

Abstract
Technologies designed to allow manipulation and modification of human embryonic stem (hES) cells are numerous and vary in the complexity of their methods, efficiency, reliability, and safety. The most commonly studied and practiced of these methods include electroporation, lipofection, nucleofection, and lentiviral transduction. However, at present, it is unclear which protocol offers the most efficient and reliable method of gene transfer to hES cells. In this study, a bi-fusion construct with ubiquitin promoter driving enhanced green fluorescent protein reporter and the firefly luciferase (pUb-eGFP-Fluc) along with neomycin selection marker was used for in vitro and in vivo studies. In vitro studies examined the transfection efficiency and viability of each technique using two hES cell lines (male H1 and female H9 cells). Lentiviral transduction demonstrated the highest efficiency (H1: 25.3 ± 4.8%; H9: 22.4 ± 6.5%) with >95% cell viability. Nucleofection demonstrated transfection efficiency of 16.1 ± 3.6% (H1) and 5.8 ± 3.2% (H9). However, minimal transfection efficiency was observed with electroporation (2.1 ± 0.4% (H1) and 1.9 ± 0.3% (H9)) and lipofection (1.5 ± 0.5% (H1) and 1.3 ± 0.2% (H9); P < 0.05 vs. lentiviral transduction). Electroporation also demonstrated the highest cell death (62 ± 11% (H1) and 42 ± 10% (H9)) followed by nucleofection (25 ± 9% (H1) and 30 ± 15 (H9)). Importantly, lentiviral transduction generated a greater number of hES cell lines stably expressing the double-fusion reporter gene (hES-DF) compared to other transfection techniques. Finally, following subcutaneous transplantation into immunodeficient nude mice, the hES-eGFP-Fluc cells showed robust proliferation as determined by longitudinal bioluminescence imaging. In summary, this study demonstrates that lentiviral transduction and nucleofection are efficient, simple, and safe techniques for reliable gene transfer in hES cells. The double-fusion construct provides an attractive approach for generating stable hES cell lines and monitoring engraftment and proliferation in vitro and in vivo.
Key Words: Human embryonic stem cell, Molecular imaging, Gene transfer, Plasmid transfection, Lentivirus transduction
Introduction
The study of embryonic stem (ES) cells will continue to yield important discoveries in the areas of cell differentiation and growth, tissue engineering, and regenerative medicine. The ES cells are derived from the inner cell mass of blastocyst embryos, have unlimited proliferation, and are the precursor to the three embryonic germ layers [1, 2]. To efficiently study ES cells and successfully expand their potential clinical applications, a rapid, reliable, and simple technique is needed to deliver genes to stem cells. Typically, gene delivery techniques have been divided into two categories: viral and non-viral gene delivery. Viral delivery uses the backbone of a viral genome, such as retrovirus, lentivirus, adenovirus, and adeno-associated virus (AAV), with insertion of the gene(s) of interest to be expressed in the stem cell. The more commonly used non-viral plasmid delivery systems include liposome, electroporation, and nucleofection-based, though newer techniques such as ultrasound [3, 4], microinjection [5], nuclear transfer [6, 7], and molecular-vibration-mediated delivery [8] have also been examined, among other techniques.
The advantages of using non-viral delivery compared to viral vector delivery include decreased immunogenicity and less insertional mutagenesis. However, the disadvantages of non-viral delivery may include decreased transfection efficiency and a shorter period of transgene expression which may limit long-term studies. For example, the transfection efficiency and viability of hES cells following electroporation has been shown to vary depending on the specific voltage and capacity settings [9, 10]. Cell survival rates following electroporation are typically low at 24 to 48 h and often not recorded, though range from >60% at 1 to 2 h [9], 73% at 2 to 3 h [11], to a median rate of 19% at 48 h [12].
In addition, the efficiency of gene transfer with cationic liposomes varies significantly based on the commercial preparation [13]. Liposome preparations may have different aliphatic chain lengths and structural compositions, as well as various associated anions. Nucleofection, a liposomal-based electroporation system that can be adapted with appropriate buffers for different cell lines, has demonstrated a significantly higher level of transfection efficiency, with results of 32% to 66% of viable H9.2 cells at 24 h [11] and 20% to 22% of viable H1 cells at 72 h [14]. The survival rate following nucleofection has been demonstrated at 74% with a control transfection at 48 h and 50% with transfection of GFP at 48 h [15]. Lastly, despite the disadvantages associated with the use of viral vectors for gene transfer, the lentiviral systems have reliably demonstrated efficient transduction into hES cells. Lentiviral transduction has demonstrated efficiencies of 20% at low viral titers to >80% at high viral titer [16, 17].
Although various studies have examined the gene-transfer efficiency to hES cells in vitro as highlighted above, few have focused on a direct comparison of these techniques. In this study, we examine lipofection, electroporation, nucleofection, and lentiviral transduction of H1 and H9 hES cell lines to identify the most efficient, reliable, and simple gene-transfer system for in vitro and in vivo applications. Using a double-fusion plasmid construct expressing eGFP and Fluc, the transfection efficiency was carefully demonstrated for each protocol. Stable cell lines expressing the bi-fusion construct were generated by lentiviral transduction and nucleofection. Various well-characterized stem cell markers, including SSEA-4 and Oct4, were assayed to ensure that the hES cells continued to express the appropriate markers following treatment. Finally, bioluminescence imaging was used to facilitate non-invasive monitoring of transplanted hES cells in vivo.
