

Abstract
In chronic wasting disease (CWD) of cervids and scrapie of sheep, prions appear to be transmitted horizontally. Oral exposure to prion-tainted blood, urine, saliva and feces has been suggested as the mode of transmission for CWD and scrapie among herbivores susceptible to these prion diseases. To explore the transmission of prions through feces, uninoculated Syrian hamsters (SHa) were cohabitated with or exposed to the bedding of SHa orally infected with Sc237 prions. Incubation times of ~140 days and 80–100% prion infection rate in exposed animals suggested transmission by feces, probably via coprophagy. We measured the disease-causing isoform of the prion protein (PrPSc) in feces by the conformation-dependent immunoassay and titrated the irradiated feces intracerebrally in transgenic mice overexpressing SHaPrP. Feces collected from infected SHa in the first 7 days after oral challenge harbored ~60 ng/g of PrPSc and prion titers of ~106.6 ID50 units/g. The excretion of infectious prions continued at lower levels throughout the asymptomatic phase of the incubation period, most likely by shedding prions from infected Peyer’s patches. Our findings suggest that horizontal transmission among herbivores may occur through the consumption of feces or foodstuff tainted with prions from feces of CWD-infected cervids and scrapie-infected sheep.
Keywords: prion, feces, horizontal transmission, chronic wasting disease, scrapie, coprophagy
INTRODUCTION
Creutzfeldt-Jakob disease (CJD) of humans, bovine spongiform encephalopathy (BSE), scrapie of sheep, and chronic wasting disease (CWD) of cervids are neurodegenerative diseases caused by prions [1]. A wealth of experimental and epidemiological data indicate that variant (v) CJD in humans results from the contamination of the food supply, probably in mechanically recovered meat, with BSE prions [2–4]. Recent evidence suggests that vCJD also may have been transmitted iatrogenically by transfusions using prion-contaminated blood [5–8]. Estimating the dose of prions to which children were exposed during ritualistic cannibalism prompted the hypothesis that another port of entry must exist, perhaps by abrasions of mucous membranes in the oral cavity or eye, or by open wounds on the hands [9].
Whereas BSE, vCJD, and sporadic (s) CJD do not seem to be horizontally transmissible by contact, both scrapie and CWD spread naturally from animals in their respective flocks and herds. The transmission rates of CWD have been reported to be as high as 30%, and can approach 100% in captive animals in endemic areas [10–13]. Despite extensive research, it is unknown how scrapie spreads between sheep or flocks [11]. The vertical transmission of scrapie—between ewes and their offspring—has not been demonstrated [14–16]. Maternal transmission also has been studied in BSE-infected ewes, but no transmission of BSE has been observed either in goats or in sheep [17]. Scrapie prions can persist in the environment up to 3 years after removal of the scrapie-affected flock [18]. Moreover, it is possible to derive a scrapie-free sheep flock from the progeny of a scrapie-afflicted flock, providing that the lambs are kept in a clean environment [19]. Similarly, circumstantial evidence suggests that CWD is transmitted by environmental contamination of CWD prions [10, 20, 21] or by contact with infected cervids [22, 23]. Regardless of the mechanisms, indirect transmission and the environmental persistence of infectious prions complicate efforts to eradicate and control the spread of both CWD and scrapie.
Oral exposure to prion-tainted blood, urine, saliva and feces have been suggested as modes of transmission for CWD and scrapie among herbivores susceptible to these diseases [10, 11, 19, 22, 23]. Both CWD and scrapie infection is thought likely to enter the body through gut-associated lymphoid tissues, in Peyer’s patches in the alimentary tract [11, 24–28]. Moreover, the presence of the infectious isoform of the prion protein, designated PrPSc, in Peyer’s patches [11, 24, 29, 30] suggests alimentary shedding of CWD and scrapie prions into feces.
To study the presence of prions in feces and their mechanism of transmission, we employed a well established, Syrian hamster model of oral prion infection [31]. The study was facilitated by the fact that Syrian hamsters are cannibalistic [31] and, like many other herbivores, practice coprophagy [32]. When noninfected Syrian hamsters were cohabitated with Syrian hamsters orally infected with Sc237 prions, we observed 80–100% infection rates within 14 days after oral challenge. Since the transmission rate of Sc237 prions from bedding was similar, the data suggest that feces could be the source of infection. Consequently, we collected feces from orally infected Syrian hamsters, irradiated them, and measured the concentration of PrPSc by the conformation-dependent immunoassay (CDI) and the prion titer by bioassay in transgenic (Tg) mice overexpressing Syrian hamster PrP. We found infectivity titers of ~106.6 ID50 units/g and corresponding levels of PrPSc in the first 7 days after oral infection. The excretion of infectious PrPSc continued at lower levels through the asymptomatic period of disease. The data presented here provide a mechanism for the horizontal transmission and environmental contamination of prions, and may help efforts to control the spread of CWD and scrapie.
MATERIALS AND METHODS
Animals and prion inoculation
Three- to five-week-old, female Golden Syrian hamsters (Mesocricetus auratus) were acquired from Charles River Laboratories and housed in polycarbonate cages with paper bedding (Paper Chip). Transgenes and production of Tg(SHaPrP+/+)7 mice, or Tg7 mice for simplicity, have been described previously [33]. All studies presented in this study were reviewed and approved by the UCSF Institutional Animal Care and Use Committee.
For intracerebral (i.c.) inoculation, hamsters and weanling mice were injected intracerebrally with 50 and 30 μl of 1% (w/v) brain homogenate prepared from Sc237-infected SHa. Inoculation was carried out with a 26-gauge, disposable hypodermic needle inserted into the right parietal lobe.
For intraperitoneal (i.p.) inoculation, five- to seven-week-old SHa were injected in the right lower quadrant of the abdominal cavity with 100 μl of 1% brain homogenate prepared from Sc237-infected SHa.
For oral challenge, five- to seven-week-old SHa were placed individually in polycarbonate cages without bedding, put on a dietary fast for 18 h, and then offered half of the brain from a Sc237-infected SHa in a petri dish. SHa were observed until the brains were consumed entirely.
After inoculation and oral challenge, animals were examined daily for neurologic dysfunction. Standard diagnostic criteria were used to identify animals affected by prion disease. Animals whose deaths were imminent were sacrificed, and their brains were removed for histological and biochemical analysis.
