

Abstract
The majority of known proteins are too large to be comprehensively examined by solution NMR methods, primarily because they tumble too slowly in solution. Here we introduce an approach to making the NMR relaxation properties of large proteins amenable to modern solution NMR techniques. The encapsulation of a protein in a reverse micelle dissolved in a low-viscosity fluid allows it to tumble as fast as a much smaller protein. The approach is demonstrated and validated with the protein ubiquitin encapsulated in reverse micelles prepared in a variety of alkane solvents.
Nuclear magnetic resonance (NMR) spectroscopy plays a central role in the characterization of the structure and dynamics of proteins, nucleic acids, and their complexes. Over the past 15 years developments in NMR techniques (1–3), especially triple resonance spectroscopy (4), and supporting technologies have made the comprehensive structural characterization of 20-kDa proteins routine. NMR spectroscopy can provide a detailed view of the solution structure of macromolecules such as proteins and allows unparalleled access to dynamic phenomena. NMR also provides a unique avenue to monitor the full structural and dynamic effects of changes in temperature, pressure, and other solution conditions, including the binding of small and large ligands. With the development of multinuclear and multidimensional NMR, the structures of proteins of significant size and spectral complexity now can be efficiently determined. However, increasing molecular size imposes several important limitations. Increasing molecular size leads to slower tumbling and correspondingly shorter spin-spin relaxation times (T2) and also leads to increasingly complex spectra. Short T2 values severely limit the power and flexibility of multiple-pulse NMR experiments in at least two ways. The signal-to-noise ratio of a Lorentzian line degrades with decreasing T2, and the effectiveness of the currently available library of multidimensional and multinuclear NMR experiments is exponentially sensitive to T2. Accordingly, the standard triple resonance experiments become unreliable at room temperature for proteins larger than 30 kDa and largely fail for proteins above 35 kDa in the absence of elevated temperature and/or extensive deuteration.
A variety of approaches have been developed to combat the limitations caused by T2. One uses extensive deuteration to reduce the dipolar field so that deuterium-decoupled triple resonance experiments are feasible (5–7). Though deuteration limits the structural information available from the nuclear Overhauser effect, selective incorporation of 1H spins holds the promise of rapidly obtaining protein topologies by solution NMR methods (8, 9). Other advances include the selection of the narrow multiplet component arising from the mutual cancellation of dipole-dipole coupling and chemical shift anisotropy in 1H-15N correlation experiments (10). The recent development of the use of residual dipolar splittings in partially aligned proteins (11) and cross-correlation effects in high-resolution triple resonance spectra (12) now provides access to an array of structural restraints that can promote the study of large biopolymers. Nevertheless, the difficulty of dealing comprehensively with large proteins significantly limits the application of solution NMR methods to the rapidly growing list of proteins being discovered by the molecular biology community. We have explored an approach that can directly increase T2 by reducing the tumbling correlation time (τm) of the protein.
MATERIALS AND METHODS
Recombinant 15N-labeled human ubiquitin was prepared as described (13). Protein in reverse micelles was prepared by first creating a solution of sodium bis(2-ethylhexyl)sulfosuccinate (AOT) reverse micelles in the desired solvent, under pressure, if necessary. This solution was transferred to the NMR cell containing lyophilized protein solvated with the volume of aqueous buffer required to give a final molar ratio of AOT to water of ≈10. The aqueous buffer was 50 mM sodium acetate, pH 5, containing 250 mM NaCl. Effective concentrations of protein in the various samples were between 0.2 and 0.4 mM.