Materials and Methods
Culture of Undifferentiated hES Cells
H1 and H9 hES cells were obtained from Wicell (Madison, WI). hES cells were maintained on irradiated mouse embryonic fibroblasts (MEFs) at 5% CO2 in medium containing Knockout-Dulbecco’s modified Eagle medium (KO-DMEM), 20% serum replacement, 1% nonessential amino acids, 1 mM l-glutamine, 0.1 mM β-mercaptoethanol, and 4 ng/ml fibroblast growth factor-2 (FGF-2; GIBCO BRL). The cells were passaged in a 4-day cycle by incubating with 1 mg/ml collagenase IV at 37°C for 45 min, maintaining the cells in small clumps. To avoid contamination by MEFs in the transfection (liposome, electroporation, nucleofection) analysis, the hES cells were cultured on Matrigel in MEF-conditioned medium for one passage prior to transfection. Approximately, 2 × 104/cm2 cells were plated uniformly in six-well plates prior to transfection. Samples of the cells were immunostained with anti-SSEA-4 (Chemicon, Temecula, CA) and anti-Oct-4 (Santa Cruz Biotechnology, Santa Cruz, CA) antibodies. Immunofluorescent labeling was analyzed using a Zeiss LSM 510 Confocal Laser Scanning Microscope equipped with a Coherent Mira 900 tunable Ti:sapphire laser for two-photon excitation (Zeiss, Minneapolis, MN).
Construction of pUb-eGFP-Fluc and pUb-eGFP-Fluc-SV40-neomycin
The pUb-eGFP-Fluc construct used for lentiviral transduction has been described previously [18]. The pUb-eGFP-Fluc-SV40-neomycin was generated by cleaving pUb-GFP-Fluc at the BamH1 restriction enzymes and blunt-end ligating the SV40-neomycin construct. This plasmid was used for stable transfection of hES cells using neomycin as a selection marker.
Plasmid Transfection Methods
(a) Lipofection: lipofection was performed 1 day after plating H1 and H9 hES cells (4–5 × 105/well) onto Matrigel at a confluency of 60–70%. Qiagen lipofection 2000 was used according to the manufacturer’s protocol. Briefly, 5 μg of plasmid DNA (pUb-eGFP-Fluc) was diluted in 250 μl of KO-DMEM followed by addition of 10 μl of liposomal reagent. The mixture was incubated for 15–20 min at room temperature. The mixture was then added to 1,000 μl of pre-warmed conditioned medium and transferred to one well of a six-well plates. (b) Electroporation: 5 μg of plasmid DNA (pUb-eGFP-Fluc) was diluted in 500 μl of phosphate-buffered saline (PBS) and added to 4–5 × 105 hES H1 and H9 cells suspended in 500 μl of hES culture medium. Electroporation was performed in a 4-mm gap cuvette using a Gene Pulser (BioRad, München, Germany) with the electric parameters set at 300 V and 220 μF. After pulsing, the cells were incubated in the cuvette at room temperature for 10 min. The cells were transferred to new medium and plated onto Matrigel-coated six-well plates at a density of 2 × 105 cells/cm2. Each plate was analyzed for eGFP expression under a fluorescence microscope at 24, 48, and 72 h post-electroporation. (c) Nucleofection: 4-5 × 105 hES H1 and H9 cells were centrifuged for 5 min at 800 rpm and 4°C. The culture medium was removed and cells were resuspended in 100 μl of the nucleofector solution (Amaxa Biosystems, Cologne, Germany). DNA (5 μg) was added, cells and DNA were gently mixed, and cells were electroporated using the Nucleofector™ device (Amaxa Biosystems). Transfections were performed with a program (A23) that was selected in a series of pilot transfection experiments with the eGFP reporter gene and proven to result in the highest transfection efficiency. Directly after transfection, hES cells were placed into the culture medium.
Lentiviral Vector Preparation and Transduction
A self-inactivating lentivirus was prepared by transient transfection of 293 T cells as described previously [16, 19]. Briefly, 12 µg of the HIV-1 packaging plasmid (peGFP-Fluc/pRRE; delta-8.9) containing the eGFP-Fluc reporter gene were cotransfected into 293 T cells with 9 µg of vesicular stomatitis virus G glycoprotein-pseudotyped envelope vector (pVSV-G), and 6.25 µg of REV plasmid (pRSV-REV). Lentivirus supernatant was collected after 72 h and concentrated by sediment centrifugation with SW29 rotor at 50,000×g for 2 hours. Concentrated virus was titrated on 293 T cells. hES H1 and H9 cells (4–5 × 105) were incubated with virus particles at MOI of 10 and 8 µg/ml polybrene (Sigma). The media was changed after 12 h by fresh medium.
Generation of Stable Cell Line
Stable hES cell lines were generated by fluorescence-activated cell sorting (FACS) in the lentiviral transduction group and by G418 selection (50–100 µg/ml) over 2 weeks in the nucleofection group. Colonies were isolated, propagated, and analyzed for eGFP expression and Fluc activity.