Collection and treatment of feces
Four i.c., i.p., or orally infected Syrian hamsters were housed together in standard cages. Ten fecal pellets were collected at 1, 8, 22, 43, 64, 85, and 106 days postinfection with sterile forceps from the bottom of the cage, in the resting area opposite of the corner used for urination. The sterile forceps were changed between cages and only fresh (dark and moist) pellets were selected. Since the i.c. inoculated animals developed symptoms starting at ~70 days postinoculation, their feces were collected only up to 63 days. The fecal pellets were stored in a plastic container at −80°C.
To prepare 10% (w/v) homogenate of feces, 1 g of frozen feces was thawed and homogenized in PBS, pH 7.4, using a standard Mini Vortexer (VWR Scientific Products). The homogenate was mixed for 10 min and filtered through folded surgical cotton gauze to remove large particles. After final homogenization in a 5-ml syringe equipped with a 21-gauge needle, a 1-ml aliquot was transferred to a sterile glass container and irradiated by Cesium-137 at a total dose of 2.9 kGy.
Transmission of prions by cohabitation and bedding
To determine the transmissibility of prions by cohabitation, 17 Syrian hamsters were orally challenged with Sc237 prions. One ear of each infected animal was surgically notched for identification purposes. One orally infected Syrian hamster was placed in a new cage with three uninoculated Syrian hamsters immediately thereafter; or 2 h; or 1, 2, 7, or 14 days after oral challenge (Supplementary Fig. 1). All animals were observed for the development of neurologic signs of prion disease. To avoid cannibalism, the sick hamsters were removed from the cages as soon as they demonstrated symptoms of prion disease.
For transmission by bedding, two Syrian hamsters infected either orally or intracerebrally with Sc237 prions were housed in standard cages with paper bedding (Paper Chip). After 3.5 or 7 days, both infected hamsters were moved to a new cage, and four noninfected hamsters were placed into the cage vacated by the two infected animals; the original bedding was maintained (Supplementary Fig. 2). This cycle was repeated until the two infected hamsters developed prion disease and were euthanized.
Incubation-time assay
The incubation-time assay was performed as previously described [34]. Titers were calculated from the incubation time using either a double-exponential function or power function to describe the relationship between incubation time and prion titer [1, 34, 35].
Sample preparation, CDI and Western blots
Preparation of brain and fecal samples, and detection of PrPSc by the CDI and Western immunoblotting, are described in the Supplementary online material.
Histopathology
Brain tissue was either immediately frozen or immersion-fixed in 10% buffered formalin for embedding in paraffin. Eight-micrometer-thick sections were stained with hematoxylin and eosin (H&E) to evaluate vacuolation. Evaluation of reactive astrocytic gliosis was performed by GFAP immunostaining using a rabbit antiserum (Dako, Carpinteria, CA). Hydrolytic autoclaving pretreatment of the formalin-fixed tissue sections was used to detect PrPSc, as described previously [36].
RESULTS
Prion transmission by cohabitation with orally infected Syrian hamsters
Syrian hamsters that were fed Sc237-infected brains were placed in cages with uninfected Syrian hamsters at 0, 0.1, 1, 2, 7, and 14 days after oral challenge (Supplementary Fig. 1). All orally infected hamsters succumbed to prion disease with an incubation time of ~130 days (Table 1). Cohabitation resulted in 100% transmission when healthy animals were exposed to infected Syrian hamsters within 2 days post-feeding. For uninoculated Syrian hamsters exposed to infected animals 7 and 14 days postinfection, we found 85% and 80% transmission rates, respectively. We conclude from these experiments that prions were transmitted from orally infected Syrian hamsters by direct animal-to-animal contact or by exposure to a high dose of prions in contaminated cages.
Table 1.
Cohabitation of orally infected Syrian hamsters with uninoculated Syrian hamsters starting at different time points post-feeding.
| Syrian hamsters | Start of cohabitation(days post-feeding) | n/n0 | Incubation time(mean days ± S.E.M.) |
|---|---|---|---|
| Orally infected | – | 17/17 | 129 ± 8 |
| Uninoculated | 0 | 6/6 | 154 ± 6 |
| 0.1 | 4/4 | 150 ± 9 | |
| 1 | 5/5 | 192 ± 17 | |
| 2 | 5/5 | 254 ± 31 | |
| 7 | 6/7 | 165 ± 20 | |
| 14 | 4/5 | 140 ± 1 |
n, number of ill animals; n0, number of animals inoculated.
Prion transmission by exposure to bedding of orally infected Syrian hamsters
To test whether exposure to the bedding of infected Syrian hamsters could result in prion transmission, two orally infected Syrian hamsters were moved out of their cages 3.5 or 7 days after oral challenge and healthy animals were moved in (Supplementary Fig. 2). All orally infected hamsters succumbed to prion disease with a mean incubation time of ~130 days. All Syrian hamsters exposed to the infected bedding developed prion disease, with mean incubation times of ~150 and 140 days, respectively (Table 2). We conclude that prions were transmitted by exposure to prions in the bedding or cages.
Table 2.
Exposure of uninoculated Syrian hamsters to bedding of orally infected Syrian hamsters starting at different time points post-feeding.
| Syrian hamsters | Start of exposure to bedding, days post-feeding | n/n0 | Incubation time |
|---|---|---|---|
| Orally infected | – | 6/6 | 120 ± 8 |
| Uninoculated | 3.5 | 10/10 | 148 ± 14 |
| 7 | 4/4 | 140 ± 1 |
n, number of ill animals; n0, number of animals inoculated. Incubation times are expressed as mean days ± standard error of the mean.
