Google Scholar]
- 23.Simon MM, Hausmann M, Tran T, Ebnet K, Tschopp J, ThaHla R, Mullbacher A. In vitro- and ex vivo-derived cytolytic leukocytes from granzyme A × B double knockout mice are defective in granule-mediated apoptosis but not lysis of target cells. J Exp. Med. 1997;186:1781–1786. doi: 10.1084/jem.186.10.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bots M, Kolfschoten IG, Bres SA, Rademaker MT, de Roo GM, Kruse M, Franken KL, Hahne M, Froelich CJ, Melief CJ, Offringa R, Medema JP. SPI-CI and SPI-6 cooperate in the protection from effector cell-mediated cytotoxicity. Blood. 2005;105:1153–1161. doi: 10.1182/blood-2004-03-0791. [DOI] [PubMed] [Google Scholar]
- 25.Ebnet K, Hausmann M, Lehmann-Grube F, Mullbacher A, Kopf M, Lamers M, Simon MM. Granzyme A-deficient mice retain potent cell-mediated cytotoxicity. EMBO J. 1995;14:4230–4239. doi: 10.1002/j.1460-2075.1995.tb00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Revell PA, Grossman WJ, Thomas DA, Cao X, Behl R, Ratner JA, Lu ZH, Ley TJ. Granzyme B and the downstream granzymes C and/or F are important for cytotoxic lymphocyte functions. J. Immunol. 2005;174:2124–2131. doi: 10.4049/jimmunol.174.4.2124. [DOI] [PubMed] [Google Scholar]
- 27.Bonville CA, Bennett NJ, Koehnlein M, Haines DM, Ellis JA, DelVecchio AM, Rosenberg HF, Domachowske JB. Respiratory dysfunction and proinflammatory chemokines in the pneumonia virus of mice (PVM) model of viral bronchiolitis. Virology. 2006;349:87–95. doi: 10.1016/j.virol.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 28.Ellis JA, Martin BV, Waldner C, Dyer KD, Domachowske JB, Rosenberg HF. Mucosal inoculation with an attenuated mouse pneumovirus strain protects against virulent challenge in wild type and interferon-gamma receptor deficient mice. Vaccine. 2007;25:1085–1095. doi: 10.1016/j.vaccine.2006.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bem RA, Farnand AW, Wong V, Koski A, Rosenfeld ME, van RN, Frevert CW, Martin TR, Matute-Bello G. Depletion of resident alveolar macrophages does not prevent Fas-mediated lung injury in mice. Am. J Physiol Lung Cell Mol. Physiol. 2008;295:L314–L325. doi: 10.1152/ajplung.00210.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cook PM, Eglin RP, Easton AJ. Pathogenesis of pneumovirus infections in mice: detection of pneumonia virus of mice and human respiratory syncytial virus mRNA in lungs of infected mice by in situ hybridization. J Gen. Virol. 1998;79(Pt 10):2411–2417. doi: 10.1099/0022-1317-79-10-2411. [DOI] [PubMed] [Google Scholar]
- 31.Mullbacher A, Waring P, Tha HR, Tran T, Chin S, Stehle T, Museteanu C, Simon MM. Granzymes are the essential downstream effector molecules for the control of primary virus infections by cytolytic leukocytes. Proc. Natl. Acad. Sci U. S. A. 1999;96:13950–13955. doi: 10.1073/pnas.96.24.13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonville CA, Percopo CM, Dyer KD, Gao J, Prussin C, Foster B, Rosenberg HF, Domachowske JB. Interferon-gamma coordinates CCL3-mediated neutrophil recruitment in vivo. BMC. Immunol. 2009;10:14. doi: 10.1186/1471-2172-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pereira RA, Simon MM, Simmons A. Granzyme A, a noncytolytic component of CD8(+) cell granules, restricts the spread of herpes simplex virus in the peripheral nervous systems of experimentally infected mice. J Virol. 2000;74:1029–1032. doi: 10.1128/jvi.74.2.1029-1032.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riera L, Gariglio M, Valente G, Mullbacher A, Museteanu C, Landolfo S, Simon MM. Murine cytomegalovirus replication in salivary glands is controlled by both perforin and granzymes during acute infection. Eur. J Immunol. 