Cell Viability Analysis
Cell viability was determined using dissociated cells prior to plating, as well as at 24 and 48 h after transfection. Viability was measured using trypan blue exclusion and expressed as a percentage of the initial number of cells used for each transfection.
Immunohistochemistry
The H1 and H9 hES cells treated with the various transfection protocols were plated onto cover slips within six-well plates, fixed in 5% paraformaldehyde for 1 min, and blocked with 1% bovine serum albumin in PBS. They were incubated with the primary antibody in PBS at 37°C for 1 h, followed by several PBS washes, and incubation with a secondary antibody at 37°C for 1 h, with final PBS washes. Slides were mounted with Vectorshield (Vector Laboratories, Inc., Burlingame, CA, USA).
Fluorescence-activated Cell Sorter Analysis
Twenty-four hours after transfection, the cells were washed twice with KO-DMEM and dissociated with Accutase II. After centrifugation, the cells were resuspended at 1 × 106 cells/ml in PBS and stored on ice for a maximum of 1 h before analysis. Acquisition was performed on a FACS Calibur system (BD Biosciences, Heidelberg, Germany) and samples were analyzed using FlowJo software (Tree Star Inc., Ashland, OR). Analysis-gating criteria for eGFP-expressing cells were set according to the level of auto-fluorescence of a non-transfected control.
Transplantation of hES Cells into Nude Mice
Harvested control hES and stable hES-DF cells (transduced with lentivirus) were kept on ice for <30 min for optimal viability. Adult immunodeficient nu/nu mice (n = 5) were injected subcutaneously with 1 × 106 hES-DF cells in 50 µL of PBS. Control animals (n = 3) received 1 × 106 non-transduced hES cells. All animals had an uneventful recovery and underwent bioluminescence imaging at various time points. Study protocols were approved by the Stanford Animal Research Committee.
Optical Bioluminescence Imaging of hES Cell Transplantation
Bioluminescence imaging was performed with the Xenogen In Vivo Imaging System (IVIS, Alameda, CA). Following intraperitoneal injection of the reporter probe d-luciferin (375 mg/kg body weight), animals were imaged for 30 min with 1-min acquisition intervals. The same mice were scanned at various time points for a 6-week period according to the specific study design. Bioluminescence was quantified in units of maximum photons per second per centimeter squared per steradian (P s−1 cm−2 sr−1), as described previously [18].
Statistical Analysis
Quantitative data were expressed as mean ± SD. Means were compared using one-way ANOVA and the Student t test. P values of less than 0.05 were considered statistically significant.
Results
hES Cells Maintained on a Feeder-free Culture System Express Stem Cell Markers
Previous studies have detailed the need to separate hES cells from contaminant feeder layer cells prior to the initiation of studies [20]. For our studies, the H9 and H1 cells were reseeded onto Matrigel-coated plates to eliminate any possible contaminating effects of the MEF feeder layer. Additional studies were performed on these hES cells to confirm that separation from the feeder layer had not changed the basic characteristics of the cells. The cell doubling time was tested by cell counting, which revealed similar results between the hES cells on feeder layer and feeder-free plates (37 ± 3.4 h vs. 36 ± 2.1 h; P = NS). The two cell lines showed the same morphology under brightfield microscopy (Fig. 1). Immunohistochemistry staining also showed similar expression pattern of Oct-4 and SSEA-4 under fluorescence microscopy for H9 and H1 hES cells. These results confirm that H9 and H1 hES cells grown in a feeder-free cell culture system maintained similar growth and surface marker expression characteristics compared to hES cells grown on conventional feeder layers.
Fig. 1.
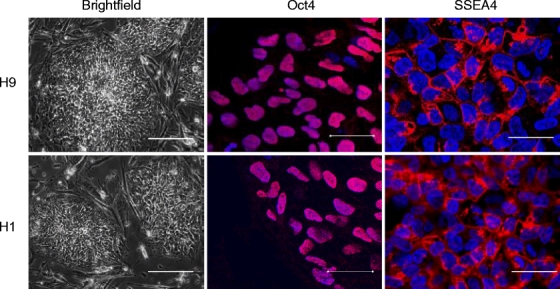
Immunohistochemistry of stem cell markers on undifferentiated H9 and H1 cell lines grown in feeder-cell free cultures in vitro. H9 and H1 cells were reseeded onto Matrigel-coated plates. Brightfield and immunohistochemistry staining with fluorescence microscopy for Oct-4 and SSEA-4 stem cell markers was performed. DAPI staining is used as a nuclear marker (scale bar = 50 µm in mid- and right fluorescence staining; 200 µm in left brightfield).
Lentivirus transduction and Plasmid Nucleofection Demonstrate Higher Efficiencies in vitro
Following a review of the literature (previously detailed) [11, 21–23], transfection conditions yielding the highest efficiencies for hES cells were utilized for each transfection protocol. Within 24 h, there was a significant difference in gene-transfer efficiencies in H9 and H1 cells. In particular, lentiviral transduction demonstrated the highest level of Fluc gene expression, with bioluminescence measurements of 0.95 ± 0.13 × 106 photons/s for H9 and 1.37 ± 0.16 × 106 for H1. Nucleofection demonstrated lower transfection efficiency compared to lentiviral transduction (3.2 ± 3.9 × 105 photons/s for H9 and 5.6 ± 0.12 × 105 for H1; P < 0.05 for both). Importantly, both electroporation and lipofection had very low transfection efficiencies compared to lentiviral transduction and nucleofection (Fig. 2). In our studies, the H1 cell line had higher transfection efficiency than the H9 hES cell line for all protocols tested, suggesting varying susceptibility to gene transfer in different hES lines.