Effect of irradiation on Sc237 prions
Because Syrian hamsters practice coprophagy, we decided to test directly whether feces of orally infected Syrian hamsters harbor prions. Since Sc237 prions are exceptionally resistant to gamma radiation, we employed conditions that would selectively inactivate bacteria and viruses in feces but not prions: a 2.9 kGy dose (0.29 Mram) has a negligible effect on prion infectivity [37, 38]. The initial cultivation experiments for fecal bacteria after different doses of radiation demonstrated that a dose of 2.9 kGy was sufficient to effectively sterilize the feces. When the 10% fecal extract from uninoculated Syrian hamsters was irradiated and inoculated intracerebrally into Tg7 mice, it did not induce any noticeable acute or late clinical symptoms; the survival rate of these feces-inoculated mice was comparable to mock-inoculated animals (Fig. 1A). Feces that were spiked with 1% brain homogenate infected with Sc237 prions, then irradiated and inoculated into Tg7 mice resulted in a mean incubation time of ~55 days, corresponding to a titer of 5.9 ± 0.2 ID50 units/ml (Fig. 1A). The same incubation time was obtained when the feces were first irradiated and then spiked with 1% Sc237-infected homogenate (data not shown). In comparison, nonirradiated 1% Sc237-infected brain homogenate inoculated into Tg7 mice resulted in a mean incubation time of ~44 days, corresponding to a titer of 8.6 ± 0.2 ID50 units/ml (Fig. 1A). The difference in prion titer between infected brain homogenate and feces spiked with infected brain homogenate was 2.7 log ID50 units/ml, as estimated from the incubation-time assay. We conclude that the decrease in titer is due to the presence of fecal material.
Figure 1.
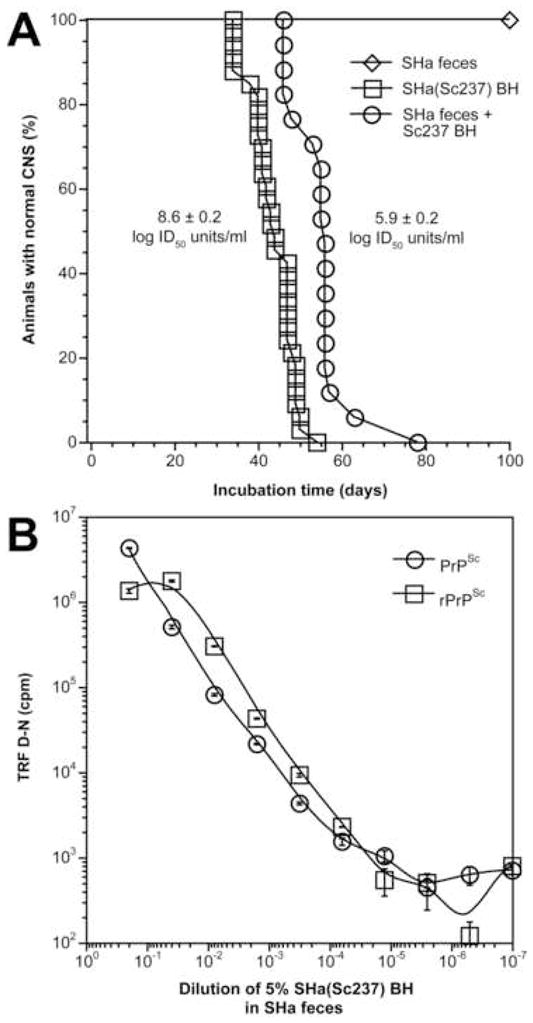
Comparable sensitivity of bioassay and CDI for Sc237 prions diluted into control SHa feces. (A) Bioassay of 1% (w/v) of Sc237-infected brain homogenate (squares), 10% (w/v) irradiated control feces (diamonds), and 10% (w/v) irradiated feces spiked with 1% (w/v) Sc237-infected brain homogenate (circles) in Tg7 mice. The data are plotted as the percentage of animals without signs of clinical disease versus time postinoculation in days. Median incubation time is extrapolated from 50% survival rate in each experiment. The titer was calculated from incubation time of individual animals and expressed as mean ± S.E.M. [35]. (B) The sensitivity of PrPSc detection by CDI in control fecal extracts spiked with 1% Sc237-infected brain homogenate and serially diluted into control feces. PrPSc was measured by CDI using recFab HuM D18 to capture and Eu-labeled mAb 3F4 to detect SHaPrP. Total PrPSc (circles) was measured after PTA precipitation without protease treatment; rPrPSc (squares) was measured after PK digestion (50 μg/ml of PK for 60 min at 37°C) and PTA precipitation. Each feces sample was tested two to four times and the results for individual samples are expressed as the mean ± S.D. The (D–N) differences of the time-resolved fluorescence measured in counts per minute (cpm) is directly proportional to concentration of PrPSc [39, 56].
CDI measurement of PrPSc in the feces of orally infected Syrian hamsters
To calibrate the CDI and to determine the cutoff value, we spiked the feces from uninfected, control Syrian hamsters with Sc237-infected, SHa brain homogenate. The spiked feces were then diluted into control feces, then tested without PK to measure total PrPSc and with PK to measure rPrPSc by CDI (Fig. 1B). Taken together with the bioassay data (Fig. 1A), the detection limit for PrPSc and rPrPSc in feces corresponds to ≤100 ID50 units/ml and ≤30 ID50 units/ml, respectively. In contrast to PK-treated, Sc237-infected, SHa brain homogenates [39], spiked feces gave ~2-fold higher readings for rPrPSc after PK treatment. This effect may suggest the presence of proteins in feces that compete with PrPSc detection or directly interact with PrPSc at the epitope for recFab D18 (residues 134–158) or 3F4 (residues 109–112).
To measure the excretion of PrPSc from orally infected Syrian hamsters, we collected their feces daily for the first 20 days and then every 3–7 days after infection (Fig. 2A), and the levels of PrPSc were determined by the CDI. Both total PrPSc and rPrPSc level peaked at 2 days postinfection, then gradually decreased over the next 16 days. At 20 days postinfection, PrPSc and rPrPSc levels in the feces of these asymptomatic Syrian hamsters increased again and fluctuated above the cutoff value until the animals became symptomatic at 116 days postinfection (Fig. 2A).
Figure 2.