2000;30:1350–1355. doi: 10.1002/(SICI)1521-4141(200005)30:5<1350::AID-IMMU1350>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 35.Domachowske JB, Rosenberg HF. Respiratory syncytial virus infection: immune response, immunopathogenesis, and treatment. Clin. Microbiol. Rev. 1999;12:298–309. doi: 10.1128/cmr.12.2.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graham BS, Bunton LA, Wright PF, Karzon DT. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J Clin. Invest. 1991;88:1026–1033. doi: 10.1172/JCI115362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hussell T, Openshaw PJ. Intracellular IFN-gamma expression in natural killer cells precedes lung CD8+ T cell recruitment during respiratory syncytial virus infection. J. Gen. Virol. 1998;79(Pt 11):2593–2601. doi: 10.1099/0022-1317-79-11-2593. [DOI] [PubMed] [Google Scholar]
- 38.Openshaw PJ. Flow cytometric analysis of pulmonary lymphocytes from mice infected with respiratory syncytial virus. Clin. Exp. Immunol. 1989;75:324–328. [PMC free article] [PubMed] [Google Scholar]
- 39.Cannon MJ, Openshaw PJ, Askonas BA. Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J Exp. Med. 1988;168:1163–1168. doi: 10.1084/jem.168.3.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frey S, Krempl CD, Schmitt-Graff A, Ehl S. Role of T cells in virus control and disease after infection with pneumonia virus of mice. J. Virol. 2008;82:11619–11627. doi: 10.1128/JVI.00375-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andrade F, Bull HG, Thornberry NA, Ketner GW, Casciola-Rosen LA, Rosen A. Adenovirus L4-100K assembly protein is a granzyme B substrate that potently inhibits granzyme B-mediated cell death. Immunity. 2001;14:751–761. doi: 10.1016/s1074-7613(01)00149-2. [DOI] [PubMed] [Google Scholar]
- 42.Sieg S, Xia L, Huang Y, Kaplan D. Specific inhibition of granzyme B by parainfluenza virus type 3. J. Virol. 1995;69:3538–3541. doi: 10.1128/jvi.69.6.3538-3541.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J. Biol. Chem. 1997;272:7797–7800. doi: 10.1074/jbc.272.12.7797. [DOI] [PubMed] [Google Scholar]
- 44.Martin TR, Hagimoto N, Nakamura M, Matute-Bello G. Apoptosis and epithelial injury in the lungs. Proc. Am. Thorac. Soc. 2005;2:214–220. doi: 10.1513/pats.200504-031AC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, Zimmerman GA, Matthay MA, Ware LB. Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am. J. Pathol. 2002;161:1783–1796. doi: 10.1016/S0002-9440(10)64455-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawasaki M, Kuwano K, Hagimoto N, Matsuba T, Kunitake R, Tanaka T, Maeyama T, Hara N. Protection from lethal apoptosis in lipopolysaccharide-induced acute lung injury in mice by a caspase inhibitor. Am. J. Pathol. 2000;157:597–603. doi: 10.1016/S0002-9440(10)64570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kitamura Y, Hashimoto S, Mizuta N, Kobayashi A, Kooguchi K, Fujiwara I, Nakajima H. Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide-induced lung injury in mice. Am. J. Respir. Crit Care Med. 2001;163:762–769. doi: 10.1164/ajrccm.163.3.2003065. [DOI] [PubMed] [Google Scholar]
- 48.Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am. J. Respir. Crit Care Med. 2000;161:237–243. doi: 10.1164/ajrccm.161.1.9810007. [DOI] [PubMed] [Google Scholar]
- 49.Buzza MS, Bird PI. Extracellular granzymes: current perspectives. Biol. Chem. 2006;387:827–837. doi: 10.1515/BC.2006.106. [DOI] [PubMed] [Google Scholar]
- 50.Kaiserman D, Bird CH, Sun J, Matthews A, Ung K, Whisstock JC, Thompson PE, Trapani JA, Bird PI. The major human and mouse granzymes are structurally and functionally divergent. J Cell Biol. 2006;175:619–630. doi: 10.1083/jcb.200606073. [DOI] [PMC free article] [PubMed] [Google Scholar]