Fig. 2.
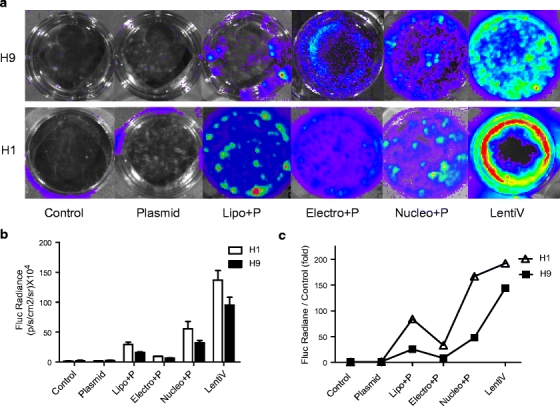
Transfection and transduction of H9 and H1 hES cells in vitro with each of the transfection protocols. (a) H9 and H1 cells were seeded onto feeder-cell free Matrigel-coated six-well plates after transfection with plasmid alone (Plasmid), lipofection (Lipo+P), electroporation (Electro+P), nucleofection (Nucleo+P), and transduction with lentivirus (LentiV). (b) The cells were examined using bioluminescence imaging of Fluc activities after transfection. The quantitative measurements are shown 24 h after gene delivery.
FACS Analysis of eGFP Expression in H1 and H9 hES Cell Lines
The double-fusion construct allows us to simultaneously analyze eGFP by FACS and Fluc by bioluminescence imaging. Consistent with the Fluc bioluminescence activity shown above, the eGFP expression was highest in lentiviral-transduced cells followed by nucleofection-transfected cells (Fig. 3). Lentiviral transduction demonstrated the highest transduction efficiency, with 25.3 ± 4.8% and 22.4 ± 6.5% positive eGFP cells for H1 and H9, respectively. Nucleofection demonstrated transfection efficiency of 16.1 ± 3.6% (H1) and 5.8 ± 3.2% (H9). Low eGFP expression was again seen in electroporation (2.1 ± 0.4% (H1) and 1.9 ± 0.3% (H9)) and lipofection (1.5 ± 0.5% (H1) and 1.3 ± 0.2% (H9)).
Fig. 3.
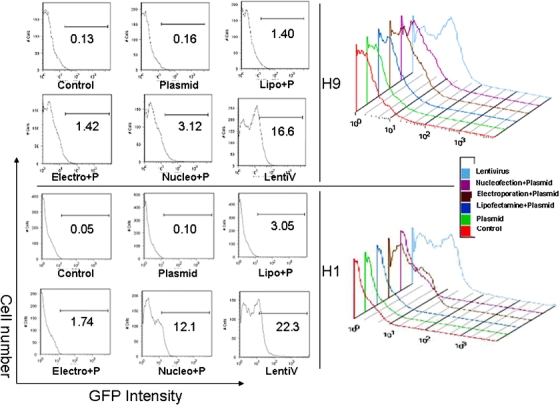
Measurement of eGFP expression using flow cytometry analysis of H1 and H9 hES cell lines transfected with each protocol. The bi-fusion pUb-eGFP-Fluc construct was transfected into H9 and H1 hES cells using lipofection (Lipo+P), electroporation (Electro+P), nucleofection (Nucleo+P), and transduction with lentivirus (LentiV). eGFP expression was measured using FACS analysis.
Higher Cell Viability Demonstrated in lentiviral-transduced Cells
The protocols and expertise to isolate and grow sufficient quantities of hES cells are complex and technically challenging. Due to this laborious process, a high hES cell viability following gene transfer is essential. It is commonly known that electroporation of hES cells results in significant cell death, despite efforts to optimize and minimize the energy delivered [9]. The use of lentivirus, on the other hand, has previously demonstrated high levels of cell viability following transduction [16, 17]. To address this issue, we examined the cell morphology and assessed the cell viability using trypan blue exclusion at 24 h following gene transfer with the various protocols. The H1 and H9 cells were examined by overlaying brightfield microscopy with eGFP fluorescence. H9 cells clearly demonstrate significantly lower viability in electroporation treated cells (Fig. 4), followed by nucleofection-treated cells. Similar results were obtained for H1 cells (data not shown). The trypan blue assay details the cell viability following electroporation at 62 ± 11% (H1) and 42 ± 10% (H9)) followed by nucleofection at 25 ± 9% (H1) and 30 ± 15% (H9) at 24 h. By contrast, the other four groups all showed greater than 95% viability.
Fig. 4.