(A) Excretion of PrPSc and rPrPSc in the feces of Syrian hamsters orally infected with Sc237 prions. Each Syrian hamster consumed half a brain of an Sc237-infected Syrian hamster. Feces were collected daily from 0 to 20 days, then every 3–7 days until Syrian hamsters showed symptoms of disease at 116 days. The concentration of PrP isoforms in fecal extracts were measured by CDI using recFab HuM D18 to capture and Eu-labeled mAb 3F4 to detect SHaPrP. Total PrPSc (circles) was measured after PTA precipitation without protease treatment; rPrPSc (squares) was measured after PK digestion and PTA precipitation as in Fig. 1. The dashed lines indicate the cut-off value calculated as mean+3*SD from age-matched, control feces. (B) Levels of PrPSc in the brains of individual, symptomatic Tg7 mice inoculated intracerebrally with irradiated fecal homogenates. The feces were collected at 1, 2, 8, 22, 43, 64, 85, and 106 days postinfection from orally infected (p.o.), intraperitoneally inoculated (i.p.), or intracerebrally inoculated (i.c.) Syrian hamsters. As a control (c), age-matched, uninoculated brains were evaluated. The dashed line indicates the cut-off value calculated as mean+3*SD from age-matched, control brain samples. For both panels, each sample was tested two to four times and the results for individual samples are expressed as the mean ± S.D. The concentration of PrPSc was calculated from the (D–N) differences of the time-resolved fluorescence measured in counts per minute (cpm) [39, 56]. The concentration of rPrPSc is directly proportional to the (D–N) value and was calculated from the published formula [39].
Titration of feces of orally infected Syrian hamsters in Tg(SHaPrP) mice
Feces from both i.c. and i.p. inoculated Syrian hamsters transmitted disease to Tg7 mice with low frequency, and the mice that became ill late in the incubation period (Table 3). In contrast, feces collected from orally infected Syrian hamsters at 1 and 8 days post-infection had a titer of 6.6 log ID50 units/g, equivalent to feces spiked with 1% brain homogenate from Syrian hamsters infected with Sc237 prions (Table 3). The feces collected at later time points were also infectious: Transmission rates in Tg7 mice were up to 50%. The titers estimated from the incubation time of infected mice were ≤2.3 log ID50 units/g of feces.
Table 3.
Bioassay of feces collected at different time points from Syrian hamsters infected with Sc237 prions. Feces were irradiated with 2.9 kGy, then inoculated intracerebrally into Tg7 mice.
| Route of infection | Feces collected days postinfection | Transmission rate n/n0 | Incubation time | Titer |
|---|---|---|---|---|
| Intracerebral inoculation | 1 | 0/5 (0%) | >500 | – |
| 2 | 0/5 (0%) | >500 | – | |
| 8 | 1/6 (17%) | 221 | – | |
| 22 | 0/7 (0%) | >500 | – | |
| 43 | 1/7 (14%) | 326 | – | |
| 64 | 1/6 (17%) | 319 | – | |
| Intraperitoneal inoculation | 1 | 0/6 (0%) | >500 | – |
| 2 | 0/7 (0%) | >500 | – | |
| 8 | 0/7 (0%) | >500 | – | |
| 22 | 0/6 (0%) | >500 | – | |
| 43 | 0/4 (0%) | >500 | – | |
| 64 | 0/7 (0%) | >500 | – | |
| 85 | 1/8 (13%) | 307 | – | |
| 106 | 2/5 (40%) | 331 ± 104 | – | |
| Oral feeding | 1 | 3/6 (50%) | 287 ± 153 | ≤2.3 |
| 2 | 3/3 (100%) | 56 ± 4 | 6.6 ± 0.3 | |
| 8 | 5/6 (83%) | 56 ± 2 | 6.6 ± 0.1 | |
| 22 | 3/6 (50%) | 248 ±112 | ≤2.3 | |
| 43 | 1/8 (13%) | 314 ± 77 | ≤2.3 | |
| 64 | 2/6 (33%) | 165 ± 102 | ≤2.3 | |
| 85 | 1/5 (20%) | 264 ± 115 | ≤2.3 | |
| 106 | 2/8 (25%) | 312 ± 41 | ≤2.3 | |
n, number of ill mice; n0, number of mice inoculated. Incubation times are expressed as mean days ± standard error of the mean. Titers are expressed as mean log ID50 units/g ± standard error of the mean.
Confirmation of diagnoses
Since half of all Tg7 mice homozygous for the SHaPrP transgene array developed disease by approximately 460 days of age [40], the clinical diagnoses of Tg7 mice inoculated with feces were verified by the CDI, Western immunoblotting, standard histopathology and immunohistochemistry (Figs. 2B, 3–4). Tg7 mice were classified as disease-positive only when tested consistently for the presence of Sc237 prions by all methods (Table 3).
Figure 3.
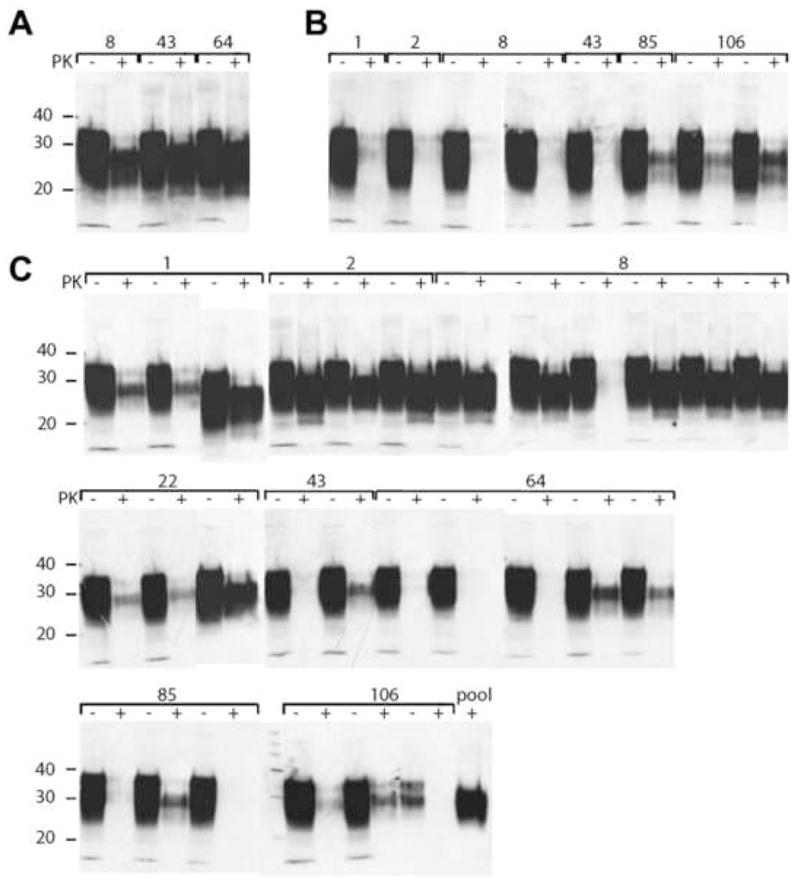
Western blot analysis of brain homogenates of Tg7 mice inoculated intracerebrally with irradiated fecal homogenates. Inocula were feces collected at 1, 2, 8, 22, 43, 64, 85, and 106 days, as indicated, from intracerebrally inoculated Syrian hamsters (A) intraperitoneally inoculated Syrian hamsters (B), and orally infected Syrian hamsters (C). PTA pellets of PK-treated brain homogenates (+) were resuspended in 150 μl of 2× sodium dodecyl sulfate (SDS) sample buffer [57]. Undigested brain homogenates (−) were diluted to 1.2% (w/v) in 1 SDS sample buffer. Equal volumes of undigested and digested samples were boiled for 5 min prior to electrophoresis. SDS gel electrophoresis and Western blotting were performed as previously described [2]. PrP was detected with the recFab HuM-P and developed with the enhanced chemiluminescent detection system (Amersham Biosciences).