Assignment of the main chain resonances of ubiquitin encapsulated in AOT reverse micelles in pentane was achieved by using HNCACB (14) and HNCA (15) triple resonance spectra supplemented with 15N-resolved total correlation (16) and nuclear Overhauser effect (17) spectra. Nitrogen spin-spin relaxation times were determined by using a standard inverse detected two-dimensional sampling method that used 15N 180° pulses spaced by 900 μs and 1H 180° pulses centered in the CPMG block to remove effects from dipolar/chemical shift anisotropy cross-correlation (18). Recycle delays were typically ≈1.1 s. Sensitivity-enhanced gradient selection of 15N coherence was used (19). Samples prepared in n-butane, n-pentane, n-hexane, and iso-octane used commercially available NMR tubes, while those prepared in propane used a custom HPLC NMR tube constructed from zirconium oxide. All NMR data were collected at 25°C on a Varian Inova spectrometer operating at 750 MHz (1H) with an 8-mm triple resonance z gradient Nalorac probe. Spectra were processed and analyzed by using the program felix (Molecular Simulations, Waltham, MA).
RESULTS AND DISCUSSION
The basic idea is to arrange for the protein to tumble more rapidly. The global tumbling correlation time of an isotropically reorienting (spherical) molecule relates to the bulk solvent viscosity through the Stokes-Einstein relation:
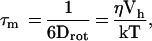 |
where Drot is the rotational diffusion constant for a sphere, η is the viscosity, Vh is the volume of the sphere, k is the Boltzman constant, and T is the absolute temperature. The correlation time for tumbling linearly relates τm to the viscosity of the solvent. If internal motion is restricted, T2 approximately varies inversely with τm. A spherical protein with molecular mass of 50 kDa and typical characteristics (partial specific volume ≈0.75 ml/g, f/fo ≈ 1.2) has a radius of ≈26 Å and tumbles in water (η ≈ 850 μPa⋅s at 300 K) with a correlation time of ≈15 ns. A much smaller 10-kDa protein tumbles with a correlation time of 3–4 ns.
Short chain alkanes and other solvents whose intermolecular interactions are restricted to London forces show viscosities in the range necessary. For example, ethane, propane, and butane are gases at standard temperature and pressure but are liquefied by modest pressures at room temperature. To “dissolve” a protein in a hydrophobic solvent such as liquid propane we use a technology that was extensively investigated in the 1980s involving encapsulation of molecules such as proteins within the water cavity formed by reverse micelles. Reverse micelles are thermodynamically stable assemblies of surfactant molecules organized around a water core and will form spontaneously as transparent solutions in a low polarity liquid. Reverse micelles have been considered for a range of applications, including molecular separations and reaction processes (20–24). Recently, perfluoropolyether microemulsions have been studied as potential hosts for various chemical reactions in solvents with low environmental impact such as supercritical carbon dioxide (25).
AOT is commonly used as a reverse micelle surfactant and has been extensively studied in a variety of organic solvents. Stable reverse micelles of AOT large enough to accommodate aqueous solutions of proteins of significant size have been demonstrated to form in a variety of liquid alkanes (26–29). Dozens of proteins, many exceeding 50 kDa in size, have been successfully solubilized by reverse micelles in organic solvents (20–25).
The radius of a reverse micelle is determined by the width of the surfactant shell (≈15 Å for AOT) and the volume of the water pool and protein enclosed within. Careful choice of absolute surfactant concentration and relative water content provides for a micelle radius corresponding to the simple sum of the hydrated protein’s effective radius and the chain length of the surfactant (8). For example, encapsulation of a spherical 50-kDa protein will result in a minimal reverse micelle with radius ≈41 Å. Using bulk viscosities (30, 31), the corresponding reorientation correlation times would be ≈60 ns in water, ≈11 ns in butane, ≈7 ns in propane, and ≈2.5 ns in ethane. The resulting increase in T2 would make available high-resolution solution NMR techniques directly applicable, the larger the protein the more pronounced the advantage. For example, encapsulation of a 100-kDa protein (spherical hydrated radius of 33 Å) would have a corresponding reverse micelle radius of ≈48 Å and would have a molecular tumbling correlation time of ≈95 ns in water, ≈18 ns in butane, ≈11 ns in propane, and ≈4 ns in ethane. These values compare favorably with correlation times of 15 ns and 31 ns for the 50- and 100-kDa proteins dissolved directly in water, respectively. Conversely, a small ≈9-kDa protein, such as ubiquitin, would have a molecular tumbling correlation time of ≈3 ns in water and the corresponding reverse micelle would tumble with an equivalent correlation time in propane.