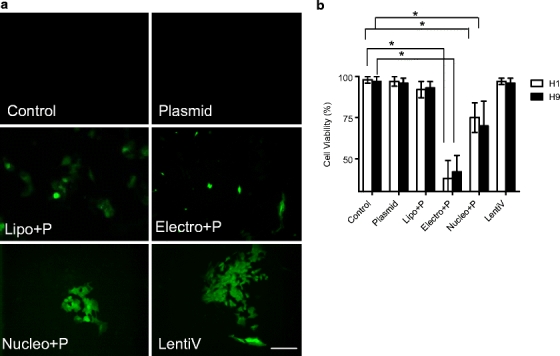
Viability of H9 and H1 hES cells in vitro following transfection with the various gene-transfer protocols. H9 and H1 hES cells were examined at 24 h following transfection with lipofection (Lipo+P), electroporation (Electro+P), nucleofection (Nucleo+P) and transduction with lentivirus (LentiV). Cell morphology and trypan blue exclusion assay was performed to measure cell viability. (a) Fluorescence microscopy of representative H1 cells. Scale bar = 100 µm. (b) Viability of each transfected cell lines at 24 h as measured using the trypan blue assay.
In vivo Imaging to Monitor hES Cell Survival, Proliferation, and Teratoma Formation
The ability to monitor transplanted hES cells in vivo without sacrificing the animals has been made possible by incorporating newer imaging technologies such as optical bioluminescence or positron-emission tomography [24]. Following subcutaneous transplantation, bioluminescence imaging was performed on days 2, 7, 14, 35, 42, and 56 (Fig. 5a). H9-DF cells (via stable lentiviral transduction) showed persistent engraftment and survival as measured by bioluminescence activities: day 2 (55.2 ± 14.3 × 106), day 7 (21.2 ± 6.1 × 106), day 14 (17.2 ± 5.0 × 106), day 35 (19.5 ± 3.2 × 106), day 42 (28.1 ± 7.2 × 106), and day 56 (65.3 ± 14.1 × 106 p/s/cm2/sr). Control H9 cells (not expressing the GFP-Fluc) cannot be detected as expected. Following imaging at day 56, animals were sacrificed and the teratoma explanted. Histologic analysis of control hES and hES-DF teratomas shows mixed regions of endodermal, mesodermal, and ectodermal differentiation (Fig. 5b). Thus, the histological data suggest that lentiviral transduction did not significantly affect the differentiation of hES cells into the three germ layer cell types in vivo.
Fig. 5.
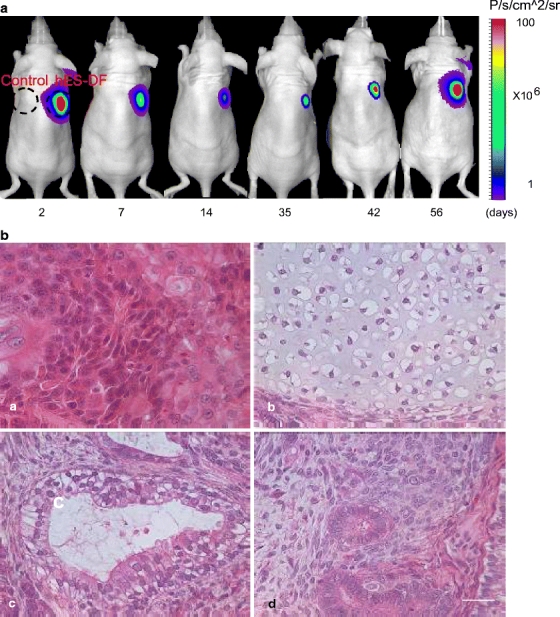
Optical bioluminescence imaging of stably expressed hES cells in vivo over time, with demonstration of proliferation and teratoma formation. The pUb-eGFP-Fluc-SV40-Puro construct was stably expressed in H9 cells by transduction with replication-incompetent lentivirus. Stable colonies (hES-DF) were selected by drug selection over 2 weeks and then injected subcutaneously into right shoulder of nude mice. Control untransduced H9 hES cells were injected into left shoulder. (a) In vivo bioluminescence imaging of the hES-DF cells on days 2, 7, 14, 35, 42, and 56. (b) Histology of the H9 hES cells stably expressing eGFP-Fluc in nude mice were examined through 56 days. Teratoma formation was demonstrated by histology at week 8 weeks after subcutaneous injection: (A) rosette consistent with neuroectodermal differentiation (ectoderm), (B) cartilage formation (mesoderm), (C) respiratory epithelium with ciliated columnar, and (D) mucin-producing goblet cells (endoderm) surrounded with mesenchymal cells (mesoderm). Scale bar = 50 µm.
Discussion
To date, the ability to manipulate stem cells for clinical applications has been limited by techniques in basic molecular and cellular biology, as well as our understanding of ES cell biology. Protocols for gene transfer that had demonstrated significant promise in mouse embryonic stem (mES) cells, such as electroporation and lipofection, are less efficient in hES cells [12]. In general, mES cells have demonstrated higher levels of transfection efficiency, whereas hES cells are notoriously difficult to transfect. This has led to optimizing existing protocols, as well as the discovery of novel and cutting-edge techniques that have increased transfection efficiency into hES cells. It is important that if these protocols are to be used routinely as an efficient, simple, and reproducible method of gene transfer, the effect on hES cell growth and differentiation should not be affected, and transgene expression should remain intact over prolonged periods of time. Using a double-fusion reporter gene that allows for non-invasive “real-time” imaging, we have examined the transfection and transduction efficiency of various protocols on the H1 and H9 hES cell lines in vitro and in vivo.