Figure 4.

Neuropathology and immunohistochemistry of brain sections from Tg7 mice inoculated i.c. with irradiated 10% fecal filtrate. Inocula were feces obtained from Syrian hamsters 1 day (G–I) and 106 days (J–L) after oral feeding of Sc237 prions; and from Syrian hamsters 42 days after i.c. inoculation with Sc237 prions (M–O). As controls, Tg7 mice were inoculated with PBS (A–C) or irradiated feces from uninfected Syrian hamsters (D–F). (A–C) Hippocampal CA3 region, (G–I) hippocampal CA1 region, (J–L) ventral pontine nucleus, (M–O) thalamus. Bar in J represents 25 μm and applies to all of the PrP-stained sections by the hydrated autoclaving method (left column). Bar in L represents 50 μm and applies to all of the H&E- and GFAP-stained sections (middle and right columns, respectively).
CDI and Western immunoblotting
Clinical diagnoses were confirmed in 81% (31 of 39) of samples by both the CDI (Fig. 2B) and Western blots (Fig. 3). Despite generating similar incubation times in Tg7 mice, feces collected from Syrian hamsters inoculated i.p. resulted in ~10-fold lower PrPSc levels in the brains of infected mice, compared to feces collected from Syrian hamsters inoculated i.c. CDI data obtained from the brains of Tg7 mice inoculated with feces collected from orally challenged Syrian hamsters also separated into two groups. One group had a high concentration of rPrPSc from Tg7 mice with short incubation times; in the second group, brain samples from Tg7 mice that had longer incubation times harbored ~10-fold less rPrPSc. Whether these differences are due to age-accelerated disease at lower levels of PrPSc or to new prion strain characteristics caused by replication in the gastrointestinal lymphoreticular system remains to be established.
Neuropathology
In asymptomatic, 544-day-old control Tg7 mice inoculated i.c. with PBS, we found oval and round intracellular deposits of PrPC that vary from 2–8 μm (Fig. 4A). Over 95% of the deposits were in the gray matter neuropil away from nerve cell bodies; some were adjacent to or within nerve cell bodies, and sparse deposits were found in white matter. PrP deposits did not bind Thioflavin S and therefore were not a form of amyloid. Neither vacuolation nor nerve cell loss was detected by H&E staining (Fig. 4B), and no reactive astrocytic gliosis was identified by GFAP IHC (Fig. 4C). In age-matched mice inoculated i.c. with irradiated feces from an uninfected hamster, we did not observe any residual material or changes that could be linked to the injection of fecal material (Fig. 4D, 4F). We conclude that the PrP deposits in aged Tg7 mice did not have the tinctorial characteristics of PrPSc and were induced by overexpression of PrPC.
In contrast, we found widespread, moderate to severe neuropathologic abnormalities in a symptomatic Tg7 mouse inoculated i.c. with irradiated fecal filtrate collected from Syrian hamsters 1 day after oral challenge. In all gray matter regions, irregularly sized deposits of SHaPrPSc were easily distinguished from the background of SHaPrPC deposits by their slightly blurred edges (Fig. 4G). We found Thioflavin S–positive amyloid plaques located between the hippocampus and the corpus callosum. Small numbers of gray matter vacuoles (Fig. 4H) and hyperintense reactive astrocytic gliosis (Fig. 4I) were observed. Similar neuropathologic changes were seen in Tg7 mice inoculated i.c. with fecal material collected from Syrian hamsters 105 days after oral challenge; PrPSc deposits were primarily in the base of the pons (Fig. 4J) in association with vacuolar degeneration of gray matter (Fig. 4K) and severe reactive astrocytic gliosis (Fig. 4L). The most intense neuropathologic changes were found in the pontine base and cerebellar cortex, and little or none in other brain regions.
Fecal material from Syrian hamsters collected 42 days after i.c. inoculation and injected i.c. into Tg7 mice resulted in disease after 362 days. Brain sections showed moderately severe neuropathologic changes in most regions, and mild changes in the neocortex and hypothalamus. While SHaPrPSc deposits were punctate and relatively difficult to distinguish from SHaPrPC aggregates, larger plaque-like masses verified the depositions as SHaPrPSc (Fig. 4M). Showing moderate fluorescence with Thioflavin S, these masses likely represent primitive PrP amyloid plaques (data not shown). Gray matter vacuoles were detected only in the thalamus. Moderate reactive astrocytic gliosis was detected in most regions, with the exceptions noted above (Fig. 4O).