To test the approach, we prepared AOT reverse micelles (70 mM) in a variety of alkane solvents. Recombinant isotopically enriched human ubiquitin was lyophilized, hydrated by a defined volume of aqueous 50 mM acetate buffer (pH 5), and then transferred to the organic phase by a passive phase transfer method. The final concentration of protein in solution was ≈0.3 mM, which appears to be the limit of solubility. The effective water loading corresponded to a molar ratio of surfactant to water of ≈10. Pulsed field gradient diffusion studies of the hydrodynamic behavior of these preparations in liquid pentane, for example, indicate that each reverse micelle has the radius expected for the encapsulation of a single minimally hydrated protein per reverse micelle.
Fig. 1 shows the 15N heteronuclear single quantum correlation spectrum of ubiquitin encapsulated in AOT reverse micelles dissolved in liquid propane. No unusual spectral artifacts are introduced by the use of the high-pressure NMR cell. The spectrum is typical of a unique folded structure and is closely similar to that obtained for ubiquitin in aqueous buffer. Standard triple resonance experiments were used to assign the main chain resonances of 13C,15N-ubiquitin solubilized by reverse micelles in pentane. Comparison of the chemical shifts of amide 1H and 15N in the AOT reverse micelle and those in water reveals minimal differences (Fig. 2). Including all data, the SDs between amide 1H and 15N chemical shifts of ubiquitin in the two states are 0.17 and 1.6 ppm, respectively. Two localized regions display perturbations. These include the C-terminal residues R72 and R74 and residues 45–48 that form a tight turn. The 15N-resolved nuclear Overhauser effect spectrum of ubiquitin in AOT/pentane shows the same pattern of nuclear Overhauser effects among main chain hydrogens as observed when the protein is dissolved directly in water (32). These observations confirm earlier optical and fluorescence studies indicating that proteins can be encapsulated in a reverse micelle environment without significant distortion of their native structure.
Figure 1.

15N- heteronuclear single quantum correlation spectrum of 15N-labeled recombinant human ubiquitin encapsulated in AOT reverse micelles dissolved in liquid propane. The protein concentration was 0.3 mM. The spectrum was acquired at 750 MHz (1H) by using an 8-mm probe in ≈30 min at 25°C and 12 bar. One hundred and fifty complex points were collected in the incremented nitrogen time domain.
Figure 2.

Correlation of amide 1H and 15N chemical shifts of recombinant human ubiquitin in aqueous buffer with those of the protein encapsulated in a reverse micelle dissolved in pentane. In both cases the protein was solvated by aqueous buffer containing 50 mM sodium acetate, pH 5, and 250 mM NaCl. 15N- heteronuclear single quantum correlation spectra were obtained at 25°C at 750 MHz (1H). (Upper) The comparison of amide 1H chemical shifts. (Lower) The comparison of 15N chemical shifts.
To study line narrowing we encapsulated 15N-ubiquitin in AOT reverse micelles prepared in iso-octane, n-heptane, n-hexane, n-pentane, n-butane, and propane. For butane and propane, NMR spectroscopy was most safely achieved by using a custom HPLC NMR cell based on a design to be described in detail elsewhere. Amide nitrogen spin-spin relaxation rates (1/T2) were measured by using standard pulse sequences (18). The main chain dynamics of ubiquitin in water have been characterized previously by NMR methods (33) and allow identification regions of the protein where T2 is largely dominated by the influence of global tumbling. The expected roughly inverse dependence of T2 on solvent viscosity is clearly revealed (Fig. 3). As predicted above, the spin-spin nitrogen relaxation times of ubiquitin in water are largely recovered when ubiquitin encapsulated in AOT reverse micelles is dissolved in liquid propane. In this range of viscosity, the reverse micelles remain in the slow tumbling limit (ωτm > 1). Extrapolation from this roughly linear region to zero viscosity ideally should yield a limiting (1/T2) value of ≈ 2 sec−1, about half of that obtained. This deviation presumably reflects differences between the effective microviscosity experienced by the reverse micelle and the viscosity of the bulk solvent as well as changes in the volume of the reverse micelle with solvent (27–29).