Few studies have performed side-by-side comparison of the efficiency of the various transfection and transduction methods available for gene transfer to hES cells. Nucleofection of hES cells has shown superior transfection efficiency compared to lipofection and electroporation for H9.2 cells [11], and in adipose-tissue-derived stem cells [21]. We have examined the protocols from these studies, as well as numerous other studies to identify the optimal experimental conditions for electroporation, lipofection, nucleofection, and lentiviral transduction using male H1 and female H9 cells, and have compared their transfection or transduction efficiency in vitro. The double-fusion construct (expressing eGFP and the Fluc) allowed for rapid, reproducible, and real-time monitoring of cells over an extended period of time. Lentiviral transduction demonstrated the highest level of gene transfer (22–25%) with the best viability following treatment (>95%). The highest efficiency using a non-viral protocol was demonstrated by nucleofection, though with a mildly decreased level of viability (70–75%). Electroporation and lipofection demonstrated very low levels of transfection efficiency, which is consistent with previous studies reported [23]. Viability following treatment with electroporation was poor (20–25%) despite attempts to optimize the specific voltage and capacity settings. For each modality, expression of the eGFP-Fluc double-fusion reporter gene did not appear to affect cell growth and viability.
Previous studies have examined transient transfection of various hES cell lines in vitro, and production of stable human ES cell lines [16, 23, 25]. However, few studies have generated stable hES cell lines and monitored their growth in vivo using noninvasive bioluminescence imaging. In this study, we have also generated stable cell lines with lentiviral transduction and plasmid nucleofection protocols using the double-fusion (DF) reporter gene construct and monitored their growth and differentiation in vivo over several months. Immunofluorescence and histological analysis demonstrates that the stable hES cell lines consist of undifferentiated cells that express numerous stem cell markers, including SSEA4 and Oct4. After several weeks, these undifferentiated cells develop into subcutaneous teratomas. In addition, the DF reporter gene did not affect hES cell viability, growth, and differentiation, similar to previous report on mES cells [24]. Of note, the higher number of stable cell lines (~twofold higher) generated by lentiviral transduction compared to nucleofection is likely secondary to a higher gene-transfer rate, in combination with a higher rate of viability.
Transfection and transduction protocols that can successfully transfer genes to the nucleus in a cell-cycle-independent manner will be most effective in hES cells. Lipofection (including Fugene and Exgen 500), transduction with many pseudotyped retroviral constructs (e.g., murine leukemia virus), and polymer- or polycation-based protocols require cell proliferation (mitosis) and breakdown of the nuclear envelope, or nuclear localization signal (NLS) directing transport from the cytoplasm to the nucleus. The trafficking of gene products to the nucleus has been challenging, with mixed success using well-characterized NLS signals, such as the SV40 NLS for example [26–28]. By contrast, lentivirus, adenovirus, and AAV can infect both dividing and non-dividing cells, have low cytogenicity, and can integrate their genome into the host chromosome. The use of vectors derived from retroviruses as gene-transfer vehicles in this setting has had limited success because of silencing of transgene expression. Two major mechanisms have been identified for retrovirus silencing: trans-acting factors that bind to the viral promoters in the long terminal repeats (LTRs) and methylation of the integrated retroviral genome and flanking host DNA sequences [29]. Similar to the lentiviral system, electroporation is independent of cell cycle and can directly transport DNA to the nucleus. Despite efforts to optimize the specific voltage and capacity settings, survival rates and levels of transfection efficiency in hES cells have been poor. Nucleofection, on the other hand, has demonstrated higher levels of transfection efficiency with significantly improved levels of cell viability over electroporation. This technique has demonstrated the ability to ubiquitously transfect various cell lines [30, 31], including CD34+ cells and H1 cells [14]. In this study, the nucleofector settings and buffer were selected based on previous studies in our lab, as well as other published reports [27, 28]. The ability to adapt the nucleofection parameters and buffers to various primary cell lines may further improve transfection efficiency. In addition, other studies using nucleofection have demonstrated efficient expression of siRNA [32], mRNA [22, 33], and various small and macromolecules [9].
In this study, both lentiviral transduction and plasmid nucleofection protocols successfully generated stable hES clones that stably expressed the DF reporter gene. Although lentivirus generated higher numbers of stable clones, nucleofection is a simple, reproducible, and efficient technique for transient as well as stable transfection. The double-fusion construct allowed for effective, non-invasive tracking of proliferating hES cells in nude mice, with no noticeable disruption of cell differentiation during prolonged gene expression. The lack of significant transgene silencing following in vivo transplantation has been previously reported [24].
In summary, the ability to study the biology of hES cells requires genetic manipulation to express various transgenes. Nucleofection is an attractive transfection protocol with minimal safety concerns. It is a simple and efficient system and can be used to generate hES cell lines stably expressing reporter genes. Although this protocol can generate stable hES cell lines, the lentiviral system is more efficient and could still play a role in various applications.