DISCUSSION
Studies of the mechanism of natural prion transmission and spread are important to understand how prion reservoirs form in nature. In most instances, prions are incapable of infecting another host by contact or cohabitation; family members in daily contact with CJD patients or sheep farmers and veterinarians in contact with scrapie appear to be immune to prion infection [41, 42]. How much of this protection is due to the specific biological characteristics of the prion strain, to the species barrier, or to the absence of a transmission mechanism is unknown. In contrast, CWD in cervids and scrapie in sheep have high incidence in endemic areas [11, 12, 22]. The source of environmental contamination, and whether these natural reservoirs are maintained by a high transmission rate from animal to animal or by contact with the contaminated environment, are subjects of debate [10–12, 22]. Also unclear is whether these prions can infect another species, as seen with BSE prions in humans, and form a new natural reservoir. Thus, studies of the natural transmission of prions have enormous implications for public health.
The data presented here clearly demonstrate that laboratory prions are transmissible by cohabitation with orally infected Syrian hamsters and that the source of infection is prion-infected feces. High PrPSc levels and prion titers were detected within 7 days after feeding Syrian hamsters prion-infected brain, indicating that prions pass intact through the digestive tract. The lower transmission rates and long incubation times in Tg7 mice inoculated with feces obtained later (>10 days postinfection) suggest that the gastrointestinal lymphatic system—tonsils, Peyer patches, or mesenteric lymph nodes—of the Syrian hamsters were infected [43, 44]. Recent findings suggest an association between the development of Peyer’s patches and susceptibility to prion infection in sheep, cattle, and humans [45]. Indeed, immunostaining for PrPSc in mesenteric lymph nodes, mucosal lymphoid follicles and Peyer’s patches were observed from the earliest time point available at 69 days postinoculation in Syrian hamsters orally infected with Sc237 prions [43]. Taken together with the results of our study in Syrian hamsters and with observations in both natural CWD and scrapie [11, 24, 29, 30], alimentary shedding of prion-infected cells or cell fragments into feces seems likely.
That Tg7 mice inoculated i.c. with irradiated, prion-free feces showed no acute toxic symptoms was surprising (Fig. 4F). In contrast, Tg7 mice inoculated i.c. with irradiated fecal filtrate collected from Syrian hamsters infected with Sc237 prions demonstrated all the classic features of prion infection, but the distribution and intensities of neuropathologic changes varied greatly. Unexpectedly, we found neuropathological changes in the pontine base and cerebellum, and little or none in other brain regions (Fig. 4J–L). Whether this change in the anatomical targeting is due to the presence of feces in the inoculum or reflects a permanent shift in the Sc237 strain characteristics due to passage through the intestinal lymphoreticular system remains to be established.
The horizontal spread of CWD and scrapie, and transmission by contaminated environments, implicate saliva, feces, or urine as vectors of infectivity. In a recent study, saliva from CWD-positive deer transmitted prions to other deer within 12 months following oral challenge. When two white-tailed deer were orally challenged with feces and urine obtained from CWD-infected deer, no transmissions were observed [23]. The two deer expressed the G/S polymorphism at PrP codon 96, which has been shown to confer longer incubation times (>18 months) for even high-titer, CWD inocula prepared from brain homogenate [46]. Given the small number of animals, expected low titers, and 12 months of observation in that report [23], conclusions about CWD infectivity in feces must be tempered.
While our studies provide evidence of transmission through feces, reports of prion infectivity in urine have been contradictory. Two studies reported the detection of PrP and low levels of prions in urine [47, 48]. However, we and others demonstrated that the reported protease-resistant PrP band detected in Western blots of the urine of diseased hamsters is due to the cross-reactivity of the anti-mouse IgG antibody with IgG light chains and possibly heavy chain fragments; therefore, this band is not likely to be PrP [49–52]. Moreover, urine was collected in metabolic cages and no attempt was made to separate feces until the clarification spin [48]. Perhaps the considerable levels of prions in feces, as our studies demonstrate, inadvertently contaminated the urine.
Considering the long-term survival of prions in soil [53–55], the data presented here have raised imperative questions about the safe disposal of excrement from BSE-infected cattle and from animals with CWD and scrapie, especially during the early, asymptomatic stage of disease. The disposal methods of vCJD and sCJD excrement in a hospital or home setting also should be reexamined. Should the presence of prions in feces be verified by bioassays or CDI, safe disposal protocols will have to be implemented. In natural reservoirs of scrapie and CWD, treatment of the feces before disposal should help to minimize long-term environmental contamination with prions.
Supplementary Material
Acknowledgments
The authors thank the staff of the Hunters Point Animal Facility for their expert Syrian hamster and mouse studies. This work was supported by grants from the National Institutes of Health (AG02132, AG010770, NS22786, and NS14069) as well as by gifts from the G. Harold and Leila Y. Mathers Foundation and Sherman Fairchild Foundation. J.G.S., S.J.D., and S.B.P. have financial interest in InPro Biotechnology, Inc.
References
- 1.Prusiner SB. Prion Biology and Diseases. 2. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2004. p. 1050. [Google Scholar]
- 2.Scott MR, Will R, Ironside J, et al. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci USA. 1999;96:15137–42. doi: 10.1073/pnas.96.26.15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wells GAH, Wilesmith JW. Bovine spongiform encephalopathy and related diseases. In: Prusiner SB, editor. Prion Biology and Diseases. 2. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2004. pp. 595–628. [Google Scholar]
- 4.Ward HJ, Everington D, Cousens SN, et al. Risk factors for variant Creutzfeldt-Jakob disease: a case-control study. Ann Neurol. 2006;59:111–20. doi: 10.1002/ana.20708. [DOI] [PubMed] [Google Scholar]
- 5.Health Protection Agency. New case of variant CJD associated with blood transfusion. 2006 http://www.hpa.org.uk/hpa/news/articles/press_releases/2006/060209_cjd.htm.