Figure 3.

Reduction of amide 15N spin-spin relaxation rate in recombinant human ubiquitin encapsulated in a reverse micelle. Spin-spin relaxation rates (1/T2) are plotted against the bulk solvent viscosity (30, 31). Data points, in order of decreasing viscosity, are for iso-octane, n-heptane, n-hexane, n-pentane, n-butane, and propane. All data were collected at 25°C. The butane and propane samples were prepared at pressures of 7 and 12 bar, respectively. All other samples were prepared at ambient pressure.
These results indicate that proteins can be encapsulated in reverse micelles dissolved in low-viscosity fluids without significant structural distortion. The preparations are reasonably stable (>1 week) and give spectroscopic results consistent with the anticipated performance. We have demonstrated the approach by using liquid propane. Even greater benefits are anticipated by using critical ethane, though significantly higher pressures will be required, perhaps up to 200 bar (34). Nevertheless, given the long history of encapsulating proteins of significant size in reverse micelle systems (20–24), this approach holds the promise of extending NMR-based structural analysis to soluble proteins of 50 kDa and beyond. The approach is also applicable to other biopolymers such as nucleic acids, carbohydrates, and, perhaps most important, membrane proteins. In addition, this approach may bring small molecule NMR methods into the arena of proteins of moderate size. Finally, it can be noted that the use of low-viscosity fluids with low freezing point temperatures could extend the study of temperature-dependent phenomena of proteins to very low temperatures.
Acknowledgments
The majority of this work was undertaken at the State University of New York at Buffalo. We are grateful to Gary Sagerman for construction of the HPLC optical and NMR cells used in this work. We thank Robert Gennis, Michael Glaser, and Eric Oldfield for helpful discussion during early phases of this work. We are grateful to Frank Bright for the loan of a HPLC fluorescence cell and for helpful discussion. We are especially indebted to Ramona Bieber Urbauer for preparation of the significant quantities of isotopically labeled ubiquitin required. This work was supported by National Institutes of Health Grant GM35940, Army Research Office Grant DAAH04-96-1-0312, and a gift from Sandra and Jacob Terner.
ABBREVIATION
- AOT
sodium bis(2-ethylhexyl)sulfosuccinate
References
- 1.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 2.Cavanagh J, Fairbrother W J, Palmer A G, Skelton N J. Protein NMR Spectroscopy. San Diego: Academic; 1996. [Google Scholar]
- 3.Wagner G. Nat Struct Biol. 1997;4:841–844. [PubMed] [Google Scholar]
- 4.Kay L E, Ikura M, Tschudin R, Bax A. J Magn Reson. 1990;89:496–505. doi: 10.1016/j.jmr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Grzesiek S, Anglister J, Ren H, Bax A. J Am Chem Soc. 1993;115:4369–4370. [Google Scholar]
- 6.Yamazaki T, Lee W, Revington M, Mattiello D L, Dahlquist F W, Arrowsmith C H, Kay L E. J Am Chem Soc. 1994;116:6464–6465. [Google Scholar]