Acknowledgments
This work was supported by CIRM RS1-00322, NIH HL089027 (JCW) as well as Shanxi Scientific and Technical Key Project (2008K13-01), NIH HL091453, and Xijing Research Boosting Program XJZT08Z04 (FC).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Thomson J, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;6(282):1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 2.Reubinoff B, Pera MF, Fong CY, Trounson A, Bongso A. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat Biotechnol. 2000;18:399–404. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]
- 3.Unger E, Hersh E, Vannan M, McCreery T. Gene delivery using ultrasound contrast agents. Echocardiography. 2001;18:355–361. doi: 10.1046/j.1540-8175.2001.00355.x. [DOI] [PubMed] [Google Scholar]
- 4.Nakashima M, Tachibana K, Iohara K, Ito M, Ishikawa M, Akamine A. Induction of reparative dentin formation by ultrasound-mediated gene delivery of growth/differentiation factor 11. Hum Gene Ther. 2003;10(14):591–597. doi: 10.1089/104303403764539369. [DOI] [PubMed] [Google Scholar]
- 5.Wakayama T, Rodriguez I, Perry AC, Yanagimachi R, Mombaerts P. Mice cloned from embryonic stem cells. Proc Natl Acad Sci U S A. 1999;21(96):14984–14989. doi: 10.1073/pnas.96.26.14984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheong H, Takahashi Y, Kanagawa H. Birth of mice after transplantation of early cell-cycle-stage embryonic nuclei into enucleated oocytes. Biol Reprod. 1993;48:958–963. doi: 10.1095/biolreprod48.5.958. [DOI] [PubMed] [Google Scholar]
- 7.Eggan K, Akutsu H, Loring J, Jackson-Grusby L, Klemm M. Hybrid vigor, fetal overgrowth, and viability of mice derived by nuclear cloning and tetraploid embryo complementation. Proc Natl Acad Sci U S A. 2001;22(98):6209–6214. doi: 10.1073/pnas.101118898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song L, Chau L, Sakamoto Y, Nakashima J, Koide M, Tuan R. Electric field-induced molecular vibration for noninvasive, high-efficiency DNA transfection. Mol Ther. 2004;9:607–616. doi: 10.1016/j.ymthe.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 9.Mohr J, de Pablo J, Palecek S. Electroporation of human embryonic stem cells: Small and macromolecule loading and DNA transfection. Biotechnol Prog. 2006;22:825–834. doi: 10.1021/bp0600334. [DOI] [PubMed] [Google Scholar]
- 10.Sukhorukov V, Reuss R, Zimmermann D, Held C, Muller KJ, Kiesel M, Gessner P, Steinbach A, Schenk WA, Bamberg E, Zimmermann U. Surviving high-intensity field pulses: strategies for improving robustness and performance of electrotransfection and electrofusion. J Membr Biol. 2005;206:187–201. doi: 10.1007/s00232-005-0791-2. [DOI] [PubMed] [Google Scholar]
- 11.Siemen H, Nix M, Endl E, Koch P, Itskovitz-Eldor J, Brustle O. Nucleofection of human embryonic stem cells. Stem Cells Dev. 2005;14:378–383. doi: 10.1089/scd.2005.14.378. [DOI] [PubMed] [Google Scholar]
- 12.Weissinger F, Reimer P, Waessa T, Buchhofer S, Schertlin T, Kunzmann V, Wilhelm M. Gene transfer in purified human hematopoietic peripheral-blood stem cells by means of electroporation without prestimulation. J Lab Clin Med. 2003;141:138–149. doi: 10.1067/mlc.2003.14. [DOI] [PubMed] [Google Scholar]
- 13.Floch V, Le Bolc'h G, Audrezet MP, Yaouanc JJ, Clement JC, Des Abbayes H, Mercier B, Abgrall JF, Ferec C. Cationic phosphonolipids as non viral vectors for DNA transfection in hematopoietic cell lines and CD34+ cells. Blood Cells Mol Dis. 1997;23:69–87. doi: 10.1006/bcmd.1997.0123. [DOI] [PubMed] [Google Scholar]
- 14.Lakshmipathy U, Pelacho B, Sudo K, Linehan J, Coucouvanis E, Kaufman D, Verfaillie C. Efficient transfection of embryonic and adult stem cells. Stem Cells. 2004;22:531–543. doi: 10.1634/stemcells.22-4-531. [DOI] [PubMed] [Google Scholar]
- 15.von Levetzow G, Spanholtz J, Beckmann J, Fischer J, Kogler G, Wernet P, Punzel M, Giebel B. Nucleofection, an efficient nonviral method to transfer genes into human hematopoietic stem and progenitor cells. Stem Cells Dev. 2006;15:278–285. doi: 10.1089/scd.2006.15.278. [DOI] [PubMed] [Google Scholar]
- 16.Ma Y, Ramezani A, Lewis R, Hawley R, Thomson J. High-level sustained transgene expression in human embryonic stem cells using lentiviral vectors. Stem Cells. 2003;21:111–117. doi: 10.1634/stemcells.21-1-111. [DOI] [PubMed] [Google Scholar]