- 6.Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet. 2004;364:527–9. doi: 10.1016/S0140-6736(04)16811-6. [DOI] [PubMed] [Google Scholar]
- 7.Llewelyn CA, Hewitt PE, Knight RS, et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417–21. doi: 10.1016/S0140-6736(04)15486-X. [DOI] [PubMed] [Google Scholar]
- 8.Wroe SJ, Pal S, Siddique D, et al. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet. 2006;368:2061–7. doi: 10.1016/S0140-6736(06)69835-8. [DOI] [PubMed] [Google Scholar]
- 9.Gajdusek DC. Unconventional viruses and the origin and disappearance of kuru. Science. 1977;197:943–60. doi: 10.1126/science.142303. [DOI] [PubMed] [Google Scholar]
- 10.Miller MW, Williams ES, Hobbs NT, Wolfe LL. Environmental sources of prion transmission in mule deer. Emerg Infect Dis. 2004;10:1003–6. doi: 10.3201/eid1006.040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunter N. Scrapie and experimental BSE in sheep. Br Med Bull. 2003;66:171–83. doi: 10.1093/bmb/66.1.171. [DOI] [PubMed] [Google Scholar]
- 12.Matthews L, Coen PG, Foster JD, Hunter N, Woolhouse ME. Population dynamics of a scrapie outbreak. Arch Virol. 2001;146:1173–86. doi: 10.1007/s007050170113. [DOI] [PubMed] [Google Scholar]
- 13.Williams ES. Chronic wasting disease. Vet Pathol. 2005;42:530–49. doi: 10.1354/vp.42-5-530. [DOI] [PubMed] [Google Scholar]
- 14.Dickinson AG, Stamp JT, Renwick CC. Maternal and lateral transmission of scrapie in sheep. J Comp Pathol. 1974;84:19–25. doi: 10.1016/0021-9975(74)90023-1. [DOI] [PubMed] [Google Scholar]
- 15.Ridley RM, Baker HF. The myth of maternal transmission of spongiform encephalopathy. Br Med J. 1995;311:1071–5. doi: 10.1136/bmj.311.7012.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hadlow WJ, Jackson TA, Race RE. Experimental infection of fetal and newborn Suffolk sheep with scrapie virus. Am J Vet Res. 1984;45:2637–9. [PubMed] [Google Scholar]
- 17.Foster J, McKelvey W, Fraser H, et al. Experimentally induced bovine spongiform encephalopathy did not transmit via goat embryos. J Gen Virol. 1999;80:517–24. doi: 10.1099/0022-1317-80-2-517. [DOI] [PubMed] [Google Scholar]
- 18.Sigurdsson B. Rida, a chronic encephalitis of sheep with general remarks on infections which develop slowly and some of their special characteristics. Br Vet J. 1954;110:341–54. [Google Scholar]
- 19.Foster J, McKenzie C, Parnham D, et al. Derivation of a scrapie-free sheep flock from the progeny of a flock affected by scrapie. Vet Rec. 2006;159:42–5. doi: 10.1136/vr.159.2.42. [DOI] [PubMed] [Google Scholar]
- 20.Williams ES, Young S. Spongiform encephalopathies in Cervidae. Rev Sci Tech. 1992;11:551–67. doi: 10.20506/rst.11.2.611. [DOI] [PubMed] [Google Scholar]
- 21.Miller MW, Wild MA, Williams ES. Epidemiology of chronic wasting disease in captive Rocky Mountain elk. J Wildl Dis. 1998;34:532–8. doi: 10.7589/0090-3558-34.3.532. [DOI] [PubMed] [Google Scholar]
- 22.Miller MW, Williams ES. Prion disease: horizontal prion transmission in mule deer. Nature. 2003;425:35–6. doi: 10.1038/425035a. [DOI] [PubMed] [Google Scholar]
- 23.Mathiason CK, Powers JG, Dahmes SJ, et al. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science. 2006;314:133–6. doi: 10.1126/science.1132661. [DOI] [PubMed] [Google Scholar]
- 24.Race R, Jenny A, Sutton D. Scrapie infectivity and proteinase K-resistant prion protein in sheep placenta, brain, spleen, and lymph node: Implications for transmission and antemortem diagnosis. J Infect Dis. 1998;178:949–53. doi: 10.1086/515669. [DOI] [PubMed] [Google Scholar]
- 25.Carp RI. Transmission of scrapie by oral route: effect of gingival scarification. Lancet. 1982;319:170–1. doi: 10.1016/s0140-6736(82)90421-4. [DOI] [PubMed] [Google Scholar]
- 26.Czub M, Braig HR, Diringer H. Pathogenesis of scrapie: study of the temporal development of clinical symptoms of infectivity titres and scrapie-associated fibrils in brains of hamsters infected intraperitoneally. J Gen Virol. 1986;67:2005–9. doi: 10.1099/0022-1317-67-9-2005. [DOI] [PubMed] [Google Scholar]
- 27.Pattison IH, Hoare MN, Jebbett JN, Watson WA. Spread of scrapie to sheep and goats by oral dosing with foetal membranes from scrapie-affected sheep. Vet Rec. 1972;90:465–8. doi: 10.1136/vr.90.17.465. [DOI] [PubMed] [Google Scholar]
- 28.Heggebo R, Press CM, Gunnes G, et al. Distribution of prion protein in the ileal Peyer’s patch of scrapie-free lambs and lambs naturally and experimentally exposed to the scrapie agent. J Gen Virol. 2000;81:2327–37. doi: 10.1099/0022-1317-81-9-2327. [DOI] [PubMed] [Google Scholar]
- 29.Gonzalez L, Martin S, Begara-McGorum I, et al. Effects of agent strain and host genotype on PrP accumulation in the brain of sheep naturally and experimentally affected with scrapie. J Comp Pathol. 2002;126:17–29. doi: 10.1053/jcpa.2001.0516. [DOI] [PubMed] [Google Scholar]
- 30.Miller MW, Williams ES. Detection of PrP(CWD) in mule deer by immunohistochemistry of lymphoid tissues. Vet Rec. 2002;151:610–2. doi: 10.1136/vr.151.20.610. [DOI] [PubMed] [Google Scholar]
- 31.Prusiner SB, Cochran SP, Alpers MP. Transmission of scrapie in hamsters. J Infect Dis. 1985;152:971–8. doi: 10.1093/infdis/152.5.971. [DOI] [PubMed] [Google Scholar]
- 32.Sakaguchi E. Digestive strategies of small hindgut fermenters. Anim Sci J. 2003;74:327–37. [Google Scholar]
- 33.Scott M, Foster D, Mirenda C, et al. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell. 1989;59:847–57. doi: 10.1016/0092-8674(89)90608-9. [DOI] [PubMed] [Google Scholar]