- 7.Farmer II B T, Venters R A, Metzler W J, Farmer B T, Spicer L D, Mueller L. J Am Chem Soc. 1995;117:9592–9593. [Google Scholar]
- 8.Gardner K H, Rosen M, Kay L E. Biochemistry. 1997;36:1389–1401. doi: 10.1021/bi9624806. [DOI] [PubMed] [Google Scholar]
- 9.Zwahlen C, Vincent S J F, Gardner K H, Kay L E. J Am Chem Soc. 1998;120:4825–4831. [Google Scholar]
- 10.Pervushin K, Riek R, Wider G, Wüthrich K. Proc Natl Acad Sci USA. 1997;94:12366–12370. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tjandra N, Bax A. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- 12.Reif B, Hennig M, Griesinger C. Science. 1997;276:1230–1233. doi: 10.1126/science.276.5316.1230. [DOI] [PubMed] [Google Scholar]
- 13.Wand A J, Urbauer J L, McEvoy R P, Bieber R J. Biochemistry. 1996;35:6116–6125. doi: 10.1021/bi9530144. [DOI] [PubMed] [Google Scholar]
- 14.Wittekind M, Mueller L. J Magn Reson. 1993;101:201–205. [Google Scholar]
- 15.Grzesiek S, Bax A. J Magn Reson. 1992;96:432–440. [Google Scholar]
- 16.Zhang O, Kay L E, Olivier J P, Forman-Kay J D. J Biomol NMR. 1994;4:845–858. doi: 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
- 17.Marion D, Kay L E, Sparks S W, Torchia D A, Bax A. J Am Chem Soc. 1989;111:1515–1517. [Google Scholar]
- 18.Palmer A G, Skelton N J, Chazin W J, Wright P E, Rance M. Mol Phys. 1992;75:699–711. [Google Scholar]
- 19.Farrow N A, Muhandiram R, Singer A U, Pascal S M, Kay C M, Gish G, Shoelson S E, Pawson T, Forman-Kay J D, Kay L E. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 20.Luisi P L. Angew Chem Intl Ed. 1985;24:439–528. [Google Scholar]
- 21.Goklen K E, Hatton T A. Biotechnol Prog. 1985;1:69–74. doi: 10.1002/btpr.5420010113. [DOI] [PubMed] [Google Scholar]
- 22.Luisi P L, Giomini M, Pileni M P, Robinson B H. Biochem Biophys Acta. 1988;947:209–246. doi: 10.1016/0304-4157(88)90025-1. [DOI] [PubMed] [Google Scholar]
- 23.De T K, Maitra A. Adv Colloid Interface Sci. 1995;59:95–193. [Google Scholar]
- 24.Goto M, Ono T, Nakashio F, Hatton T A. Biotechol Bioeng. 1997;54:26–32. doi: 10.1002/(SICI)1097-0290(19970405)54:1<26::AID-BIT3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 25.Johnston K P, Harrison K L, Clarke M J, Howdle S M, Heitz M P, Bright F V, Carlier C, Randolph T W. Science. 1996;271:624–626. [Google Scholar]
- 26.Frank S G, Zografi G. J Colloid Interface Sci. 1969;29:27–35. doi: 10.1016/0021-9797(69)90342-7. [DOI] [PubMed] [Google Scholar]
- 27.Gale R W, Fulton J L, Smith R D. J Am Chem Soc. 1987;109:920–921. [Google Scholar]
- 28.Fulton J L, Smith R D. J Phys Chem. 1988;92:2903–2907. [Google Scholar]
- 29.Fulton J L, Blitz J P, Tingey J M, Smith R D. J Phys Chem. 1989;93:4198–4202. [Google Scholar]
- 30.Younglove B A, Ely J F. J Phys Chem Ref Data. 1987;16:577–769. [Google Scholar]
- 31.Weast R C. Handb Chem Phys. 1974;55:F49–F55. [Google Scholar]
- 32.Di Stefano D L, Wand A J. Biochemistry. 1987;26:7272–7282. doi: 10.1021/bi00397a012. [DOI] [PubMed] [Google Scholar]
- 33.Schneider D M, Dellwo M J, Wand A J. Biochemistry. 1992;31:3645–3652. doi: 10.1021/bi00129a013. [DOI] [PubMed] [Google Scholar]
- 34.Smith R D, Fulton J L, Blitz J P, Tingey J M. J Phys Chem. 1990;94:781–787. [Google Scholar]