- 17.Gropp M, Itsykson P, Singer O, Ben-Hur T, Reinhartz E, Galun E, Reubinoff B. Stable genetic modification of human embryonic stem cells by lentiviral vectors. Mol Ther. 2003;7:281–287. doi: 10.1016/S1525-0016(02)00047-3. [DOI] [PubMed] [Google Scholar]
- 18.Cao F, Drukker M, Lin S, Sheikh AY, Xie X, Li Z, Connolly AJ, Weissman IL, Wu JC. Molecular imaging of embryonic stem cell misbehavior and suicide gene ablation. Cloning Stem Cells. 2007;9:107–117. doi: 10.1089/clo.2006.0E16. [DOI] [PubMed] [Google Scholar]
- 19.Follenzi A, Naldini L. HIV-based vectors. Preparation and use. Methods Mol Med. 2002;69:259–274. [PubMed] [Google Scholar]
- 20.Xu C, Inokuma MS, Denham J, Golds K, Kundu P, Gold JD, Carpenter MK. Feeder-free growth of undifferentiated human embryonic stem cells. Nat Biotechnol. 2001;19:971–974. doi: 10.1038/nbt1001-971. [DOI] [PubMed] [Google Scholar]
- 21.Zaragosi L, Billon N, Ailhaud G, Dani C. Nucleofection is a valuable transfection method for transient and stable transgene expression in adipose tissue-derived stem cells. Stem Cells. 2007;25:790–797. doi: 10.1634/stemcells.2006-0235. [DOI] [PubMed] [Google Scholar]
- 22.Ponsaerts P, van der Sar S, Van Tendeloo V, Jorens P, Berneman Z, Singh P. Highly efficient mRNA-based gene transfer in feeder-free cultured H9 human embryonic stem cells. Cloning Stem Cells. 2004;6:211–216. doi: 10.1089/clo.2004.6.211. [DOI] [PubMed] [Google Scholar]
- 23.Kim J, Do H-J, Choi S-J, Cho H-J, Park K-H, Yang H-M, Lee S-H, Kim D-K, Kwack K, Oh S-K, et al. Efficient gene delivery in differentiated human embryonic stem cells. Exp Mol Med. 2005;28(37):36–44. doi: 10.1038/emm.2005.5. [DOI] [PubMed] [Google Scholar]
- 24.Cao F, Lin S, Xie X, Ray P, Patel M, Zhang X, Drukker M, Dylla S, Connolly A, Chen X, et al. In vivo visualization of embryonic stem cell survival, proliferation, and migration after cardiac delivery. Circulation. 2006;113:1005–1014. doi: 10.1161/CIRCULATIONAHA.105.588954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hohenstein K, Pyle A, Chern J, Lock L, Donovan P. Nucleofection mediates high-efficiency stable gene knockdown and transgene expression in human embryonic stem cells. Stem Cells. 2008;26:1436–43. doi: 10.1634/stemcells.2007-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Aa MA, Koning GA, d'Oliveira C, Oosting RS, Wilschut KJ, Hennink WE, Crommelin DJ. An NLS peptide covalently linked to linear DNA does not enhance transfection efficiency of cationic polymer based gene delivery systems. J Gene Med. 2005;7:208–217. doi: 10.1002/jgm.643. [DOI] [PubMed] [Google Scholar]
- 27.Ciolina C, Byk G, Blanche F, Thuillier V, Scherman D, Wils P. Coupling of nuclear localization signals to plasmid DNA and specific interaction of the conjugates with importin alpha. Bioconjug Chem. 1999;10:49–55. doi: 10.1021/bc980061a. [DOI] [PubMed] [Google Scholar]
- 28.Neves C, Escriou V, Byk G, Scherman D, Wils P. Intracellular fate and nuclear targeting of plasmid DNA. Cell Biol Toxicol. 1999;15:193–202. doi: 10.1023/A:1007693805849. [DOI] [PubMed] [Google Scholar]
- 29.Pfeifer A, Ikawa M, Dayn Y. Verma IM: transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. PNAS USA. 2002;99(4):2140–5. doi: 10.1073/pnas.251682798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gresch O, Engel FB, Nesic D, Tran TT, England HM, Hickman ES, Korner I, Gan L, Chen S, Castro-Obregon S, et al. New non-viral method for gene transfer into primary cells. Methods. 2004;33:151–163. doi: 10.1016/j.ymeth.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Hamm A, Krott N, Breibach I, Blindt R, Bosserhoff A. Efficient transfection method for primary cells. Tissue Eng. 2002;8:235–245. doi: 10.1089/107632702753725003. [DOI] [PubMed] [Google Scholar]
- 32.Vallier L, Rugg-Gunn P, Bouhon I, Andersson F, Sadler A, Pedersen R. Enhancing and diminishing gene function in human embryonic stem cells. Stem Cells. 2004;22:2–11. doi: 10.1634/stemcells.22-1-2. [DOI] [PubMed] [Google Scholar]
- 33.Wiehe J, Ponsaerts P, Rojewski MT, Homann JM, Greiner J, Kronawitter D, Schrezenmeier H, Hombach V, Wiesneth M, Zimmermann O, Torzewski J. mRNA-mediated gene delivery into human progenitor cells promotes highly efficient protein expression. J Cell Mol Med. 2007;11:521–530. doi: 10.1111/j.1582-4934.2007.00038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]