- 34.Peretz D, Supattapone S, Giles K, et al. Inactivation of prions by acidic sodium dodecyl sulfate. J Virol. 2006;80:322–31. doi: 10.1128/JVI.80.1.322-331.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prusiner SB, Cochran SP, Groth DF, Downey DE, Bowman KA, Martinez HM. Measurement of the scrapie agent using an incubation time interval assay. Ann Neurol. 1982;11:353–8. doi: 10.1002/ana.410110406. [DOI] [PubMed] [Google Scholar]
- 36.Muramoto T, DeArmond SJ, Scott M, Telling GC, Cohen FE, Prusiner SB. Heritable disorder resembling neuronal storage disease in mice expressing prion protein with deletion of an a-helix. Nat Med. 1997;3:750–5. doi: 10.1038/nm0797-750. [DOI] [PubMed] [Google Scholar]
- 37.Bellinger-Kawahara CG, Kempner E, Groth DF, Gabizon R, Prusiner SB. Scrapie prion liposomes and rods exhibit target sizes of 55,000 Da. Virology. 1988;164:537–41. doi: 10.1016/0042-6822(88)90569-7. [DOI] [PubMed] [Google Scholar]
- 38.Miekka SI, Forng RY, Rohwer RG, et al. Inactivation of viral and prion pathogens by gamma-irradiation under conditions that maintain the integrity of human albumin. Vox Sang. 2003;84:36–44. doi: 10.1046/j.1423-0410.2003.00256.x. [DOI] [PubMed] [Google Scholar]
- 39.Safar J, Wille H, Itri V, et al. Eight prion strains have PrPSc molecules with different conformations. Nat Med. 1998;4:1157–65. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 40.Westaway D, DeArmond SJ, Cayetano-Canlas J, et al. Degeneration of skeletal muscle, peripheral nerves, and the central nervous system in transgenic mice overexpressing wild-type prion proteins. Cell. 1994;76:117–29. doi: 10.1016/0092-8674(94)90177-5. [DOI] [PubMed] [Google Scholar]
- 41.Chatelain J, Cathala F, Brown P, Raharison S, Court L, Gajdusek DC. Epidemiologic comparisons between Creutzfeldt-Jakob disease and scrapie in France during the 12-year period 1968–1979. J Neurol Sci. 1981;51:329–37. doi: 10.1016/0022-510x(81)90111-8. [DOI] [PubMed] [Google Scholar]
- 42.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–39. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- 43.Beekes M, McBride PA. Early accumulation of pathological PrP in the enteric nervous system and gut-associated lymphoid tissue of hamsters orally infected with scrapie. Neurosci Lett. 2000;278:181–4. doi: 10.1016/s0304-3940(99)00934-9. [DOI] [PubMed] [Google Scholar]
- 44.Bergstrom AL, Jensen TK, Heegaard PM, et al. Short-term study of the uptake of PrP(Sc) by the Peyer’s patches in hamsters after oral exposure to scrapie. J Comp Pathol. 2006;134:126–33. doi: 10.1016/j.jcpa.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 45.St Rose SG, Hunter N, Matthews L, et al. Comparative evidence for a link between Peyer’s patch development and susceptibility to transmissible spongiform encephalopathies. BMC Infect Dis. 2006;6:5. doi: 10.1186/1471-2334-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Rourke KI, Spraker TR, Hamburg LK, Besser TE, Brayton KA, Knowles DP. Polymorphisms in the prion precursor functional gene but not the pseudogene are associated with susceptibility to chronic wasting disease in white-tailed deer. J Gen Virol. 2004;85:1339–46. doi: 10.1099/vir.0.79785-0. [DOI] [PubMed] [Google Scholar]
- 47.Shaked GM, Shaked Y, Kariv-Inbal Z, Halimi M, Avraham I, Gabizon R. A protease-resistant prion protein isoform is present in urine of animals and humans affected with prion diseases. J Biol Chem. 2001;276:31479–82. doi: 10.1074/jbc.C100278200. [DOI] [PubMed] [Google Scholar]
- 48.Kariv-Inbal Z, Ben-Hur T, Grigoriadis NC, Engelstein R, Gabizon R. Urine from scrapie-infected hamsters comprises low levels of prion infectivity. Neurodegener Dis. 2006;3:123–8. doi: 10.1159/000094770. [DOI] [PubMed] [Google Scholar]
- 49.Serban A, Legname G, Hansen K, Kovaleva N, Prusiner SB. Immunoglobulins in urine of hamsters with scrapie. J Biol Chem. 2004;279:48817–20. doi: 10.1074/jbc.M409107200. [DOI] [PubMed] [Google Scholar]
- 50.Miyazawa K, Shiga Y, Matsuzaki M, Takeda A, Itoyama Y. The detection of protease-resistant prion protein isoform in urine of Creutzfeldt-Jakob disease patients. Ann Neurol. 2002;52:S54. [Google Scholar]
- 51.Kariv-Inbal Z, Halimi M, Dayan Y, Engelstein R, Gabizon R. Characterization of light chain immunoglobulin in urine from animals and humans infected with prion diseases. J Neuroimmunol. 2005;162:12–8. doi: 10.1016/j.jneuroim.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 52.Head MW, Kouverianou E, Taylor L, Green A, Knight R. Evaluation of urinary PrPSc as a diagnostic test for sporadic, variant, and familial CJD. Neurology. 2005;64:1794–6. doi: 10.1212/01.WNL.0000161842.68793.8A. [DOI] [PubMed] [Google Scholar]
- 53.Brown P, Gajdusek DC. Survival of scrapie virus after 3 years’ interment. Lancet. 1991;337:269–70. doi: 10.1016/0140-6736(91)90873-n. [DOI] [PubMed] [Google Scholar]
- 54.Johnson CJ, Pedersen JA, Chappell RJ, McKenzie D, Aiken JM. Oral transmissibility of prion disease is enhanced by binding to soil particles. PLoS Pathog. 2007;3:e93. doi: 10.1371/journal.ppat.0030093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seidel B, Thomzig A, Buschmann A, et al. Scrapie Agent (Strain 263K) can transmit disease via the oral route after persistence in soil over years. PLoS ONE. 2007;2:e435. doi: 10.1371/journal.pone.0000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Safar JG, Scott M, Monaghan J, et al. Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat Biotechnol. 2002;20:1147–50. doi: 10.1038/nbt748. [DOI] [PubMed] [Google Scholar]
- 57.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T-4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.